

Significance
T-cell genome engineering holds great promise for cancer immunotherapies and cell-based therapies for HIV, primary immune deficiencies, and autoimmune diseases, but genetic manipulation of human T cells has been inefficient. We achieved efficient genome editing by delivering Cas9 protein pre-assembled with guide RNAs. These active Cas9 ribonucleoproteins (RNPs) enabled successful Cas9-mediated homology-directed repair in primary human T cells. Cas9 RNPs provide a programmable tool to replace specific nucleotide sequences in the genome of mature immune cells—a longstanding goal in the field. These studies establish Cas9 RNP technology for diverse experimental and therapeutic genome engineering applications in primary human T cells.
Keywords: CRISPR/Cas9, genome engineering, Cas9 ribonucleoprotein, RNP, primary human T cells
Abstract
T-cell genome engineering holds great promise for cell-based therapies for cancer, HIV, primary immune deficiencies, and autoimmune diseases, but genetic manipulation of human T cells has been challenging. Improved tools are needed to efficiently “knock out” genes and “knock in” targeted genome modifications to modulate T-cell function and correct disease-associated mutations. CRISPR/Cas9 technology is facilitating genome engineering in many cell types, but in human T cells its efficiency has been limited and it has not yet proven useful for targeted nucleotide replacements. Here we report efficient genome engineering in human CD4+ T cells using Cas9:single-guide RNA ribonucleoproteins (Cas9 RNPs). Cas9 RNPs allowed ablation of CXCR4, a coreceptor for HIV entry. Cas9 RNP electroporation caused up to ∼40% of cells to lose high-level cell-surface expression of CXCR4, and edited cells could be enriched by sorting based on low CXCR4 expression. Importantly, Cas9 RNPs paired with homology-directed repair template oligonucleotides generated a high frequency of targeted genome modifications in primary T cells. Targeted nucleotide replacement was achieved in CXCR4 and PD-1 (PDCD1), a regulator of T-cell exhaustion that is a validated target for tumor immunotherapy. Deep sequencing of a target site confirmed that Cas9 RNPs generated knock-in genome modifications with up to ∼20% efficiency, which accounted for up to approximately one-third of total editing events. These results establish Cas9 RNP technology for diverse experimental and therapeutic genome engineering applications in primary human T cells.
The CRISPR/Cas9 system has been used increasingly to edit mammalian germline sequence and cell lines (1, 2). Considerable efforts are underway to use this powerful system directly in primary human tissues, but efficiency has been limited, especially in human CD4+ T cells. Plasmid delivery of cas9 and single-guide RNAs (sgRNAs) was efficient in other cell types, but ablated only 1–5% of target protein expression in CD4+ T cells (3). Improved ability to ablate key targets and correct pathogenic genome sequence in human T cells would have direct therapeutic applications, eventually allowing T cells to be edited ex vivo and then reintroduced into patients.
Multiple scientific and clinical trials are underway to manipulate T-cell genomes with available technologies, including gene deletions with transcription activator-like effector nucleases and zinc finger nucleases and exogenous gene introduction by viral transduction (4, 5). Genetic manipulations have been attempted to “knock out” HIV coreceptors CXCR4 and CCR5 in T cells to gain resistance to HIV infection (6–8). There also has been marked success in engineering T cells to recognize and kill hematological malignancies, but additional genetic modifications appear necessary for solid organ tumor immunotherapy (9–11). Deletion of genes that encode key immune checkpoints such as PD-1 could prove useful for these efforts (12, 13). Further therapeutic opportunities would be possible if targeted T-cell genomic loci could be corrected with specific replacement sequence, rather than deleted (14). Efficient technology to promote homologous recombination in T cells could eventually allow therapeutic correction of mutations that affect specialized T-cell functions.
Recent reports in mammalian cell lines demonstrate that Cas9 ribonucleoproteins (RNPs; recombinant Cas9 protein complexed with an in vitro-transcribed single-guide RNA) can accomplish efficient and specific genome editing (15–17). Here we show that electroporation of Cas9 RNPs leads to efficient genome editing of CD4+ T cells. We were able to ablate a target gene with the random insertion and deletion mutations that likely result from nonhomologous end joining (NHEJ) repair of a Cas9-induced double-stranded DNA break (DSB). Cells with genomic edits in CXCR4 could be enriched by sorting based on low CXCR4 expression. We were also able to introduce precisely targeted nucleotide replacements in primary T cells at CXCR4 and PD-1 by homology-directed repair (HDR) using Cas9 RNPs and exogenous single-stranded DNA templates. This technology enabled Cas9-mediated generation of “knock-in” primary human T cells. Deep sequencing of a target site confirmed that Cas9 RNPs promoted knock-in genome modifications with up to ∼20% efficiency (∼22% was achieved with 50 pmol and ∼18% with 100 pmol of HDR template), which accounted for up to approximately one-third of the total editing events. These findings suggest that Cas9 RNP-mediated nucleotide replacement could eventually prove useful for therapeutic correction of disease-associated mutations. Our study establishes Cas9 RNP technology for experimental and therapeutic knock-out and knock-in editing of the genome in primary human T cells.
