

Abstract
Across eukaryotes, mitochondria exhibit staggering diversity in genomic architecture, including the repeated evolution of multichromosomal structures. Unlike in the nucleus, where mitosis and meiosis ensure faithful transmission of chromosomes, the mechanisms of inheritance in fragmented mitochondrial genomes remain mysterious. Multichromosomal mitochondrial genomes have recently been found in multiple species of flowering plants, including Silene noctiflora, which harbors an unusually large and complex mitochondrial genome with more than 50 circular-mapping chromosomes totaling ∼7 Mb in size. To determine the extent to which such genomes are stably maintained, we analyzed intraspecific variation in the mitochondrial genome of S. noctiflora. Complete genomes from two populations revealed a high degree of similarity in the sequence, structure, and relative abundance of mitochondrial chromosomes. For example, there are no inversions between the genomes, and there are only nine SNPs in 25 kb of protein-coding sequence. Remarkably, however, these genomes differ in the presence or absence of 19 entire chromosomes, all of which lack any identifiable genes or contain only duplicate gene copies. Thus, these mitochondrial genomes retain a full gene complement but carry a highly variable set of chromosomes that are filled with presumably dispensable sequence. In S. noctiflora, conventional mechanisms of mitochondrial sequence divergence are being outstripped by an apparently nonadaptive process of whole-chromosome gain/loss, highlighting the inherent challenge in maintaining a fragmented genome. We discuss the implications of these findings in relation to the question of why mitochondria, more so than plastids and bacterial endosymbionts, are prone to the repeated evolution of multichromosomal genomes.
Keywords: meiosis, mitochondrial DNA, mitosis, multichromosomal, multipartite
The diversity in mitochondrial genome architecture across eukaryotes (1, 2) tells a history of highly divergent evolutionary trajectories since the origin of mitochondria from an alphaproteobacterial endosymbiont (3–5). At one extreme, many Jakobid protists have highly conserved mitochondrial genome architectures with reduced but still very much bacterial-like genomes (6, 7). In contrast, mitochondrial genomes in many other eukaryotes have been radically transformed or even lost entirely. One recurring evolutionary pattern is the fragmentation of mitochondrial DNA (mtDNA) into multichromosomal genomes, which has occurred independently in a wide variety of taxa, including kinetoplastids (8), diplonemids (9), dinoflagellates (10), the green alga Polytomella parva (11), ichthyosporean protists (12), chytrid fungi (13), and multiple lineages of both metazoans (14–20) and angiosperms (21–23). The fragmentation of genomes into chromosomes is a very general property of eukaryotic nuclear genomes. In the nucleus, the complex molecular machinery of mitosis and meiosis ensures the reliable partitioning of nuclear chromosomes into daughter cells. By contrast, little is known about the basic mechanisms of inheritance and evolutionary maintenance of multichromosomal mitochondrial genomes.
Flowering plants are a prime example of a eukaryotic lineage with extreme and highly diverse mitochondrial genome structures. Angiosperm mitochondrial genomes are characterized by a number of unusual properties, including their large and variable sizes (ranging from ∼200 kb to over 10 Mb), low gene densities, extensive posttranscriptional modifications, and propensity to capture foreign DNA sequences (24, 25). They generally exhibit very low rates of nucleotide substitution, but their structures are highly dynamic, varying extensively among and even within species as a result of frequent rearrangements and gain/loss of intergenic sequences (26–30). In most angiosperm mitochondria, there is also a high frequency of recombination between large repeated sequences within the genome, interconverting between a suite of alternative conformations that coexist within an individual (31). As a result, angiosperm mitochondrial genomes typically produce multipartite maps that can be represented either as a single “master circle” or as a set of subgenomes (32, 33).
Multichromosomal mitochondrial genomes have recently been identified in three independent angiosperm lineages (21–23). These genomes differ from the typical multipartite mitochondrial genome structures in angiosperms in that at least some of the chromosomes do not share large recombining repeats with the other chromosomes and are, therefore, considered to be autonomous. These genomes cannot be mapped as a single master circle. The first multichromosomal mitochondrial genome to be identified in angiosperms was found in cucumber (Cucumis sativus), in which two small circular-mapping chromosomes (45 and 84 kb) were assembled independently from the main 1.6-Mb chromosome (21). A multichromosomal mitochondrial genome has since been identified in Amborella trichopoda (23). The most extreme examples of multichromosomal mitochondrial genomes have been found within the genus Silene (Caryophyllaceae). In this genus, species with accelerated rates of mitochondrial substitution harbor exceptionally large mitochondrial genomes that have been fragmented into dozens of circular-mapping chromosomes (22). For example, the sequenced mitochondrial genome of Silene noctiflora (6.7 Mb) is larger than most free-living bacterial genomes and consists of 59 chromosomes ranging in size from 66 to 192 kb. Remarkably, many of these chromosomes do not contain a single identifiable gene. It is, therefore, unclear what functional significance they might have and whether they are maintained by selection.
One impediment to understanding the ongoing evolutionary forces that act on multichromosomal mitochondrial genomes is the lack of data on intraspecific variation in the sequence and structure of these genomes. Here, we used a combination of whole-genome sequencing and quantitative PCR (qPCR) to identify natural variation in the extreme mitochondrial genome of S. noctiflora. Although we found little variation in sequence, structure, or relative abundance of most chromosomes, we show that mitochondrial genomes within this species differ by the presence or absence of entire chromosomes. In light of this surprising finding, we discuss hypotheses for the origins and ongoing evolution of the extreme mitochondrial genome architecture in S. noctiflora and implications for the repeated evolution of multichromosomal mitochondrial genomes throughout eukaryotes.
Results
The S. noctiflora Blue Ridge Parkway Mitochondrial Genome.
