

Abstract
Kaempferol, a natural dietary flavonoid, is well known to possess chemopreventive and therapeutic anticancer efficacy; however, its antimetastatic effects have not been mechanistically studied so far in any cancer model. This study was aimed to investigate the inhibitory effect and accompanying mechanisms of kaempferol on epithelial-to-mesenchymal transition (EMT) and cell migration induced by transforming growth factor-β1 (TGF-β1). In human A549 non–small lung cancer cells, kaempferol strongly blocked the enhancement of cell migration by TGF-β1–induced EMT through recovering the loss of E-cadherin and suppressing the induction of mesenchymal markers as well as the upregulation of TGF-β1–mediated matrix metalloproteinase-2 activity. Interestingly, kaempferol reversed TGF-β1–mediated Snail induction and E-cadherin repression by weakening Smad3 binding to the Snail promoter without affecting its C-terminus phosphorylation, complex formation with Smad4, and nuclear translocation under TGF-β1 stimulation. Mechanism study revealed that the phosphorylation of Smad3 linker region induced by TGF-β1 was required for the induction of EMT and cell migration, and selective downregulation of the phosphorylation of Smad3 at Thr179 residue (not Ser204, Ser208, and Ser213) in the linker region was responsible for the inhibition by kaempferol of TGF-β1–induced EMT and cell migration. Furthermore, Akt1 was required for TGF-β1–mediated induction of EMT and cell migration and directly phosphorylated Smad3 at Thr179, and kaempferol completely abolished TGF-β1–induced Akt1 phosphorylation. In summary, kaempferol blocks TGF-β1–induced EMT and migration of lung cancer cells by inhibiting Akt1-mediated phosphorylation of Smad3 at Thr179 residue, providing the first evidence of a molecular mechanism for the anticancer effect of kaempferol.
Introduction
Non–small-cell lung cancer (NSCLC) is the most common type of lung cancer. NSCLC shows a poor prognosis and accounts for the leading cause of cancer-related death every year worldwide [1]. Because of the lack of robust and dependable molecular markers for the early diagnosis, most patients with NSCLC present locally advanced and metastatic cancer disease at the time of diagnosis. Moreover, the metastasis of lung cancer cells is a major contributor in aggressiveness of NSCLC and is responsible for the main cause of deaths in lung cancer patients. Therefore, identifying of the key factors that contribute to the lung metastatic process and a better understanding of the molecular mechanisms underlying lung cancer metastasis are crucial in providing a promising approach for lung cancer therapy that target metastasis.
Tumor metastasis is a dynamic multistep cascade process. In the metastatic process, epithelial-to-mesenchymal transition (EMT) is an important morphogenetic event for triggering metastasis from primary tumors and is characterized by the loss of E-cadherin–mediated cell-cell junction and the upregulation of mesenchymal markers including N-cadherin, vimentin, and fibronectin [2,3]. Therefore, changes during EMT lead to the transition of a polarized epithelial phenotype to a migratory mesenchymal phenotype, and then cells degrade extracellular matrix by activating matrix metalloproteinases (MMPs) and have invasive characteristics.
Transforming growth factor-β1 (TGF-β1), a prototypical member of the TGF-β superfamily, is a multifunctional cytokine that regulates cell proliferation, differentiation, apoptosis, and migration [4]. In normal physiologic condition, TGF-β1 has tumor-suppressive functions through inhibiting cell proliferation and promoting apoptosis. However, TGF-β1 acts also as a metastatic inducer by promoting EMT in late-stage tumor progression [5]. Molecular signaling mechanism studies on TGF-β1–induced EMT indicate crucial roles of Smad3 signaling pathway. Depletion of Smad3 completely abolishes TGF-β1 induction of EMT [6–8]. Smad3 acts as a transcriptional activator of E-cadherin repressors such as Snail, Slug, and Twist [9–11]. Smad3 also negatively regulates E-cadherin by upregulating ZEB1 and ZEB2 via microRNA-200 pathway [12].