Results
We aimed to overcome long-standing challenges in genetic manipulation of primary T cells and establish an efficient genome engineering toolkit. Recent reports in mammalian cell lines suggest that Cas9 RNPs can accomplish efficient and specific genome editing (15–18). Given the significant challenges of efficient genome editing of T cells with DNA delivery of Cas9, we tested the efficacy of Cas9 RNP delivery for targeted genome editing in primary human T cells (Fig. 1A).
Fig. 1.
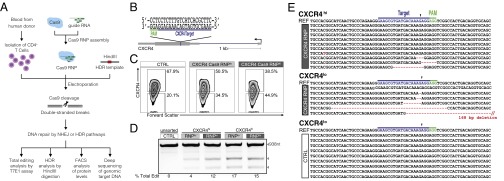
Efficient editing of CXCR4 in primary human CD4+ T cells. (A) Experimental scheme of Cas9:single-guide RNA ribonucleoprotein (Cas9 RNP) delivery to primary human CD4+ T cells for genome editing, followed by genetic and phenotypic characterization. (B) Schematic representation of sgRNA target (blue) and PAM (green) sequence designed to edit coding sequence in the human CXCR4 locus. (C) FACS plots show increasing percentages of cells with low CXCR4 expression (CXCR4lo) with higher concentrations of CXCR4 Cas9 RNP (Cas9 RNP lo: 0.9 µM; Cas9 RNP hi: 1.8 µM) compared with control-treated cells (Cas9 without sgRNA, CTRL; final concentration: 1.8 µM). (D) T7 endonuclease I (T7E1) assay demonstrates genome editing in the CXCR4 locus with more editing observed in FACS-sorted CXCR4lo cells than in CXCR4hi cells. Expected PCR product size (938 nt) and approximate expected sizes of T7E1-digested fragments are indicated. The total editing frequencies are indicated as percentage of Total Edit below the agarose gel image. (E) Mutation patterns detected by cloning and Sanger sequencing of the CXCR4 locus in sorted Cas9 RNP (1.8 µM)-treated CXCR4hi and CXCR4lo cells are compared with the sequence from CXCR4lo control-treated cells (CTRL). Reference (REF) sequence is shown on top of clonal sequences from each population with sgRNA target (blue) and PAM (green) sequences indicated. Red dashes denote deleted bases, and red sequences indicate mutated nucleotides. Arrowhead indicates the predicted Cas9 cut site. Poor quality sequences obtained from three additional CXCR4lo clones were removed from the sequence alignment.
Ablation of HIV Coreceptor CXCR4 with Cas9 RNPs.
A major goal in T-cell engineering is targeted ablation of specific cell-surface receptors, including coreceptors for HIV infection and coinhibitory immune checkpoints that impair tumor immune response. Here, we programmed the Cas9 RNPs to target the exonic sequence of CXCR4, which encodes a chemokine receptor with multiple roles in hematopoiesis and cell homing that is expressed on CD4+ T cells and serves as a coreceptor for HIV entry (19–21). We purified recombinant Streptococcus pyogenes Cas9 carrying two nuclear localization signal sequences fused at the C terminus. This Cas9 protein was incubated with in vitro-transcribed sgRNA designed to uniquely recognize the human CXCR4 genomic sequence (Fig. 1B). These preassembled Cas9 RNP complexes were electroporated into human CD4+ T cells isolated from healthy donors.