Mitochondrial DNA was isolated from a single maternal family of S. noctiflora derived from a collection on the Blue Ridge Parkway in Virginia, United States (hereafter, referred to as BRP). The genome was sequenced with a 454 shotgun library (493,183 reads, 184.7 Mb) and a 454 paired-end library with an average span of 2.8 kb (153,974 reads, 34.0 Mb). De novo genome assembly revealed an enormous mitochondrial genome that has been fragmented into dozens of circular-mapping chromosomes. The assembled BRP mitochondrial genome is 7,137,666 bp in length and comprises 63 chromosomes ranging in size from 64,875 bp to 191,194 bp (Table S1). The genome contains 32 protein, tRNA, and rRNA genes (Table 1), but many of these genes are present in multiple copies (Table S2), resulting in a total gene count of 54.
Table 1.
Comparison of S. noctiflora BRP and OSR mitochondrial genomes
| BRP | OSR | |
| Genome size, bp | 7,137,666 | 6,727,869 |
| No. of chromosomes | 63 | 59 |
| Empty chromosomes | 25 | 20 |
| Unique chromosomes | 11 | 8 |
| GC content, % | 40.82 | 40.77 |
| Total no. of genes | 32 (54) | 32 (54) |
| Protein genes | 26 (32) | 26 (33) |
| rRNA genes | 3 (16) | 3 (15) |
| tRNA genes | 3 (6) | 3 (6) |
| Total no. of introns | 18 | 18 |
| cis-Spliced introns | 12 | 12 |
| trans-Spliced introns | 6 | 6 |
| Repetitive content, % | 12.1 | 10.9 |
Gene counts are reported as the number of unique genes annotated in each genome, with the total gene number (including duplicates) indicated in parentheses.
The BRP mitochondrial genome was compared with a previously published mitochondrial genome from S. noctiflora (22). The original genome was derived from a roadside population of S. noctiflora on Old Schoolhouse Road (OSR) in Eggleston, VA. It was sequenced with a combination of 454 and Illumina technologies and assembled de novo with the same methodology applied in this study.
S. noctiflora OSR and BRP Mitochondrial Genomes Vary in the Presence/Absence of Entire Chromosomes.
The two sequenced mitochondrial genomes from S. noctiflora (OSR and BRP) differ in their number of chromosomes (59 and 63, respectively). They share 50 chromosomes with conserved synteny (Fig. 1). In addition, the entire content of OSR chromosome 7 is present in the BRP genome but divided between two chromosomes, BRP 25 and BRP 33 (with the latter also containing 78.8 kb of additional sequence that is not found in the OSR mitochondrial genome). The remaining 8 chromosomes in OSR and 11 chromosomes in BRP are unique to their respective genomes (Fig. 1).
Fig. 1.

Comparison of S. noctiflora OSR and BRP mitochondrial genomes. Blocks on the outer ring represent the individual chromosomes in each genome, with the width and height of the blocks representing chromosome length and sequencing coverage depth, respectively. Chromosomes are ordered in decreasing size from top to bottom. The internal ribbons connect regions of homologous sequence shared between the two genomes. Chromosomes shown in green and red are conserved between OSR and BRP, respectively. Chromosomes shown in lighter shading are unique to their respective genomes whereas chromosomes shown in blue have experienced the structural rearrangement described in the text. Chromosomes that lack any identifiable genes are indicated with short black lines just inside the circle of blocks. This figure was generated with Circos v0.66 (60).
In the mitochondrial genomes of both BRP and OSR, the number of chromosomes exceeds the number of genes (Table 1), resulting in a large number of seemingly “empty” chromosomes that lack any annotated functional element (20 chromosomes in OSR and 25 in BRP). Of the 19 chromosomes that are unique to one of the two genomes, 14 fall in this category of lacking any identifiable genes. The remaining five lack any unique genes but contain genes that are duplicated on one of the chromosomes that is shared between the genomes. Therefore, the variation in chromosome number between OSR and BRP does not seem to translate into a difference in overall functional gene content between the two genomes. Instead, like the rest of the genome, these chromosomes consist mostly or entirely of noncoding sequence. Some of this noncoding sequence can be traced to insertions of nuclear or plastid DNA, but the vast majority lacks detectable similarity to any known DNA sequence (22).
The total genome size of BRP is 410 kb larger than that of OSR (Table 1). This entire size difference results from the greater number and length of unique chromosomes in BRP. The lengths of the 50 chromosomes that are shared between BRP and OSR are similar in the two genomes; in fact, their total length is 13 kb shorter in BRP than in OSR.
Some of the 19 chromosomes that are unique to either OSR or BRP contain large repeats that are duplicated elsewhere in the genome. In the most extreme example, although BRP chromosome 61 does not have a full-length homolog in the OSR genome, it contains a 22-kb sequence that is shared with BRP chromosome 32 and the corresponding homolog in OSR (chromosome 27) (Fig. 2). Overall, the BRP mitochondrial genome has a slightly higher percentage of repetitive sequence content than OSR (Table 1).
Fig. 2.
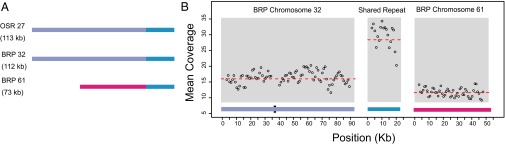
Repeated regions and variation in sequencing coverage depth between chromosomes. (A) OSR chromosome 27 and BRP chromosome 32 share sequence homology across their entire lengths (purple and blue). BRP chromosome 61 shares a duplicated 22-kb fragment (blue) with these chromosomes, but the rest of the chromosome 61 (magenta) is unique to the BRP genome. (B) Mean sequencing coverage is plotted as a sliding window, with a window size of 2 kb and a step size of 1 kb. As indicated by the red-dashed lines, single copy regions of BRP chromosomes 32 and 61 differ in their average coverage (16× and 12×, respectively). As expected, the average coverage (28×) for the repeat shared between these two chromosomes is approximately equal to the sum of the coverages in the two single-copy regions. The black box in chromosome 32 represents a 1.15-kb sequence that is duplicated elsewhere in the genome and was, therefore, excluded from the analysis of single-copy read depth.
Conservation of Mitochondrial Sequence and Structure in S. noctiflora.