Smad3 is a key mediator of TGF-β signaling pathway. Upon TGF-β1 stimulation, TβRI is activated by TβRII and mediates the phosphorylation of the conserved COOH-tail serine residues of Smad3. The phosphorylated Smad3 interacts with Smad4 and translocates from the cytosol into the nucleus, where it regulates transcription of target genes [13,14]. However, the regulatory mechanisms by which Smad3 determines the functional outcome of TGF-β1 responses under physiologic and pathologic conditions have yet to be fully elucidated. Smad3 linker region is a less conserved intermediate region that connects between conserved Mad-homology (MH) 1 and MH2 domains and contains several threonine and serine residues (Thr179, Ser204, Ser208, and Ser213) that phosphorylated by fundamental signaling kinases in a strongly cell context-dependent manner [15–19]. Several lines of recent evidence indicate the phosphorylation of the linker region of Smad3 as a crucial determinant of distinct cellular responses to TGF-β1 in normal and cancer cells. For example, in early carcinogenic process, hepatitis B virus X protein shifts hepatocytic TGF-β signaling from the tumor-suppressive Smad3 pathway to the oncogenic Smad3 pathway through inducing c-Jun NH2-terminal kinase–mediated phosphorylation of Smad3 linker region [20]. Constitutively activated Ras confers a selective advantage on benign tumors by inducing the phosphorylation of Smad3 linker region, leading to carcinoma in situ [21]. Hydrogen peroxide–mediated phosphorylation of Smad3 linker region opposes the antiproliferative responses of epithelial cells induced by TGF-β1 [22]. Therefore, the pharmacologic inhibition of Smad3 phosphorylation in the linker region may prevent progression to advanced carcinoma by suppressing oncogenic TGF-β signaling.
Kaempferol (3,5,7-trihydroxy-2-(-4-hydroxyphenyl)-4H-1-benzopyran-4-one) is a common natural flavonoid (chemical structure as shown in Figure 1A) that is abundant in tea, grapes, berries, and cruciferous vegetables [23]. In several types of human cancer cells, kaempferol acts as a potent antitumor growth agent that inhibits phosphatidylinositol-3-kinase (PI3K) and ribosomal S6 kinase activities [24–26] and increases the expression of tumor suppressor, phosphatase and tensin homolog [27]. Kaempferol also inhibits tumor angiogenesis and expression of MMP-2 [28,29]. However, the effect of kaempferol on the cancer metastasis of NSCLC, as well as the underlying mechanisms of the effect, has not yet been reported. In the present study, we investigated the molecular mechanisms by which kaempferol elicits its anticancer activity against metastatic potentials induced by TGF-β1 in human NSCLC cell lines. Our data clearly demonstrate that kaempferol inhibits TGF-β1–induced EMT, migration, and invasiveness of A549 lung cancer cells by suppressing Akt1-mediated phosphorylation of Smad3 at Thr179 residue in the linker region.
Figure 1.

Kaempferol (KF) inhibits TGF-β1–induced EMT in A549 lung cancer cells. (A) Chemical structure of KF. (B) Phase contrast images of cells treated with DMSO, 5 ng/ml of TGF-β1, or 5 ng/ml of TGF-β1 plus 25 μM of KF for 24 hours. (C) A549 cells were pretreated with 25 μM of KF at the indicated concentrations for 30 minutes and then stimulated with 5 ng/ml of TGF-β1 for 48 hours. Then, the cells were subjected to Western blot analysis for E-cadherin, N-cadherin, and smooth muscle α-actin (SMα actin). β-Actin levels were monitored as a loading control for whole-cell extracts. (D) A549 cells were pretreated with DMSO or 25 μM of KF for 30 minutes before 5 ng/ml of TGF-β1 treatment for 48 hours and then subjected to immunofluorescence staining for E-cadherin (green), N-cadherin (red), and vimentin (red). DAPI (blue) was used to stain nuclei.
Experimental Procedures
Materials
Recombinant TGF-β1 was purchased from R&D systems (Minneapolis, MA). Kaempferol was obtained from Sigma-Aldrich (St. Louis, MO). AktIV, LY294002, U0126, and SB431542 were purchased from Calbiochem (San Diego, CA). Small interfering RNAs for control, Smad2, and Smad3 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell Culture
A549, human non–small lung cancer cell line, was obtained from American Type Culture Collection (Manassas, VA). The cells were cultured in monolayers at 37°C in a 5% CO2 incubator in RPMI1640 medium supplemented with 2 mM l-glutamine, 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin, and 100 mg/ml streptomycin.
Lentiviral Vector Production and Infection
The lentiviral vectors carrying human Smad3 wild type, Smad3 (EPSM), and Smad3 (T179V) were cloned from pCMV-Myc-Smad3 wild type, pCMV-Myc-Smad3 (EPSM), and pCMV-Myc-Smad3 (T179V) (a gift from Dr. Fang Liu; The State University of New Jersey). The lentiviral vector pCAG was digested with Mlu5 and Nhe3, and the two primers used are as follows: forward, 5ʹ-GATCACGCGTGGATCCCATCGATTTAAAGCT-3ʹ; reverse, 5ʹ-GATCGCTAGCCCCTCTAGATGCATG-3ʹ. For the production of lentivirus, 293T cells were co-transfected with pCAG vector together with psPAX2 and pMD2. G (Addgene, Cambridge, MA) by FuGENE6. The viral supernatant was collected 72 hours after transfection, cleared by centrifugation (2000 rpm/min, 10 minutes, and 4°C), and then filtered through a 0.45-μm filter (Millipore, Billerica, MA). Target cells (1 × 105/well) were seeded in six-well plate, and after incubation at 37°C for 24 hours, the medium of each well was replaced with 1-ml viral suspension supplemented with 8 mg/ml Polybrene (Sigma-Aldrich). Then, the plates were centrifuged at 1200 rpm for 30 minutes at room temperature, followed by 12-hour incubation in standard cell culture condition, and media were replaced with fresh Dulbecco’s modified Eagle’s medium. After 48 hours of additional incubation, the protein levels of each gene were analyzed by immunoblotting.