Electroporation of CXCR4 Cas9 RNPs caused efficient, site-specific editing of genomic DNA. The Cas9 RNP-induced DSBs in the CXCR4 gene were likely repaired by NHEJ, a predominant DNA repair pathway in cells that gives rise to variable insertions and deletions (indels) and often results in frameshift mutations and loss of gene function (22). Flow cytometry revealed a Cas9 RNP dose-dependent increase in the percentage of T cells expressing low levels of CXCR4, consistent with mutation of the CXCR4 gene (Fig. 1C). The T7 endonuclease I (T7E1) assay is a convenient method to assess editing at specific sites in the genome. Here, T7E1 assay confirmed genomic DNA editing at the CXCR4 locus in cells treated with CXCR4 Cas9 RNPs, but not in control cells treated with Cas9 protein alone (no sgRNA; CTRL). Cas9 RNP-treated cells were separated based on CXCR4 expression with fluorescence-activated cell sorting (FACS). Using the T7E1 assay, we found an enrichment of editing in the CXCR4lo cells (15–17%) compared with CXCR4hi cells (4–12% with varying doses of Cas9 RNP) (Fig. 1D). Sanger sequencing of the target CXCR4 genomic site, performed to directly identify editing events, suggested that the T7E1 assay may have underestimated editing efficiency. The T7E1 assay uses denaturation and hybridization of the wild-type and mutant sequences to create a mismatch DNA duplex, which is then digested by T7 endonuclease. However, hybridization of the mismatch duplex may be inefficient, especially when the indel mutation is drastically different from the wild-type sequence, making self-hybridization an energetically more favorable product. Other potential reasons for observed underestimation of editing efficiency with endonuclease assays include incomplete duplex melting, inefficient cleavage of single-base-pair indels, and deviation from the expected 300- and 600-bp products on the agarose gel as a result of large genome edits (23). Sequencing of the CXCR4 gene in CXCR4lo cells showed that 5/6 clones had mutations/deletions whereas such mutations/deletions were observed in only 4/10 clones and 0/9 clones in CXCR4hi and CTRL-treated CXCR4lo cells, respectively. Importantly, none of the observed edits in the CXCR4hi population terminated the coding sequence (one missense mutation and three in-frame deletions), consistent with the maintenance of protein expression. By contrast, the CXCR4lo population was enriched for cells with a more extensive mutational burden in the locus (Fig. 1E). These findings demonstrated successful genomic targeting with Cas9 RNPs and a functional effect on protein expression in human CD4+ T cells. Importantly, FACS was able to enrich the population of edited cells, providing an additional useful tool for Cas9 RNP applications in primary T cells.
Efficient Genetic Knock-In with Homology-Directed Repair.
Exogenous template-mediated HDR is a powerful technique for precise gene modifications that would enable experimental and therapeutic editing of specific variant sequences. Given the high editing efficiency of Cas9 RNPs, we next tested whether we could achieve exogenous template-mediated HDR in primary T cells. We used a single-stranded oligonucleotide DNA template (HDR template) with 90 nucleotide homology arms to recombine with the CXCR4 locus at the Cas9 RNP cleavage site (16). The CXCR4 HDR template was designed to replace 12 nucleotides from the human reference genome, including the protospacer adjacent motif (PAM) sequence required for CRISPR-mediated DNA cleavage, and to introduce a HindIII restriction enzyme cleavage site (Fig. 2A). Cas9 RNPs were electroporated into primary CD4+ T cells in the presence of four different concentrations of CXCR4 HDR template (0, 50, 100, and 200 pmol; SI Materials and Methods). Cas9 RNP without HDR template again reduced the percentage of CXCR4hi cells. Notably, in this experiment, addition of the CXCR4 HDR template improved the efficacy of CXCR4 ablation, although this effect on cell-surface expression was not seen in all experiments (Fig. S1A). In the experiment shown here, ∼60% of cells lost high-level cell-surface CXCR4 expression with 100 pmol HDR template and Cas9 RNP (1 vs. 60% in control-treated cells) (Fig. 2 B and C).
Fig. 2.

Efficient homology-directed repair allows targeted DNA replacement in primary human T cells. (A) Schematic representation of single-stranded oligonucleotide HDR template with 90-nt homology arms designed to replace 12 nt, including the PAM sequence, and introduce a novel HindIII restriction enzyme cleavage site (red) at the CXCR4 locus, where the Cas9 RNP cleaves. sgRNA target (blue) and PAM (green) sequence are indicated. (B) Histograms of CXCR4 cell-surface staining assessed by flow cytometry in CXCR4 Cas9 RNP-treated cells in the presence of varying concentrations of single-stranded HDR template (compared with control Cas9 protein-treated cells and unstained cells). (C) FACS plots (corresponding to histograms in B) show maximal ablation of CXCR4 with Cas9 RNP treatment and 100 pmol of HDR template. (D) T7E1 assay was used to estimate the “% Total Edit” (defined as the sum of all NHEJ and HDR events that gives rise to indels at the Cas9 cleavage site), whereas HDR frequency was determined by HindIII digestion, which specifically cleaved the newly integrated HindIII site, and was calculated as the ratio of DNA product to DNA substrate. Expected PCR product size (938 nt) and approximate expected T7E1 and HindIII digestion fragments are indicated.