In contrast to the variation associated with the presence/absence of entire chromosomes, the rest of the mitochondrial genome in OSR and BRP exhibits a remarkable degree of similarity in both sequence and structure. The BRP and OSR genomes do not differ by any inversions, and there are few examples of other large-scale structural rearrangements. Indels and SNPs are far more common than inversions and structural rearrangements but still contribute little overall sequence divergence between the OSR and BRP mitochondrial genomes. Alignments of the 50 shared chromosomes with a total length of 5.8 Mb revealed 2,242 indels, but the majority (1,879) of these indels are associated with single-nucleotide repeat regions (i.e., homopolymers), which are subject to a high rate of length errors with 454 sequencing technology. The 363 indels that remained after excluding homopolymer regions represented a total of 40.5 kb (0.70% of the aligned chromosome length) and ranged in size from 1 bp to 14.9 kb.
The chromosomal alignments contained a total of 2,344 SNPs (excluding those associated with homopolymer regions), corresponding to an overall nucleotide sequence identity of 99.96%. Although consistent with our previous findings (22), the low level of nucleotide polymorphism within S. noctiflora mtDNA is surprising given the history of rapid nucleotide substitution in this species. The lack of polymorphism may reflect a very low effective population size and/or a recent reversion to lower mitochondrial mutation rates in S. noctiflora (22). Of the identified SNPs, 498 are transitions (Ti), and 1,846 are transversions (Tv), producing a Ti:Tv ratio of 0.27. Most of the identified SNPs were in noncoding regions. In a total of 25,338 bp of protein-coding sequence in the entire genome (after excluding duplicate gene copies), we found only nine SNPs in four genes (ccmFn, cox1, mttB, and nad2). Of these nine SNPs, five were nonsynonymous (0.0003 polymorphisms per nonsynonymous site) and four were synonymous (0.0006 polymorphisms per synonymous site). SNPs are not evenly distributed among chromosomes, with nucleotide divergence ranging from 0.01% to 0.41% among pairs of homologous chromosomes (Table S3).
Variation in Relative Abundance of Mitochondrial Chromosomes.
The existence of a fragmented mitochondrial genome in S. noctiflora, along with the finding that entire chromosomes can be subject to gain/loss, raises intriguing questions about variation in the relative abundance of chromosomes. Sequencing coverage of single-copy regions varied approximately twofold among chromosomes in the BRP mitochondrial genome, ranging from a median of 12× in chromosome 61 to 25× in chromosome 30 (Table S4). As expected, large repeat sequences exhibited higher total coverage resulting from their presence on two or more chromosomes (Fig. 2). We found that longer chromosomes tended to have a higher average sequencing coverage (r = 0.50; P < 0.0001) but that there was no significant difference in coverage between the sets of chromosomes with and without genes (t = 1.66; P = 0.10). We used absolute qPCR measurements to validate the differences in relative abundance inferred from variation in sequencing read depth among chromosomes. Analysis of chromosomes 30 and 61 confirmed a significantly higher abundance of markers on chromosome 30 in two independent DNA samples derived from the S. noctiflora BRP lineage (P < 0.0001) (Fig. S1 and Table S5).
Although many of the mitochondrial chromosomes in S. noctiflora were present in only one of the two sequenced genomes (8 unique chromosomes in OSR and 11 in BRP), the relative abundances of the remaining chromosomes were generally stable across the two populations. The 50 shared mitochondrial chromosomes exhibited a strong positive correlation in relative abundance, as measured by average sequencing read depth, between OSR and BRP (r = 0.75; P < 0.0001) (Fig. 3 and Table S6). Chromosomes with and without identifiable genes exhibited similar correlations and ranges of relative abundance (Fig. 3). This finding was validated with qPCR using S. noctiflora families collected from two additional populations (BDA and BWT). After normalization with a nuclear control gene, mitochondrial qPCR markers exhibited a strong linear relationship between samples from the BDA and BWT families (Fig. S2), with no evidence of a chromosome × family interaction (P > 0.5) (Table S7). This pattern is consistent with the observation that chromosomes shared across S. noctiflora families maintain similar relative abundances.
Fig. 3.

Correlation in relative abundance of shared chromosomes between S. noctiflora OSR and BRP mitochondrial genomes. Filled and open circles represent BRP chromosomes with or without annotated genes, respectively.
We expanded our qPCR survey to include individuals from a total of 12 geographically dispersed populations of S. noctiflora (Table S8) and a sample of markers from seven different chromosomes, including two that were specific to OSR and two that were specific to BRP. Most individuals shared an identical pattern of chromosome presence/absence with either OSR or BRP. However, one of the 12 individuals exhibited a third pattern that included a mixture of BRP-specific and OSR-specific chromosomes (Fig. 4).
Fig. 4.

Presence/absence survey of a sample of seven mitochondrial chromosomes in families from 12 different populations of S. noctiflora (Table S8). Gray shading indicates that the chromosome was present. For each chromosome, presence/absence was confirmed with three different qPCR markers, which were all in agreement.
Discussion
Intraspecific Variation in the Massive Mitochondrial Genome of S. noctiflora.
Angiosperm mitochondrial genomes harbor tremendous variation in genome size (22, 23, 34, 35). Previous examples of variation within and between species have often been attributed to large sequence duplications (27) or the insertions of large quantities of foreign DNA (23, 36, 37). In contrast, a fundamentally different mechanism seems to be at play in S. noctiflora, with the presence or absence of entire chromosomes explaining the size variation between genomes within this species (Fig. 1). Although the S. noctiflora BRP and OSR mitochondrial genomes differ by only 6.1% in size, this difference represents more than 400 kb or roughly the equivalent of an entire mitochondrial genome from a “typical” angiosperm (24, 25). In total, the chromosomes that are unique to either of the two genomes represent more than 1.8 Mb of sequence. This variation at the level of entire chromosomes is in striking contrast to the overall pattern of structural conservation in the set of shared chromosomes. The lack of inversions and rearrangements is highly unusual for angiosperm mitochondrial genomes, which are often structurally dynamic even within species (27–30). This difference is likely caused by a reduced rate of intragenomic recombination between small repeats in S. noctiflora mtDNA (22).