Luciferase Reporter Assay
The pSG5-p110α (K227E), which encodes a constitutively active p110α (CA-p110α), the catalytic subunit of PI3K, and pSG5-p85ΔiSH2-N (deleted for amino acids 478-513 and widely referred to as pSG5-Δp85), which encodes a dominant-negative mutant of p85 (DN-p85), a regulator of PI3K, were gifts from Dr. J. Downward (Cancer Research, UK). The pCMV2-Akt1 (E40K), which encodes a constitutively active Akt1 (CA-Akt1), was provided by Dr. Naoya Fujita (University of Tokyo, Japan). The pcDNA3-HA-Akt1 (K179M), which encodes a dominant-negative mutant of Akt1 (DN-Akt1), was a gift from Dr. Kwon Young-Guen (Yeonse University, Korea). A549 cells in six-well plates (5 × 105 cells per plate) were co-transfected with 0.5 μg of a luciferase reporter plasmid containing the human E-cadherin promoter (gift from Dr. Kyung Lib Jang, Pusan National University, Korea), the human Snail promoter (gift from Dr. Guhung Jung, Seoul National University), or an artificial SBE4-Luc reporter plasmid containing four tandem repeats of Smad-binding elements together with 0.2 μg of the β-galactosidase expression plasmid pCMV-LacZ with the use of FuGENE 6 reagent (Roche, Mannheim) according to the manufacturer’s instructions. Luciferase reporter activity was assessed on a luminometer with a dual-luciferase Reporter Assay System (Promega, Madison, WI). The activity of β-galactosidase was also measured and was used to normalize luciferase activity. The results represent the average and SD of three independent experiments.
Gelatin Zymography
The activity of MMP2 in culture medium was assessed by gelatin zymography. After serum starvation for 24 hours, A549 cells were subjected to treatment and then incubated further for 24 hours. The supernatants were collected, centrifuged at 3000 ×g for 10 minutes, concentrated using Amicon Ultra Centrifugal Filter Units (Millipore), and quantified using Bio-Rad protein assay reagent. Proteins (30 μg) were subjected to SDS-PAGE on a 7.5% gel containing gelatin (2 mg/ml). For detection of gelatinolytic activity, the gel was incubated for overnight at 37°C in a solution containing 50 mM Tris-HCl, pH 7.5, 10 mM CaCl2, 150 mM NaCl, and 0.02% sodium azide; stained with solution containing 0.1% Comassie Brilliant Blue, 25% methanol, and 0.7% acetic acid for 30 minutes; and destained with a solution of 30% methanol and 10% acetic acid.
Scratch Wound Migration Assay
Confluent monolayers of A549 cells were scratched using a sterile 200-μl pipette tip to generate a cell-free gap between two adjoining areas. After removal of the loose cells by twice washes with PBS, cells were subjected to treatment. Scratched wound healing was monitored under phase-contrast microscopy immediately after incision and after 24 hours of treatment. The area of migrating cells was randomly photographed at three separate sites along the length of the scratch.
Transwell Migration Assay
A549 cells uninfected or infected with lentivirus were serum starved (0.1% FBS) overnight and then trypsinized and resuspended in RPMI1640 (serum free) medium at 2 × 105 cells/ml. A total of 250 μl of suspension was applied to the upper chamber of Transwell insert (8-μm pore size, Corning). Cells were immediately exposed to treatment and allowed to migrate for 24 hours toward chemoattracting complete medium present in the bottom chamber. Unmigrated cells on the upper surface were removed, and migrated cells at the bottom of the insert were fixed in methanol and stained with eosin and hematoxylin for cell counting. The number of migrated cells was counted in five randomly selected fields per filter. The results represent the average and SD of three independent experiments.