Remarkably efficient HDR was observed in cells treated with Cas9 RNP and the single-stranded oligonucleotide HDR template (Fig. 2D). Up to 33% total editing (defined as the sum of all NHEJ and HDR events that give rise to indels at Cas9 cleavage site) was observed in the presence of 50 pmol CXCR4 HDR template, as estimated by T7E1 assays. At this concentration, 14% HDR was estimated by HindIII digest of the target locus, indicating that a high fraction of editing resulted from HDR (see results below for further quantification). The nearly complete loss of CXCR4 staining with addition of the HDR template suggests that the mutation introduced by HDR (84DLLFV88 → 84ESLDP88) strongly affected the cell-surface expression of CXCR4 or its recognition by the antibody (Fig. 2 B and C). The editing efficiency was reduced with 200 pmol HDR template, perhaps as a result of cellular toxicity.
Both total editing and HDR could be enriched by sorting the CXCR4lo population, although the effect was less pronounced than in Fig. 1, consistent with the larger fraction of CXCR4lo cells in the unsorted population. Note that in these experiments a more stringent gate was applied to separate the cells with the highest expression of CXCR4, and in this CXCR4hi population no editing was observed. These studies collectively demonstrated the power of Cas9 RNPs coupled with single-stranded oligonucleotide HDR template to precisely replace targeted DNA sequences in primary human T cells.
Deep Sequencing of Target Genomic DNA.
Deep sequencing of the targeted CXCR4 locus allowed more detailed and quantitative analysis of genome-editing events. The results highlighted in Fig. 3 show the frequency of insertions, deletions, and HDR-mediated nucleotide replacement in CXCR4 Cas9 RNP-treated cells with or without CXCR4 HDR template compared with control-treated cells. In CXCR4 Cas9 RNP-treated cells, we found 55% of reads overlapping the CXCR4 target site containing at least one indel within a 200-nucleotide window centered around the expected cut site (Fig. 3 A and B and Dataset S1). As discussed above, the T7E1 assays are useful for identifying edited loci, but may underestimate actual editing efficiency (quantitation of the T7E1 assay in Fig. 2D suggested 33% editing compared with the 55% editing efficiency computed by deep sequencing). We also sequenced the two top predicted “off-target” sites for the CXCR4 Cas9 RNP (Fig. 3B). Rare indels were observed at both off-target sites (∼1–2%), but at a rate comparable to that observed for those sites in the control cells treated with Cas9 protein only (∼1–2%) (Dataset S1).
Fig. 3.
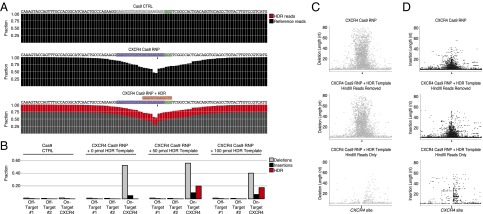
Quantitative analysis of Cas9 RNP-mediated editing and HDR by deep sequencing. (A) CXCR4 Cas9 RNP-mediated indels and HDR from experiments in Fig. 2 were analyzed by targeted deep sequencing of the CXCR4 locus. A total of 100 nt centered on the predicted cut site are shown with sgRNA target (blue), PAM (green), and predicted sequence after HDR genome targeting (red). At each position, the fraction of reads that correctly aligned to the reference genome (black) or HDR template-derived sequence (red) are shown. Although rare (∼1–2%), edits were detected with Cas9-only control treatment, including at the predicted CXCR4 cut site, potentially indicating trace amounts of experimental contamination of the Cas9 RNPs. (B) Bar graph summarizes the fractions of reads edited with deletions (gray), insertions (black), or successful HDR targeting (red) in Cas9 CTRL, CXCR4 Cas9 RNP, and CXCR4 Cas9 RNP cells with 50 or 100 pmol CXCR4 HDR template at the CXCR4 site and two predicted off-target sites. Reads with HDR template-derived sequence incorporated were removed to calculate fractions with deletions and insertions (Dataset S1). Scatter plots show the genomic localization (±100 nt around the expected Cas9 cut side; chromosome 2: 136873140–136873340) and the length of (C) deletions and (D) insertions. (Top) Deletions/insertions for CXCR4 RNP-treated cells. (Middle) Deletions/insertions in reads without HDR template sequence incorporated in cells treated with CXCR4 RNP and CXCR4 HDR template. (Bottom) Deletions/insertions in reads with HDR template-derived sequence incorporated. Arrowheads indicate approximate location of expected Cas9 cut site.