The Functional Significance of Empty Chromosomes.
One of the most perplexing findings from our analysis of the multichromosomal mitochondrial genomes in Silene is that they contain numerous chromosomes that lack any identifiable genes. Such seemingly empty chromosomes have also been found in other mitochondria with multichromosomal genomes, including those of cucumber (21) and ichthyosporean protists (12). The existence of these chromosomes suggests two alternative scenarios. One possibility is that they contain some, as of yet, unidentified elements that are important for cell function. For example, the numerous minicircles in kinetoplastid mitochondrial genomes code for guide RNAs that regulate the RNA editing process (38). Alternatively, empty chromosomes may persist as genomic parasites or perhaps commensals that impose little or no fitness cost on the organism. Our finding that the presence of these empty chromosomes is highly variable within a species supports the latter possibility and suggests that they do not serve any important biological function. In light of the renewed debate over the prevalence (and definition) of junk DNA in eukaryotic genomes (39–41), plant mitochondria continue to provide examples of genomes that are seemingly awash in nonfunctional DNA (23).
Are S. noctiflora Mitochondrial Chromosomes Being Gained or Lost?
Although the BRP and OSR mitochondrial genomes differ in their number of chromosomes, it is not immediately clear whether these differences reflect chromosome gains, losses, or some combination of both. Mitochondrial genomes that have been sequenced from other Silene species are so divergent in content and structure that they cannot be used to infer the ancestral state for S. noctiflora. Nevertheless, based on the overall stability in the rest of these genomes, we speculate that divergence in chromosome number reflects an ongoing process of whole-chromosome loss. We propose a “big-bang” model in which the S. noctiflora mitochondrial genome underwent massive expansion and fragmentation, possibly associated with disruption in recombinational machinery (22), resulting in a genome size and chromosome number even larger than currently observed. Under this hypothesis, our findings would reflect an ongoing “filtering” stage in which chromosomes with essential genes are maintained by selection, but other chromosomes are subject to stochastic loss. The alternative model would be that novel chromosomes are constantly being formed within lineages. Testing these hypotheses will require more extensive intraspecific sampling of S. noctiflora mitochondrial genomes to map the history of chromosome gains/losses onto a genealogy, ideally in combination with genomic data from a close outgroup such as Silene turkestanica (42).
The in Vivo Structure and Inheritance of Multichromosomal Mitochondrial Genomes.
The field of plant mitochondrial genomics has long dealt with uncertainties about the extent to which assembled genome sequences or other forms of genome mapping are consistent with biological reality. These uncertainties can be summarized by two underlying questions: (i) Are genome maps an accurate depiction of the in vivo physical structure of mtDNA? (ii) To what extent is the analysis of genomic content in vegetative tissue applicable to the inherited form of the genome that is found in meristematic tissue?
The answer to the first question seems to be no. Whereas plant mitochondrial genomes typically map as circles, they exist in more complex structural forms in vivo (43, 44). Across eukaryotes, a variety of techniques have been used to identify and characterize multichromosomal mitochondrial genomes. As in this study, the identification of multichromosomal structures in angiosperms has been based almost exclusively on mapping/sequencing (21–23), but, in some other cases, sequence data have been supplemented with electrophoretic separation of chromosomes and electron microscopy-based observations (11, 12, 14, 15, 17, 18, 20, 45, 46). To date, the only direct characterization of chromosomal structure in an angiosperm multichromosomal mitochondrial genome was performed in Silene vulgaris (26–30). In this case, a Southern blot analysis of the smallest (∼5 kb) mitochondrial chromosome confirmed the existence of the predicted circular molecules but found that most of the DNA was present in multimeric forms that varied in susceptibility to exonuclease digestion, indicating diversity in molecular size and structure. Based on these results, we predict that the circular-mapping chromosomes found in S. noctiflora predominantly exist as high-molecular weight, multimeric structures in vivo.
The question of whether mtDNA from vegetative tissue is representative of the inherited form of the mitochondrial genome is relevant to all multicellular eukaryotes. For example, dicyemid mtDNA has been found to contain single-gene minicircles that accumulate in somatic cells over the course of development, but it has been proposed that the genome transmitted in the germ line is actually a higher molecular weight structure (47). Addressing this question in S. noctiflora and plants in general will require technologies that can directly assay mitochondrial genome structure in meristematic tissue. Nevertheless, the most parsimonious explanation for variation in the presence or absence of entire mitochondrial chromosomes in S. noctiflora is that they are relevant units of inheritance.
Why Have Multichromosomal Mitochondrial Genomes Evolved Repeatedly in Diverse Eukaryotic Lineages?
Few subjects stir the embers of the neutralist–selectionist debate more than interpretations of the extreme and often bizarre variations in genomic architecture observed across the tree of life. Variation in complexity among eukaryotic genomes, including differences in genome size and structure, has attracted both adaptive and nonadaptive interpretations that center on the long-standing debate over the functional role of noncoding (or “junk”) DNA.
Many adaptive hypotheses have been advanced to explain the origins of multichromosomal mitochondrial genomes, including the effects of genome fragmentation on replication time (18, 48) and regulation of gene expression (49). One of the more provocative hypotheses is that multichromosomal mitochondrial genomes could enable a form of sex to combat the accumulation of deleterious mutations (49). Because mitochondrial genomes occur at high copy numbers within each cell, distributing genes across multiple chromosomes could reduce selective interference between mutations in different genes, allowing mutations to spread (or be lost) independently of mutations on other chromosomes. In the absence of biparental inheritance, the effect of this mechanism in counteracting Muller’s ratchet will be limited because it would operate only on new mutations before they spread to fixation within a cell. Nevertheless, it is intriguing that the highly multichromosomal genomes analyzed in this study have evolved in a species with an apparent history of accelerated mitochondrial mutation rates and reduced frequency of inter- and intragenomic recombination (22). Given that many multichromosomal mitochondrial genomes are found in eukaryotic lineages with accelerated evolutionary rates (50), the hypothesis that multichromosomal architectures can play a role in mutational clearance merits further investigation.