Western Blotting and Immunoprecipitation
Cellular contents were extracted in lysis buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, 5 mM EDTA, 2 mM DTT, 1 mM sodium orthovanadate, 10 mM β-glycerphosphate, 1 mM sodium fluoride, 1 μM microcystin, 1 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 10 μg/ml aprotinin] at 4°C. Cell lysates were clarified by microcentrifugation at 12,000 rpm/min at 4°C for 10 minutes. The supernatants were recovered, and total protein concentrations were measured using Bio-Rad protein assay reagent. Proteins in the cell lysates (500 μg) were immunoprecipitated with 2 μg of a monoclonal anti-Smad3 antibody, followed by incubation with protein A/G-Sepharose beads. The immunoprecipitates were washed with lysis buffer four times. Immunoprecipitates or equivalent amounts of total cellular proteins were heated at 95°C for 3 minutes and then separated by SDS-PAGE, followed by electrotransfer to polyvinylidene difluoride membranes. The membranes were subsequently blocked for 1 hour with Tris-buffered saline containing 0.05% (v/v) Tween 20 and 5% (w/v) nonfat dry milk and then incubated with appropriate antibodies [anti-myc and anti-vimentin (Santa Cruz Biotechnology), anti-Smad2 and anti-Smad3 (Zymed), anti-phospho-Smad3 (Ser423/425) and anti-phospho-Akt (Ser426) (Cell Signaling Technology, Danvers, MA), and anti-E-cadherin and anti–N-cadherin (BD Transduction laboratories, Lexington, KY), anti–smooth muscle α-actin and anti–β-actin (Sigma-Aldrich)]. Primary antibody binding was detected using horseradish peroxidase–conjugated goat anti-mouse or anti-rabbit immunoglobulin G secondary antibody (Santa Cruz Biotechnology) visualized with Supersignal West Pico Chemiluminscent Substrate (Pierce/ThermoScientific, Rockford, IL).
Chromatin Immunoprecipitation (ChIP) Assay
Cells were cross-linked with 1% formaldehyde at room temperature for 15 minutes, rinsed with ice-cold PBS, and collected. After centrifugation, the precipitate was washed sequentially with 1 ml of buffer I (0.25% Triton X-100, 10 mM EDTA, 0.5 mM EGTA, 10 mM HEPES, pH 6.5) and buffer II (200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 10 mM HEPES, pH 6.5). Cells were then resuspended in 0.3 ml of lysis buffer [1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1, 1 × protease inhibitor cocktail (Roche Molecular Biochemicals, Indianapolis, IN)] and sonicated 10 times for 10 seconds, followed by centrifugation for 10 minutes at 13,000 rpm. The sheared chromatin was incubated on a rolling shaker overnight at 4°C with antibody (2 μg per reaction) and protein A-Sepharose beads [45 μl of a 50% slurry in 10 mM Tris-HCl (pH 8.1) and 1 mM EDTA]. The following rabbit or mouse polyclonal antibodies were used: c-Myc (9E10) and control IgG from Santa Cruz Biotechnology and Smad3 from Invitrogen (Carlsbad, CA). The antibody-bound protein/DNA complexes were precipitated and washed sequentially for 10 minutes each in TSE I [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl (pH 8.1), 150 mM NaCl], TSE II [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl (pH 8.1), 500 mM NaCl], and buffer III [0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl (pH 8.1)]. The captured genomic DNA was eluted, cross-linking was reversed at 94°C for 15 minutes, and proteins were removed by treatment with Proteinase K at 37°C for 1 hour. Ten percent of total genomic DNA from the nuclear extract was used as input. The primer used for detection of Snail promoter sequence as follows: forward primer, 5ʹ-CGCTCCGTAAACACTGGATAA-3ʹ; reverse primer, 5ʹ-GAAGCGAGGAAAGGGACAC-3ʹ. The samples were amplified by conventional polymerase chain reaction (PCR) using the above Smad3 Snail promoter-specific forward and reverse primers and Platinium Taq DNA polymerase (Invitrogen).
Semiquantitative Reverse Transcriptase (RT)-PCR
Total cellular RNA was extracted from cells using the phenol-guanidinium isothiocyanate method [30]. Two micrograms of RNA was reverse transcribed for 1 hour at 42°C and amplified by PCR using specific primers for human Snail (sense, 5ʹ-GGGCAGGTATGGAGAGGAAGA-3ʹ; antisense, 5ʹ-TTCTTCTGCGCTACTGCTGCG-3ʹ) and β-actin (sense, 5ʹ-ACGTTGCTATCCAGGCTGTG-3ʹ; antisense, 5ʹ-GCGACGTAGCACAGCTTCTC-3ʹ; internal control). The detailed PCR condition for Snail involved 30 cycles of denaturation at 94°C for 30 seconds, annealing at 55°C for 30 seconds, and elongation at 72°C for 60 seconds. Amplified PCR products were subjected to electrophoresis on 1% agarose gel and were visualized by ethidium bromide staining and photographed with Polaroid 667 film (Cambridge, MA).