The deep-sequencing results allowed quantitative analysis of observed indel mutations and their spatial distribution in the target region. Consistent with reports that S. pyogenes Cas9 cuts about three nucleotides upstream from the PAM sequence, we found the highest frequency of indels at four nucleotides upstream of the PAM (Fig. 3A). Indels were distributed throughout the sequenced region (Fig. 3 C and D) with the majority of events near cut sites (>94% within 40 nucleotides). In CXCR4 Cas9 RNP-treated cells within ±100 nucleotides from the cut site, we observed that 95% of reads with indels contained a deletion event whereas 10% contained an insertion event (Dataset S1). Interestingly, of the reads with insertion events, ∼50% also contained at least one deletion. We observed a wide range of insertion and deletion sizes, with many reads exhibiting deletions up to ∼80 nucleotides in length (mean: 18 nucleotides; SD: 15 nucleotides) and some insertions up to ∼55 nucleotides in length (mean: 4.4 nucleotides; SD: 4.8 nucleotides) (Fig. 3 C and D and Fig. S2). This range of indel sizes and locations was consistent with the extensive mutational burden observed in Sanger sequencing of CXCR4lo selected cells in Fig. 1.
Deep sequencing verified the successful targeted replacement of 12 nt at the CXCR4 locus, but only in cells treated with both Cas9 RNPs and CXCR4 HDR template. We observed 25% incorporation of HDR template sequence with 50 pmol HDR template and 21% with 100 pmol HDR template (Fig. 3A). Of the reads with HDR template sequence incorporated, ∼14% of the detected HDR template reads had additional nonspecific indels surrounding the incorporated HindIII site or other imperfect forms of editing within the 200-nucleotide window centered at the predicted cut site. However, the frequency of indels in reads with the HindIII site incorporated was reduced compared with reads where the HindIII site was not detected (Fig. 3 C and D and Fig. S2). Interestingly, there was a consistent pattern of deletion events between CXCR4 Cas9 RNP with and without CXCR4 HDR template with an enrichment of deletions of 2 nucleotides (11%) and 22 nucleotides (5.4%) (Fig. S2). Replacement of the PAM sequence likely helped to limit recutting of knock-in sequence. Overall, 18–22% of reads (with varying concentrations of HDR template) had correctly replaced nucleotides throughout the sequenced genomic target site, suggesting that this approach could prove useful for generation of experimental and therapeutic nucleotide knock-in primary human T cells.
Specific Knock-In Targeting of Key Cell-Surface Receptors.
To confirm that Cas9 RNPs mediate HDR at other genomic sites, we designed a guide RNA and HDR template to target the PD-1 (PDCD1) locus. PD-1 is an “immune checkpoint” cell-surface receptor found on the surface of chronically activated or exhausted T cells that can inhibit effective T-cell–mediated clearance of cancers. Monoclonal antibody blockade of PD-1 is approved for treatment of advanced malignancy, and genetic deletion of PD-1 may prove useful in engineering T cells for cell-based cancer immunotherapies (12). Primary human T cells were electroporated with a PD-1 Cas9 RNP and a PD-1 HDR template designed to generate a frameshift mutation and a knock-in a HindIII restriction site in the first exon of PD-1, thereby replacing the PAM sequence (Fig. 4A).
Fig. 4.