There are also alternative nonadaptive explanations for the origins of multichromosomal mitochondrial genomes. From the perspective of constructive neutral evolution (51, 52), one hypothesis is that selection could allow the breakup of a genome into multiple chromosomes as long as preexisting mechanisms were in place to replicate each. After genome fragmentation, there would then be strong selection to maintain every chromosome that carries an essential gene. Therefore, rather than asking why selection has favored multichromosomal mitochondrial genomes in many eukaryotic lineages, the more pertinent question may be why mitochondria have been able to tolerate the repeated evolution of fragmented genomes.
One feature of mitochondria that may have allowed them to mitigate the challenges associated with transmitting multiple chromosomes is their ability to fuse (53). Mitochondria can undergo large-scale fusion events in coordination with the cell cycle (54), and the pooling of mitochondrial genome copies that results may reduce the probability of segregational loss of chromosomes caused by stochastic sampling. In support of this hypothesis, the evolution of multichromosomal genomes seems to be rare in plastids, which, unlike mitochondria, do not fuse. To our knowledge, the only examples of multichromosomal plastid genomes are found in dinoflagellates (55), and these genomes are associated with extensive gene loss and transfer to the nucleus (56), suggesting that the prevention of segregational loss of chromosomes may be problematic in these systems (57). In addition, a recent analysis of an organelle-like bacterial endosymbiont in cicadas has found that the genome has split into two largely complementary chromosomes (58). However, these endosymbiotic bacteria presumably cannot fuse, and, rather than maintain a multichromosomal genome, they have instead split into two interdependent cellular lineages, each housing one of the chromosomes. Finally, although multichromosomal mitochondrial genomes have been found in some eukaryotes that harbor only a single mitochondrion per cell (9), which clearly do not have the opportunity to fuse, these species would seem to have alternative mechanisms to ensure transmission. For example, in kinetoplastids, the mitochondrial genome’s enormous copy number and complex structure of interlinked circular molecules (8) may facilitate the reliable inheritance of the entire mitochondrial genome.
Further analysis of variation in multichromosomal genomes within species, within individuals, and within cells, along with associated mechanisms of transmission, should provide insight into this fundamental problem of genome inheritance and how it has been solved outside of the eukaryotic nucleus.
Materials and Methods
S. noctiflora BRP mtDNA Extraction and Sequencing.
Seeds from the S. noctiflora BRP population were originally collected at milepost 8 south of Humpback Rocks on the Blue Ridge Parkway in Virginia, United States. A total of 200 g of leaf tissue from a single maternal family derived from this collection was used for purification of mtDNA and generation of shotgun and 3-kb paired-end 454 sequencing libraries as described previously (22). This population was chosen for mitochondrial genome sequencing because it was known to differ from the originally sequenced S. noctiflora OSR mitochondrial genome at the one mitochondrial (cox1) SNP previously identified in the species (22).
S. noctiflora BRP Mitochondrial Genome Assembly and Annotation.
Assembly of 454 sequence data was performed de novo and generally followed protocols used for assembly of the original S. noctiflora OSR mitochondrial genome (22). However, the largest repeats in the S. noctiflora BRP genome (up to 22 kb) greatly exceeded the average span (2.8 kb) of the paired-end library used for assembly. These large repeats are known to undergo recombination and be associated with alternative genome conformations. Therefore, in these cases, we followed the convention of reporting chromosomes as minimal possible “subcircular” maps rather than as larger combined circular maps. Chromosomes were numbered in decreasing order of size. Assembly of 454 data produced closed circular maps for each chromosome with the exception of chromosome 61, which had the lowest sequence coverage of any chromosome in the genome (Table S4) and contained a small gap in an AT-rich region. Gap closing was performed by PCR amplification of total cellular DNA with primers in the regions flanking the gap (Table S9), followed by direct Sanger sequencing of the resulting PCR product.
Annotation of the assembled S. noctiflora BRP mitochondrial genome was conducted using Mitofy (35) and direct comparison with the previously generated annotations for S. noctiflora OSR (22). The annotated S. noctiflora BRP mitochondrial chromosome sequences were deposited in GenBank (accession nos. KP053825–KP053887).
Comparative Analysis of S. noctiflora Mitochondrial Genomes.
To identify regions of homology and conserved synteny, the newly sequenced BRP mitochondrial genome was compared with the previously sequenced OSR mitochondrial genome with NCBI-BLASTN v2.2.24+ (blastn). The 50 pairs of homologous chromosomes shared between the two genomes were aligned with MAFFT v7.150b (FFT-NS-2), followed by manual adjustment (59). Five unalignable regions within otherwise conserved chromosomes were excluded from subsequent analysis. All SNPs and indels were parsed from the resulting alignments using a custom program written in C (available from the authors upon request). Because of the high error rate associated with homopolymers in 454 sequence data, SNPs and indel analyses were also performed after excluding all sites located within 5 bp of any single nucleotide repeat of 5 bp or longer. Repetitive content was identified by searching each genome against itself with NCBI-BLASTN (megablast) and applying a minimum score cutoff of 30 as described previously (22). Sequencing read depth for each chromosome was determined using site-specific coverage reported in the 454AlignmentInfo.tsv file produced by the GS de novo Assembler. Because sequencing coverage estimates are inflated for repeat regions, we restricted this analysis to single-copy regions by excluding all repeats longer than 300 bp and greater than 75% nucleotide identity.
Quantitative PCR Analysis.
Conventional and quantitative PCR analyses were performed using total cellular DNA extracted from rosette leaf tissue with the Qiagen Plant DNeasy Kit following the manufacturer’s protocols. All qPCR amplifications were performed in 10-μL volumes with SsoAdvanced SYBR Green 2× Supermix (Bio-Rad), 2 pmol of each primer, and 1 ng of template DNA. Amplification was performed on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad) with an initial 3-min incubation at 95 °C and 40 cycles of 3 s at 95 °C and 20 s at 60 °C, followed by a melt curve analysis.