Indirect Immunofluorescence Microscopy
Cells were grown on coverslips, rinsed in PBS, fixed with 3.7% formaldehyde in PBS for 10 minutes at 4°C, and permeabilized with PBS supplemented with 0.5% Triton X-100 for 10 minutes at room temperature. Permeabilized cells were rinsed three times in PBS supplemented with 0.5% Tween 20 and blocked with blocking solution (0.1% BSA in PBS, pH 7.4) for 90 minutes. Cells were rinsed three times in PBS and incubated with primary antibodies against E-cadherin, N-cadherin, vimentin, or Smad3 in humidified chamber overnight at 4°C. After rinsing three times in PBS supplemented with 0.5% Tween 20, cover slips were incubated with the secondary antibodies Alexa-488–conjugated goat anti-rabbit or anti-mouse IgGs (Invitrogen Molecular Probes, Eugene, OR) for 1 hour at 37°C. Cell nuclei were counterstained with 4ʹ, 6ʹ-diamidino-2-phenylindole (DAPI). The intracellular distribution of the protein was analyzed using confocal fluorescence microscopy (Fluoview, FV-300; Olympus, Melville, NY) with excitation at 488 nm (for Alexa-488) and 340 nm (for DAPI).
Statistical Analysis
The data are representative of three independent experiments. Results are presented as the means ± SD. Analyses were performed with Student’s t test for paired data using SigmaPlot 8.0 software. P values less than .05 were considered significant.
Results
Inhibition of TGF-β1–induced EMT, Cell Migration, and MMP2 Activation by Kaempferol
Because TGF-β1 has been reported to induce metastatic responses, including EMT, motility, and invasiveness, in A549 lung cancer cells [31], we used it as an experimental system to evaluate the antimetastatic potential of kaempferol in cancer cells. As previously reported, treatment with TGF-β1 dramatically induced a loss of cell contact and acquired spindle-like shape in A549 cells, but this morphological change was clearly blocked by pretreatment with kaempferol (Figure 1B). In line with this result, kaempferol completely abolished TGF-β1–mediated repression of E-cadherin and upregulation of N-cadherin and smooth muscle α-actin in a concentration-dependent manner (Figure 1C). Immunofluorescence staining of E-cadherin, N-cadherin, and vimentin in A549 cells also revealed that the changes of those EMT marker expression induced by TGF-β1 were significantly reduced by treatment with kaempferol (Figure 1D).
EMT is a key event in metastatic tumor progression by which epithelial cancer develops toward more aggressive phenotype with enhanced motile and invasive capabilities [2,3]. We thus examined whether kaempferol affects the migration of A549 cells induced by TGF-β1. TGF-β1–induced cell migration (Figure 2A) and wound closure (Figure 2B) were substantially attenuated in cells pretreated with kaempferol compared with kaempferol-untreated cells. Upregulation of MMP2 is also known to promote the invasion of cancer cells through the degradation of basement membrane and extracellular matrix [32]. Gelatin zymography and reporter gene assay revealed that TGF-β1–induced increases of secreted MMP2 (Figure 2C) and MMP2 gene promoter activities (Figure 2D) were considerably abolished by treatment with kaempferol. All these results confirm the potential of kaempferol as an inhibitor of TGF-β1–induced EMT, cell migration, and invasiveness in A549 lung cancer cells.
Figure 2.

Kaemferol inhibits TGF-β1–induced migration and MMP2 activation in A549 lung cancer cells. (A) A549 cells treated as in Figure 1D were subjected to wound closure assay, and the wells were imaged every 12 hours with Nikon90i microscope, 100 ×. (B) A549 cells (1 × 104) seeded on 8-mm porous Transwell chambers were treated as in Figure 1D. Transmigrating cells were stained with hematoxylin and eosin and counted for each of the indicated cells. Quantitative data are shown as the mean ± SD of three independent experiments. *P < .05, compared with vehicle-treated controls; **P < .01, compared with TGF-β1–treated cells. (C) A549 cells were treated as in Figure 1C, the culture medium from indicated cells was collected, and MMP2 activities were measured by gelatin zymography. (D) A549 cells were transiently transfected with MMP2 promoter-reporter construct. The cells were pretreated with 25 μM of KF at the indicated concentrations for 30 minutes and then stimulated with 5 ng/ml of TGF-β1 for 24 hours. All quantitative data are the mean ± SD of three independent experiments. *P < .05; **P < .01, compared with TGF-β1–treated cells.