Cas9 RNPs can be programmed for knock-in editing of PD-1 or CXCR4. (A) Schematic representation of the single-stranded PD-1 HDR template with 90-nt homology arms designed to replace 12 nt with 11 nt, introducing a novel HindIII restriction enzyme cleavage site to replace the PAM sequence (red). sgRNA target (blue) and PAM (green) sequences are indicated. (B) Histograms of PD-1 cell-surface expression levels assessed by flow cytometry. All cells were treated with 100 pmol of PD-1 HDR template. PD-1 Cas9 RNP-treated cells are shown in blue, CXCR4 Cas9 RNP-treated cells in light gray, and scrambled guide (no predicted cut within the human genome) Cas9 RNP-treated cells in dark gray. (C) Histograms of CXCR4 cell-surface expression levels assessed by flow cytometry. All cells were treated with 100 pmol of CXCR4 HDR template. CXCR4 Cas9 RNP-treated cells are shown in red, PD-1 Cas9 RNP-treated cells in light gray, and scrambled guide Cas9 RNP-treated in dark gray. (B and C) Results of four experiments with two differently in vitro-transcribed and purified CXCR4 and PD-1 sgRNAs (SI Materials and Methods) tested in two different blood donors. For each blood donor, experiments done with phenol/chloroform-extracted sgRNAs are shown on Top and experiments with PAGE-purified sgRNAs are shown at the Bottom; scrambled guides were prepared for both experiments with phenol/chloroform extraction. Dotted line indicates gating on PD-1 high-expressing or CXCR4 high-expressing cells, respectively. The percentage of PD-1 high-expressing cells was significantly lower with PD-1 Cas9 RNP treatment compared either CXCR4 Cas9 RNP treatment (P < 0.001) or scrambled guide Cas9 RNP treatment (P < 0.001). The percentage of CXCR4 high-expressing cells was significantly lower with CXCR4 Cas9 RNP treatment compared with either PD-1 Cas9 RNP treatment (P < 0.001) or scrambled guide Cas9 RNP treatment (P < 0.001) (Pearson’s χ2). (D) Genome editing was analyzed by T7E1 assay, whereas HDR was detected by HindIII digestion, which specifically cleaved the newly integrated HindIII site; cleavage products for both assays are indicated with arrowheads. Concentrations of various HDR templates are indicated above the agarose gels. CTRL HDR template refers to a scrambled version of the original CXCR4 HDR template, including a HindIII restriction site. A nonspecific second gel band of unclear significance was noted in the T7E1 of the PD-1 amplicon under all conditions. Total editing and HDR frequencies were calculated and are displayed below agarose gel images.
To examine the specificity of Cas9 RNP-mediated targeting, we compared PD-1 cell-surface expression following treatment with PD-1 Cas9 RNP vs. CXCR4 Cas9 RNP (which should not target the PD-1 locus) or scrambled guide Cas9 RNP (no predicted cut within the human genome). We performed replicate experiments side by side with two different blood donors and with sgRNAs generated with two different in vitro transcription protocols (SI Materials and Methods). PD-1 Cas9 RNPs electroporated with PD-1 HDR template significantly reduced the percentage of cells with high PD-1 cell-surface expression relative to both CXCR4 Cas9 RNPs and scrambled guide Cas9 RNPs delivered with PD-1 HDR template (Fig. 4B). Similarly, CXCR4 Cas9 RNPs and CXCR4 HDR template caused a decrease in the CXCR4hi cell population relative to both PD-1 and scrambled guide Cas9 RNP treatments with CXCR4 HDR template (Fig. 4C). Loss of CXCR4 was not a nonspecific effect of single-stranded DNA delivered along with CXCR4 Cas9 RNP; we observed a higher percentage of CXCR4-expressing cells after treatment with CXCR4 Cas9 RNP and scrambled HDR template than with CXCR4 Cas9 RNP and CXCR4 HDR template (Fig. S1A). These findings confirmed the target-specific modulation of cell-surface receptor expression in primary T cells with the programmable Cas9 RNP and HDR template treatments.
We then tested the specificity of HDR templates for nucleotide replacement (Fig. 4D; examples of corresponding cell-surface expression data are shown in Fig. S1B). As expected, we observed efficient PD-1 editing by PD-1 Cas9 RNPs regardless of whether they were delivered with PD-1 HDR template, CXCR4 HDR template, or without any HDR template. In contrast, the HindIII site was incorporated into PD-1 only in the presence of both PD-1 Cas9 RNP and PD-1 HDR template, but not with CXCR4 HDR template, which should not be recombined at PD-1 locus due to the lack of sequence homology. Similarly, a HindIII site was incorporated only into CXCR4 following treatment with CXCR4 Cas9 RNP and CXCR4 HDR template; HDR was not observed at the CXCR4 locus with PD-1 HDR template, control scrambled HDR template (with a HindIII site), or without HDR template (Fig. 4D). Taken together, these studies established that specific pairing of a programmed Cas9 RNP and corresponding HDR template is required for targeted nucleotide replacement in primary human T cells.
Discussion
Cas9-mediated genome engineering has enormous potential to experimentally and therapeutically target DNA elements crucial for T-cell function. We report here successful genome engineering in human CD4+ T cells by delivery of in vitro-assembled Cas9 RNPs. Electroporation of Cas9 RNPs allowed targeted knock-out of the CXCR4 cell-surface receptor. Cas9 RNPs also promoted successful Cas9-mediated genetic knock-in of specific nucleotides to CXCR4 and PD-1 in primary human T cells. The efficient targeted DNA replacement in mature immune cells achieves a longstanding goal in the immunology field to enable diverse research and therapeutic applications. These studies collectively establish a broadly applicable toolkit for genetic manipulation of human primary T cells.