To validate the variation in chromosome abundance that was inferred from sequence coverage data, we performed absolute qPCR measurements of the two BRP chromosomes (30, 60) with the most highly divergent read depths (Table S4). Three markers were assayed on each chromosome. For each marker, a fragment of ∼650 bp in length was amplified from total cellular DNA by conventional PCR, cleaned with Agencourt AMPure XP PCR Purification beads (Beckman-Coulter), and quantified with the Qubit dsDNA BR Assay kit (Life Technologies). These fragments were used as standards for qPCR to amplify an internal fragment of ∼120 bp in length using nested primers (Table S9). A standard curve was generated with a tenfold dilution series, using a range of 108 to 102 copies of the 650-bp PCR fragment as template. Samples from two biological replicates (different BRP individuals) were compared against this standard curve to estimate copy number for each marker. All assays were performed with three technical replicates. Statistical analyses of the resulting copy-number estimates were compared by ANOVA in R v2.15.3 (61), with biological replicate and chromosome as main effects and marker nested within chromosome.
To test for differences in relative abundance of mitochondrial chromosomes within and between families, we compared samples from three individuals from each of two maternal families derived from different S. noctiflora populations (BDA and BWT) (Table S8). Each sample was assayed with qPCR markers on seven different chromosomes, including three that are present in both OSR and BRP, two that are present in OSR but absent in BRP, and two that are present in BRP but absent in OSR (Fig. 4). Three different markers were used for each chromosome, and two technical replicates were performed for each assay. BDA and BWT both lacked the BRP-specific chromosomes (all three markers on each produced no amplification) so these chromosomes were excluded from further analysis. For each sample, a nuclear gene (XY4) was also assayed as a control. Cycle threshold (Ct) values for the nuclear control gene were subtracted from each mitochondrial Ct value to create ΔCt values. The resulting ΔCt data were compared by ANOVA in R, with family and chromosome as main effects, biological replicate nested within family, and marker nested within chromosome.
The same set of qPCR markers from the sample of seven chromosomes was used to screen for presence/absence variation in a single individual from 10 additional populations of S. noctiflora (Table S8). Product specificity was verified by inspection of melt curves. All positive samples amplified within 26 cycles whereas negative samples failed to produce specific amplification within the full 40 cycles of the run.
Supplementary Material
Acknowledgments
We thank two anonymous reviewers and the laboratory groups of Rachel Mueller and D.B.S. for insightful comments on an earlier version of this manuscript. We also thank Laura Bergner for assistance with mitochondrial DNA extraction and Peng Chen for assistance with computational analysis. Multiple S. noctiflora seed collections were generously provided by the Kew Royal Botanic Gardens Millennium Seed Bank. This research was supported by Colorado State University and the National Science Foundation (Grants MCB-1022128 and MCB-1412260).
Footnotes
The authors declare no conflict of interest.
This paper results from the Arthur M. Sackler Colloquium of the National Academy of Sciences, “Symbioses Becoming Permanent: The Origins and Evolutionary Trajectories of Organelles,” held October 15–17, 2014, at the Arnold and Mabel Beckman Center of the National Academies of Sciences and Engineering in Irvine, CA. The complete program and video recordings of most presentations are available on the NAS website at www.nasonline.org/Symbioses.
This article is a PNAS Direct Submission. J.P.M. is a guest editor invited by the Editorial Board.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. KP053825–KP053887). Raw 454 sequencing reads were deposited in the National Center for Biotechnology Information (NCBI) Sequencing Read Archive (SRP049332).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1421397112/-/DCSupplemental.
References
- 1.Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science. 1999;283(5407):1476–1481. doi: 10.1126/science.283.5407.1476. [DOI] [PubMed] [Google Scholar]
- 2.Lynch M, Koskella B, Schaack S. Mutation pressure and the evolution of organelle genomic architecture. Science. 2006;311(5768):1727–1730. doi: 10.1126/science.1118884. [DOI] [PubMed] [Google Scholar]
- 3.Margulis L. Origin of Eukaryotic Cells: Evidence and Research Implications for a Theory of the Origin and Evolution of Microbial, Plant, and Animal Cells on the Precambrian Earth. Yale Univ. Press; New Haven, CT: 1970. [Google Scholar]
- 4.Gray MW, Archibald JM. 2012. Origins of mitochondria and plastids. Genomics of Chloroplasts and Mitochondria, Advances in Photosynthesis and Respiration, eds Bock R, Knoop V (Springer, Dordrecht, The Netherlands), pp 1–30.