Kaempferol Reverses TGF-β1–Mediated E-Cadherin Repression by Weakening Smad3 Binding to Snail Promoter Without Affecting Canonical Smad3 Activation
Receptor-activated Smads, including Smad2 and Smad3, act independently as the major intracellular mediators of TGF-β signal transduction pathway [14]. Therefore, we analyzed the role of Smad2 and Smad3 in TGF-β1–mediated down-regulation of E-cadherin in A549 cells. Western blot analysis revealed that depletion of Smad3 with siRNA markedly reduced the changes in E-cadherin and Snail expression induced by TGF-β1, whereas depletion of Smad2 with siRNA had no such effect (Figure 3A). Similar to this result, overexpression of Smad3 (3SA), but not Smad2 (3SA), mutant lacking phosphorylation sites in the COOH-terminal region significantly reduced TGF-β1–induced Snail promoter activity (Figure 3B). TGF-β1–induced Snail expression at the mRNA level was also completely abolished by treatment with Smad3 siRNA (Figure 3C). We next examined whether kaempferol affects the binding of Smad3 to Snail promoter in response to TGF-β1. ChIP was applied to measure the enrichment of Smad3 on Snail promoter with two primers spanning its binding site. As shown in Figure 3D, kaempferol treatment completely inhibited TGF-β1–induced recruitment of Smad3 on the − 616/− 623 region of Snail promoter, and the inhibitory effect was comparable to that of SB431542, a specific TβRI inhibitor. Consistent with this result, RT-PCR showed that TGF-β1–induced Snail expression was significantly attenuated by treatment with kaempferol (Figure 3E). Furthermore, overexpression of Smad3 (3SA) markedly attenuated TGF-β1–induced migration of A549 cells compared with TGF-β1–treated control cells (data not shown). These results strongly suggest that Smad3 has a functional importance in TGF-β1–induced EMT and migration and can be a target of kaempferol.
Figure 3.

Kaempferol inhibits TGF-β1–induced Smad3-mediated Snail induction and E-cadherin repression in A549 lung cancer cells. (A) A549 cells were transfected with control (siCon), Smad2 (siS2), or Smad3 (siS3) siRNAs for 24 hours before 5 ng/ml of TGF-β1 treatment for 48 hours, after which the expressions of E-cadherin, Snail, Smad2, and Smad3 were determined by Western blot analysis. β-Actin levels were monitored as a loading control for whole extracts. (B) A549 cells were transiently co-transfected with Snail promoter-reporter construct (Snail-Luc) and vector for Smad2-3SA (S23SA), Smad3-3SA (S33SA), or the corresponding empty vector for 16 hours and then stimulated with 5 ng/ml of TGF-β1 for 24 hours. All cells were then assayed for relative luciferase activity. All quantitative data are the mean ± SD of three independent experiments. **P < .01. (C) A549 cells were transfected with control (siCont) or Smad3 (siS3) siRNAs for 24 hours before treatment with 5 ng/ml of TGF-β1 for 3 hours, after which the expressions of Snail and Smad3 mRNAs were determined by RT-PCR analysis. β-Actin levels were monitored as a loading control for total RNA. (D) A549 cells were pretreated with DMSO, 1 μM of SB431542, or 25 μM of KF for 30 minutes before treatment with 5 ng/ml of TGF-β1 for 24 hours and then subjected to ChIP assay using anti-Smad3 antibody. The purified DNA was analyzed by RT-PCR using primers spanning the Smad3-binding sites at Snail promoter. (E) A549 cells were pretreated with DMSO or 25 μM of KF for 30 minutes before treatment with 5 ng/ml of TGF-β1 for 3 hours. Expression of Snail mRNA was analyzed as in C. β-Actin levels were monitored as a loading control for total RNA.
In the canonical TGF-β signaling pathway, activation of Smad3 by TGF-β1 is a multistep process involving its phosphorylation in the COOH-terminal SSXS motif, complex formation with Smad4, and translocation into the nucleus [13]. We therefore evaluated the effect of kaempferol on TGF-β1–induced Smad3 activation in A549 cells. Interestingly, no difference on TGF-β1–induced Smad3 C-terminus phosphorylation was observed in cells pretreated with kaempferol compared with kaempferol-untreated cells (Figure 4, A and B). Immunoprecipitation followed by Western blotting also showed that TGF-β1–induced endogenous Smad3/Smad4 complex formation was not inhibited by treatment with kaempferol (Figure 4C). Furthermore, TGF-β1 treatment caused the nuclear translocation of Smad3 in kaempferol-treated cells without remarkable difference to that seen in kaempferol-untreated cells (Figure 4, D and E). Taken together, these results indicate that kaempferol suppresses TGF-β1–induced E-cadherin repression by blocking Smad3 binding to Snail promoter without affecting the canonical process for Smad3 activation.
Figure 4.