There are notable advantages to genome engineering with transient Cas9 RNP delivery compared with other CRISPR/Cas9 delivery methods. Recent work reported ablation of cell-surface markers in human CD4+ T cells by transfection of plasmid carrying the cas9 gene and guide RNA-coding sequence (3). Although successful, efficiency was notably low in CD4+ T cells compared with other cell types, possibly due to suboptimal levels of Cas9 or sgRNA, suboptimal nuclear translocation, or suboptimal intracellular Cas9 RNP complex formation (or some combination of these factors). Cas9 RNP-based delivery circumvents these challenges. Delivery of Cas9 RNPs offers fast editing action and rapid protein turnover in the cells as they are reportedly degraded within 24 h of delivery (15). This limited temporal window of Cas9 editing may make Cas9 RNPs safer for therapeutic applications than other delivery modes where cells are exposed to Cas9 for a longer time frame. Further testing will be needed to assess the purity of Cas9 RNPs and potential off-target effects before any clinical use. Our findings now suggest that Cas9 RNPs are able to rapidly and efficiently edit human T cells.
We were able to achieve remarkably efficient HDR here with almost complete loss of the CXCR4hi cell population with Cas9 RNPs and an HDR template targeting CXCR4 in one experiment. Up to 25% of the reads showed incorporation of HDR template sequence in the CXCR4 locus, and ∼20% of the reads showed correctly replaced sequence throughout the target site with no additional indels. Future studies will optimize remaining variables that affect editing and HDR efficiency in primary T cells. We recently demonstrated in cultured cell lines that variation in cell type and cell cycle dynamics significantly alter Cas9 RNP efficiency (16). In primary human T cells, editing efficiency could also be affected by T-cell donor-specific factors (e.g., genetics, recent infection), in vitro T-cell activation status, and characteristics of the targeted genomic locus (e.g., DNA sequence, chromatin state). Characterizing these variables and further optimizing genome engineering efficiency will accelerate the experimental and therapeutic editing of T cells using Cas9 RNPs.
The ability to edit specific DNA sequences in human T-cell subsets will enable experimental investigation of transcription factors, cis-regulatory elements, and target genes implicated in T-cell inflammatory and suppressive functions. Extensive efforts have mapped key gene regulatory circuitry controlling the development and function of diverse and specialized T-cell subsets (24). We recently reported that many causal genetic variants contributing to risk of human autoimmune diseases map to key regulatory elements in T cells (25). Looking forward, genome editing of primary T cells will now provide a powerful perturbation test to assess the function of regulatory elements and characterize the effects of disease-associated coding and noncoding variation.
Therapeutic editing requires improved techniques to identify successfully edited cells in a population. Selection of edited cells is notably challenging in primary cells that cannot be maintained indefinitely in culture, unlike transformed cell lines. Here we demonstrate FACS enrichment of edited cells based on expected phenotypic changes in cell-surface receptor expression. The success of Cas9 RNP-mediated HDR should also allow introduction of genetic markers to purify homogenously edited cells for further functional characterization and potentially also for therapeutic applications.
Therapeutic T-cell engineering requires efficient and precisely targeted genome editing in primary cells. The Cas9 RNP technology reported here should accelerate efforts to correct genetic variants and engineer human T-cell function for the treatment of infection, autoimmunity, and cancer.
Materials and Methods
Human T-cell Isolation and Culture.
Human primary T cells were either isolated from fresh whole blood or buffy coats. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll gradient centrifugation. CD4+ T cells were pre-enriched with an Easysep Human CD4+ T-cell enrichment kit (Stemcell Technologies) according to the manufacturer’s protocol. Pre-enriched CD4+ T cells were stained with following antibodies: αCD4-PerCp (SK3; Becton Dickinson), αCD25-APC (BC96; TONBO Biosciences), αCD127-PE (R34-34; TONBO Biosciences), αCD45RA-violetFluor450 (HI100; TONBO Biosciences), and αCD45RO-FITC (UCHL1; TONBO Biosciences). CD4+CD25loCD127hi T effectors (Teffs) were isolated using a FACS Aria Illu (Becton Dickinson).
Cas9 RNP Assembly and Electroporation.