- 5.Wang Z, Wu M. Phylogenomic reconstruction indicates mitochondrial ancestor was an energy parasite. PLoS ONE. 2014;9(10):e110685. doi: 10.1371/journal.pone.0110685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lang BF, et al. An ancestral mitochondrial DNA resembling a eubacterial genome in miniature. Nature. 1997;387(6632):493–497. doi: 10.1038/387493a0. [DOI] [PubMed] [Google Scholar]
- 7.Burger G, Gray MW, Forget L, Lang BF. Strikingly bacteria-like and gene-rich mitochondrial genomes throughout jakobid protists. Genome Biol Evol. 2013;5(2):418–438. doi: 10.1093/gbe/evt008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lukes J, Hashimi H, Zíková A. Unexplained complexity of the mitochondrial genome and transcriptome in kinetoplastid flagellates. Curr Genet. 2005;48(5):277–299. doi: 10.1007/s00294-005-0027-0. [DOI] [PubMed] [Google Scholar]
- 9.Vlcek C, Marande W, Teijeiro S, Lukeš J, Burger G. Systematically fragmented genes in a multipartite mitochondrial genome. Nucleic Acids Res. 2011;39(3):979–988. doi: 10.1093/nar/gkq883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slamovits CH, Saldarriaga JF, Larocque A, Keeling PJ. The highly reduced and fragmented mitochondrial genome of the early-branching dinoflagellate Oxyrrhis marina shares characteristics with both apicomplexan and dinoflagellate mitochondrial genomes. J Mol Biol. 2007;372(2):356–368. doi: 10.1016/j.jmb.2007.06.085. [DOI] [PubMed] [Google Scholar]
- 11.Fan J, Lee RW. Mitochondrial genome of the colorless green alga Polytomella parva: Two linear DNA molecules with homologous inverted repeat Termini. Mol Biol Evol. 2002;19(7):999–1007. doi: 10.1093/oxfordjournals.molbev.a004180. [DOI] [PubMed] [Google Scholar]
- 12.Burger G, Forget L, Zhu Y, Gray MW, Lang BF. Unique mitochondrial genome architecture in unicellular relatives of animals. Proc Natl Acad Sci USA. 2003;100(3):892–897. doi: 10.1073/pnas.0336115100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burger G, Lang BF. Parallels in genome evolution in mitochondria and bacterial symbionts. IUBMB Life. 2003;55(4-5):205–212. doi: 10.1080/1521654031000137380. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe KI, Bessho Y, Kawasaki M, Hori H. Mitochondrial genes are found on minicircle DNA molecules in the mesozoan animal Dicyema. J Mol Biol. 1999;286(3):645–650. doi: 10.1006/jmbi.1998.2523. [DOI] [PubMed] [Google Scholar]
- 15.Armstrong MR, Blok VC, Phillips MS. A multipartite mitochondrial genome in the potato cyst nematode Globodera pallida. Genetics. 2000;154(1):181–192. doi: 10.1093/genetics/154.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pont-Kingdon G, et al. Mitochondrial DNA of Hydra attenuata (Cnidaria): A sequence that includes an end of one linear molecule and the genes for l-rRNA, tRNA(f-Met), tRNA(Trp), COII, and ATPase8. J Mol Evol. 2000;51(4):404–415. doi: 10.1007/s002390010103. [DOI] [PubMed] [Google Scholar]
- 17.Suga K, Mark Welch DB, Tanaka Y, Sakakura Y, Hagiwara A. Two circular chromosomes of unequal copy number make up the mitochondrial genome of the rotifer Brachionus plicatilis. Mol Biol Evol. 2008;25(6):1129–1137. doi: 10.1093/molbev/msn058. [DOI] [PubMed] [Google Scholar]
- 18.Shao R, Kirkness EF, Barker SC. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res. 2009;19(5):904–912. doi: 10.1101/gr.083188.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith DR, et al. First complete mitochondrial genome sequence from a box jellyfish reveals a highly fragmented linear architecture and insights into telomere evolution. Genome Biol Evol. 2012;4(1):52–58. doi: 10.1093/gbe/evr127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lavrov DV, et al. Mitochondrial DNA of Clathrina clathrus (Calcarea, Calcinea): Six linear chromosomes, fragmented rRNAs, tRNA editing, and a novel genetic code. Mol Biol Evol. 2013;30(4):865–880. doi: 10.1093/molbev/mss274. [DOI] [PubMed] [Google Scholar]
- 21.Alverson AJ, Rice DW, Dickinson S, Barry K, Palmer JD. Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell. 2011;23(7):2499–2513. doi: 10.1105/tpc.111.087189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sloan DB, et al. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012;10(1):e1001241. doi: 10.1371/journal.pbio.1001241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rice DW, et al. Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science. 2013;342(6165):1468–1473. doi: 10.1126/science.1246275. [DOI] [PubMed] [Google Scholar]
- 24.Knoop V, Volkmar U, Hecht J, Grewe F. 2011. Mitochondrial genome evolution in the plant lineage. Plant Mitochondria, Advances in Plant Biology, ed Kempken F (Springer, New York), pp 3–29.
- 25.Mower JP, Sloan DB, Alverson AJ. 2012. Plant mitochondrial genome diversity: The genomics revolution. Plant Genomes, Their Residents, and Their Evolutionary Dynamics, Plant Genome Diversity, ed Wendel JF (Springer, Vienna), Vol 1, pp 123–144.
- 26.Palmer JD, Herbon LA. Plant mitochondrial DNA evolves rapidly in structure, but slowly in sequence. J Mol Evol. 1988;28(1-2):87–97. doi: 10.1007/BF02143500. [DOI] [PubMed] [Google Scholar]
- 27.Allen JO, et al. Comparisons among two fertile and three male-sterile mitochondrial genomes of maize. Genetics. 2007;177(2):1173–1192. doi: 10.1534/genetics.107.073312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darracq A, et al. Structural and content diversity of mitochondrial genome in beet: a comparative genomic analysis. Genome Biol Evol. 2011;3:723–736. doi: 10.1093/gbe/evr042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davila JI, et al. Double-strand break repair processes drive evolution of the mitochondrial genome in Arabidopsis. BMC Biol. 2011;9:64. doi: 10.1186/1741-7007-9-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sloan DB, Müller K, McCauley DE, Taylor DR, Storchová H. Intraspecific variation in mitochondrial genome sequence, structure, and gene content in Silene vulgaris, an angiosperm with pervasive cytoplasmic male sterility. New Phytol. 2012;196(4):1228–1239. doi: 10.1111/j.1469-8137.2012.04340.x. [DOI] [PubMed] [Google Scholar]
- 31.Arrieta-Montiel MP, Mackenzie SA. 2011. Plant mitochondrial genomes and recombination. Plant Mitochondria, Advances in Plant Biology, ed Kempken F (Springer, New York), pp 65–82.