Kaempferol does not inhibit Smad3 C-terminal phosphorylation and its complex formation with Smad4 and translocation into the nucleus upon TGF-β1 stimulation. (A) A549 cells were pretreated with DMSO or 25 μM of KF for 30 minutes before treatment with 5 ng/ml of TGF-β1 for the indicated periods, after which the extent of Smad3 phosphorylation at Sre423/425 was determined by Western blot analysis. (B) A549 cells were pretreated with 25 μM of KF at the indicated concentrations for 30 minutes and then stimulated with 5 ng/ml of TGF-β1 for 1 hour. Western blot analysis was performed as in A. β-Actin levels were monitored as a loading control for whole extracts. (C) A549 cells were pretreated with DMSO or 25 μM of KF for 30 minutes before treatment with 5 ng/ml of TGF-β1 for 30 minutes, and cell lysates were immunoprecipitated with anti-Smad3 antibody and immunoblotted as indicated. β-Actin levels were monitored as a loading control for whole extracts. (D) A549 cells were treated as in A except for the TGF-β1 treatment for 10 and 90 minutes. (E) A549 cells were treated as in B. All cells were then subjected to immunofluorescence staining for Smad3 (green). DAPI (blue) was used to stain nuclei.
Kaempferol Inhibits TGF-β1–Induced EMT and Cell Migration Through Downregulation of the Phosphorylation of Smad3 at Thr179 in the Linker Region
There is a growing body of evidence pointing to an important role of Smad3 linker region phosphorylation (pSmad3L) in the regulation of Smad3 function under physiologic and pathologic conditions [15,20]. To verify the requirement of pSmad3L in TGF-β1–mediated induction of EMT and migration of A549 cells, we used recombinant lentivirus constitutively expressing Smad3EPSM mutant, in which all four phosphorylation sites of linker region, including Thr179, Ser204, Ser208, and Ser213, are replaced with Val or Ala (Figure 5A). Western blotting and immunofluorescence staining revealed that lentivirus-mediated overexpression of Smad3EPSM greatly reversed the changes in epithelial and mesenchymal marker expression induced by TGF-β1 (Figure 5, B and C). TGF-β1–induced cell migration was also significantly attenuated in cells infected with lentivirus carrying Smad3EPSM gene compared with cells infected with lentivirus carrying empty vector or Smad3WT gene (Figure 5D). In addition, TGF-β1–mediated increases of secreted MMP2 (Figure 5E) and MMP2 gene promoter activity (Figure 5F) were fully impaired by overexpression of Smad3EPSM but not by overexpression of Smad3WT. These results indicate that pSmad3L is critically required for metastatic responses induced by TGF-β1 in A549 cells.
Figure 5.

Expression of Smad3 linker mutant that lacks phosphorylation sites attenuates TGF-β1–induced EMT, migration, and MMP2 activation in A549 lung cancer cells. (A) Schematic representation of Smad3 wild type (S3WT) and mutant lacking phosphorylation sites in the linker region (S3EPSM). A549 cells infected with lentivirus carrying expression vector for Myc-tagged Smad3-WT (Myc-S3WT) or Myc-tagged Smad3-EPSM (Myc-S3EPSM), or its corresponding empty vector (pCAG) were stimulated with 5 ng/ml of TGF-β1 for 36 hours and then subjected to Western blot analysis using anti-E-cadherin, anti-N-cadherin, anti-vimentin, and anti-Myc antibodies (B) and immunofluorescence staining for E-cadherin and Myc-S3EPSM (C). (D) A549 cells (1 × 104) that were infected as in B were seeded on 8-mm porous Transwell chambers and then stimulated with treatment with 5 ng/ml of TGF-β1 for 36 hours. Transmigrating cells were stained and counted as in Figure 2B. Quantitative data are shown as the mean ± SD of three independent experiments. **P < .01. (E) A549 cells infected with lentivirus as in B were stimulated with 5 ng/ml of TGF-β1 for 24 hours, the culture medium from indicated cells was collected, and MMP2 activities were measured by gelatin zymography. (F) A549 cells were transiently transfected with MMP2 promoter-reporter construct and vector for Smad3-EPSM (S3EPSM) or Smad3T179V (S3T179V) or the corresponding empty vector (pcDNA) and then stimulated with 5 ng/ml of TGF-β1 for 24 hours. All cells were then assayed for relative luciferase activity. All quantitative data are the mean ± SD of three independent experiments. **P < .01.