Cas9 RNPs were prepared immediately before experiments by incubating 20 µM Cas9 with 20 µM sgRNA at 1:1 ratio in 20 μM Hepes (pH 7.5), 150 mM KCl, 1 mM MgCl2, 10% (vol/vol) glycerol, and 1 mM Tris(2-chloroethyl) phosphate (TCEP) at 37 °C for 10 min to a final concentration of 10 µM. T cells were electroporated with a Neon transfection kit and device (Invitrogen).
Analysis of Genome Editing.
Editing efficiency was estimated by T7 endonuclease I assay. HDR templates were designed to introduce a HindIII restriction site into the targeted gene loci; successful HDR was confirmed with HindIII restriction enzyme digestion. The genomic DNA library, flanking the regions of Cas9 target sites for the CXCR4 on-target and two predicted off-target genes, was assembled by 2-step PCR method and sequenced with the Illumina HiSeq 2500.
See SI Materials and Methods for further details.
Supplementary Material
Acknowledgments
We thank Mary Rieck, Jacqueline Howells, Amy Putnam, and Caroline Raffin in the J.A.B. laboratory; Richard Lao and the University of California at San Francisco (UCSF) Institute for Human Genetics Genomics Core; Michael Lee, Vinh Nguyen, and the UCSF Flow Cytometry Core; Amy Lee in the Cate laboratory; all members of the A.M., J.A.B., and J.A.D. laboratories for suggestions and technical assistance; and K. M. Ansel for critical reading of the manuscript. This research was supported by the UCSF Sandler Fellowship (to A.M.); a gift from Jake Aronov (to A.M.); NIH funding for the HIV Accessory & Regulatory Complexes Center (P50GM082250) (to A.M. and J.A.D.); a National MS Society Collaborative MS Research Center Award (to A.M. and J.A.D.); and the Howard Hughes Medical Institute (HHMI) (J.A.D.). S.L. is an HHMI Fellow of the Damon Runyon Cancer Research Foundation [DRG-(2176-13)], and G.E.H. is supported by an NIH training grant to UCSF Diabetes, Endocrinology and Metabolism (T32 DK741834).
Footnotes
Conflict of interest statement: J.A.D. is a co-founder of Caribou Biosciences Inc., Editas Medicine, and Intellia and is on the scientific advisory board of Caribou Biosciences Inc. The A.M. laboratory receives sponsored research support from Epinomics. A patent has been filed based on the findings described here.
Data deposition: The sequence reported in this paper has been deposited in the National Center for Biotechnology Information (NCBI) BioSample database, www.ncbi.nlm.nih.gov/biosample/ (accession no. SAMN03850749).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1512503112/-/DCSupplemental.
References
- 1.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213):1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 2.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157(6):1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mandal PK, et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 2014;15(5):643–652. doi: 10.1016/j.stem.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maus MV, et al. Adoptive immunotherapy for cancer or viruses. Annu Rev Immunol. 2014;32:189–225. doi: 10.1146/annurev-immunol-032713-120136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Passerini L, et al. CD4⁺ T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci Transl Med. 2013;5(215):215ra174. doi: 10.1126/scitranslmed.3007320. [DOI] [PubMed] [Google Scholar]
- 6.Hütter G, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360(7):692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 7.Didigu CA, et al. Simultaneous zinc-finger nuclease editing of the HIV coreceptors ccr5 and cxcr4 protects CD4+ T cells from HIV-1 infection. Blood. 2014;123(1):61–69. doi: 10.1182/blood-2013-08-521229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tebas P, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370(10):901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat Rev Immunol. 2012;12(4):269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moon EK, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. 2014;20(16):4262–4273. doi: 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.John LB, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19(20):5636–5646. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 14.Genovese P, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510(7504):235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24(6):1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 2014;3:e04766. doi: 10.7554/eLife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zuris JA, et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol. 2015;33(1):73–80. doi: 10.1038/nbt.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sung YH, et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Res. 2014;24(1):125–131. doi: 10.1101/gr.163394.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393(6685):595–599. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 20.Berson JF, et al. A seven-transmembrane domain receptor involved in fusion and entry of T-cell-tropic human immunodeficiency virus type 1 strains. J Virol. 1996;70(9):6288–6295. doi: 10.1128/jvi.70.9.6288-6295.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272(5263):872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 22.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 23.Guschin DY, et al. A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol. 2010;649:247–256. doi: 10.1007/978-1-60761-753-2_15. [DOI] [PubMed] [Google Scholar]
- 24.Vahedi G, et al. Helper T-cell identity and evolution of differential transcriptomes and epigenomes. Immunol Rev. 2013;252(1):24–40. doi: 10.1111/imr.12037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farh KK, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518(7539):337–343. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.