- 32.Palmer JD, Shields CR. Tripartite structure of the Brassica campestris mitochondrial genome. Nature. 1984;307:437–440. [Google Scholar]
- 33.Sugiyama Y, et al. The complete nucleotide sequence and multipartite organization of the tobacco mitochondrial genome: Comparative analysis of mitochondrial genomes in higher plants. Mol Genet Genomics. 2005;272(6):603–615. doi: 10.1007/s00438-004-1075-8. [DOI] [PubMed] [Google Scholar]
- 34.Ward BL, Anderson RS, Bendich AJ. The mitochondrial genome is large and variable in a family of plants (cucurbitaceae) Cell. 1981;25(3):793–803. doi: 10.1016/0092-8674(81)90187-2. [DOI] [PubMed] [Google Scholar]
- 35.Alverson AJ, et al. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae) Mol Biol Evol. 2010;27(6):1436–1448. doi: 10.1093/molbev/msq029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goremykin VV, Salamini F, Velasco R, Viola R. Mitochondrial DNA of Vitis vinifera and the issue of rampant horizontal gene transfer. Mol Biol Evol. 2009;26(1):99–110. doi: 10.1093/molbev/msn226. [DOI] [PubMed] [Google Scholar]
- 37.Goremykin VV, Lockhart PJ, Viola R, Velasco R. The mitochondrial genome of Malus domestica and the import-driven hypothesis of mitochondrial genome expansion in seed plants. Plant J. 2012;71(4):615–626. doi: 10.1111/j.1365-313X.2012.05014.x. [DOI] [PubMed] [Google Scholar]
- 38.Sturm NR, Simpson L. Kinetoplast DNA minicircles encode guide RNAs for editing of cytochrome oxidase subunit III mRNA. Cell. 1990;61(5):879–884. doi: 10.1016/0092-8674(90)90198-n. [DOI] [PubMed] [Google Scholar]
- 39.Doolittle WF. Is junk DNA bunk? A critique of ENCODE. Proc Natl Acad Sci USA. 2013;110(14):5294–5300. doi: 10.1073/pnas.1221376110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graur D, et al. On the immortality of television sets: “Function” in the human genome according to the evolution-free gospel of ENCODE. Genome Biol Evol. 2013;5(3):578–590. doi: 10.1093/gbe/evt028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kellis M, et al. Defining functional DNA elements in the human genome. Proc Natl Acad Sci USA. 2014;111(17):6131–6138. doi: 10.1073/pnas.1318948111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sloan DB, Oxelman B, Rautenberg A, Taylor DR. Phylogenetic analysis of mitochondrial substitution rate variation in the angiosperm tribe Sileneae. BMC Evol Biol. 2009;9:260. doi: 10.1186/1471-2148-9-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sloan DB. One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 2013;200(4):978–985. doi: 10.1111/nph.12395. [DOI] [PubMed] [Google Scholar]
- 44.Bendich AJ. Reaching for the ring: The study of mitochondrial genome structure. Curr Genet. 1993;24(4):279–290. doi: 10.1007/BF00336777. [DOI] [PubMed] [Google Scholar]
- 45.Marande W, Lukeš J, Burger G. Unique mitochondrial genome structure in diplonemids, the sister group of kinetoplastids. Eukaryot Cell. 2005;4(6):1137–1146. doi: 10.1128/EC.4.6.1137-1146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roy Chowdhury A, et al. The killing of African trypanosomes by ethidium bromide. PLoS Pathog. 2010;6(12):e1001226. doi: 10.1371/journal.ppat.1001226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Awata H, Noto T, Endoh H. Differentiation of somatic mitochondria and the structural changes in mtDNA during development of the dicyemid Dicyema japonicum (Mesozoa) Mol Genet Genomics. 2005;273(6):441–449. doi: 10.1007/s00438-005-1157-2. [DOI] [PubMed] [Google Scholar]
- 48.Egan ES, Waldor MK. Distinct replication requirements for the two Vibrio cholerae chromosomes. Cell. 2003;114(4):521–530. doi: 10.1016/s0092-8674(03)00611-1. [DOI] [PubMed] [Google Scholar]
- 49.Rand DM. ‘Why genomes in pieces?’ revisited: Sucking lice do their own thing in mtDNA circle game. Genome Res. 2009;19(5):700–702. doi: 10.1101/gr.091132.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith DR, Keeling PJ. 2015. Mitochondrial and plastid genome architecture: Reoccurring themes, but significant differences at the extremes. Proc Natl Acad Sci USA 112:10177–10184.
- 51.Stoltzfus A. On the possibility of constructive neutral evolution. J Mol Evol. 1999;49(2):169–181. doi: 10.1007/pl00006540. [DOI] [PubMed] [Google Scholar]
- 52.Gray MW, Lukeš J, Archibald JM, Keeling PJ, Doolittle WF. Cell biology: Irremediable complexity? Science. 2010;330(6006):920–921. doi: 10.1126/science.1198594. [DOI] [PubMed] [Google Scholar]
- 53.Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11(12):872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- 54.Sheahan MB, McCurdy DW, Rose RJ. Mitochondria as a connected population: ensuring continuity of the mitochondrial genome during plant cell dedifferentiation through massive mitochondrial fusion. Plant J. 2005;44(5):744–755. doi: 10.1111/j.1365-313X.2005.02561.x. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Z, Green BR, Cavalier-Smith T. Single gene circles in dinoflagellate chloroplast genomes. Nature. 1999;400(6740):155–159. doi: 10.1038/22099. [DOI] [PubMed] [Google Scholar]
- 56.Howe CJ, Nisbet RE, Barbrook AC. The remarkable chloroplast genome of dinoflagellates. J Exp Bot. 2008;59(5):1035–1045. doi: 10.1093/jxb/erm292. [DOI] [PubMed] [Google Scholar]
- 57.Dorrell RG, Howe CJ. 2015. Integration of plastids with their hosts: Lessons learned from dinoflagellates. Proc Natl Acad Sci USA 112:10247–10254.
- 58.Van Leuven JT, Meister RC, Simon C, McCutcheon JP. Sympatric speciation in a bacterial endosymbiont results in two genomes with the functionality of one. Cell. 2014;158(6):1270–1280. doi: 10.1016/j.cell.2014.07.047. [DOI] [PubMed] [Google Scholar]
- 59.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krzywinski M, et al. Circos: An information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.R Development Core Team . R Foundation for Statistical Computing; Vienna: 2014. R: A Language and Environment for Statistical Computing. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.