We next evaluated the effects of TGF-β1 and kaempferol on pSmad3L in A549 cells. As expected, treatment with TGF-β1 strongly induced the phosphorylation of Smad3 at Thr179, Ser203, Ser208, and Ser213 in the linker region (Figure 6, A and B). Interestingly, we found that kaempferol specifically inhibited TGF-β1–induced pSmad3L at Thr179 without affecting the phosphorylation of the remaining linker region sites (Figure 6, A and B). To assess the role of Smad3 phosphorylation at Thr179 in TGF-β1–induced EMT and migration of A549 cells, we used recombinant lentivirus constitutively expressing Smad3T179V mutant, in which Thr179 phosphorylation site is replaced with Val. Western blotting (Figure 6C) and immunofluorescence staining (Figure 6D) of E-cadherin showed that TGF-β1–induced repression of E-cadherin was significantly abolished in cells infected with lentivirus carrying Smad3T179V gene compared with cells infected with empty vector lentivirus. Consistent with this result, TGF-β1–mediated suppression of E-cadherin promoter activity was markedly rescued by co-transfection with Smad3T179V, but not with Smad3WT, plasmid DNA (Figure 6E). In line with these results, ChIP assay revealed that TGF-β1–induced enrichment of Smad3 on the -616/-623 region of Snail promoter was completely abolished in cells infected with lentivirus carrying Smad3T179V mutant gene compared with cells infected with lentivirus carrying empty vector or Smad3 wild-type gene (Figure 6F). In parallel, TGF-β1–induced Snail promoter activity was markedly decreased by overexpression of Smad3T179V, but not of Smad3WT, plasmid DNA (Figure 6G). Furthermore, lentivirus-mediated ectopic expression of Smad3T179V in A549 cells resulted in significant attenuation of cell migration (Figure 6H) induced by TGF-β1. TGF-β1–induced increases of MMP2 gene promoter activity were also fully impaired by overexpression of Smad3T179V (Figure 6I). These results clearly suggest that the ablation of Smad3 phosphorylation at Thr179 is responsible for the suppression by kaempferol of TGF-β1–induced EMT, cell migration, and MMP2 activation in A549 lung cancer cells.
Figure 6.

Kaemferol specifically inhibits the phosphorylation of Smad3 at Thr179 that is required for TGF-β1–mediated induction of EMT, cell migration, and MMP2 activation. (A) A549 cells were treated as in Figure 4B. (B) A549 cells treated as in Figure 4A except for the TGF-β1 treatment for 10 minutes. All cells then were subjected to Western blot analysis using specific phosphopeptide antibodies against the phosphorylated Thr179, Ser203, Ser208, and Ser213 in the Smad3 linker region. A549 cells infected with lentivirus carrying expression vector for Myc-tagged Smad3-T179V (Myc-S3T179V) or its corresponding empty vector (pCAG) were stimulated with 5 ng/ml of TGF-β1 for 36 hours and then subjected to Western blot analysis (C) and immunofluorescence staining (D) using anti–E-cadherin antibody. β-Actin levels were monitored as a loading control for whole extracts. DAPI (blue) was used to stain nuclei. (E) A549 cells were transiently transfected with E-cadherin promoter-reporter construct (E-cadherin-Luc) and Smad3-WT (S3WT), Smad3-T179V (S3T179V), or the corresponding empty vector (pcDNA) plasmid DNA and then stimulated with 5 ng/ml of TGF-β1 for 24 hours. All quantitative data are the mean ± SD of three independent experiments. **P < .01. (F) A549 cells infected with lentivirus carrying expression vector for Myc-tagged Smad3 wild type (S3WT), Smad3-T179V (Myc-S3T179V), or its corresponding empty vector (pCAG) were stimulated with 5 ng/ml of TGF-β1 for 24 hours and then subjected to ChIP assay using anti-Myc antibody. The purified DNA was analyzed as in Figure 3D. (G) A549 cells were transiently co-transfected with Snail promoter-reporter (Snail-Luc) and Smad3-WT (S3WT), Smad3-T179V (S3T179V), or the corresponding empty vector (pcDNA) plasmid DNA and then stimulated with 5 ng/ml of TGF-β1 for 24 hours. All quantitative data are the mean ± SD of three independent experiments. **P < .01. (H) A549 cells (1 × 104) that were infected as in Figure 5B were seeded on 8-mm porous Transwell chambers and then stimulated with 5 ng/ml of TGF-β1 for 36 hours. Transmigrating cells were stained and counted as in Figure 2B. Quantitative data are shown as the mean ± SD of three independent experiments. **P < .01. (I) A549 cells were transiently transfected with MMP2 promoter-reporter construct (MMP2-Luc) and Smad3-WT (S3WT), Smad3-T179V (S3T179V), or the corresponding empty vector (pcDNA) plasmid DNA and then stimulated with 5 ng/ml of TGF-β1 for 24 hours. All quantitative data are the mean ± SD of three independent experiments. **P < .01.
Kaempferol Suppresses TGF-β1–Induced pSmad3L at Thr179 and Metastatic Responses Through Akt Inhibition
Previous study had identified PI3K as a direct target molecule for the inhibitory effects of kaempferol on neoplastic cell transformation [25]. It has been also reported that Akt, a downstream effector of PI3K, plays an important role in the regulation of TGF-β signaling through cross talk with canonical Smad pathway [33]. These prompted us to ask whether the ablation of PI3K-Akt pathway by kaempferol is responsible for the inhibition of TGF-β1–induced pSmad3L and metastatic cellular responses. Treatment of A549 cells with TGF-β1 resulted in rapid phosphorylation of Akt, but the effect was completely abrogated by kaempferol (Figure 7A). Pretreatment with AktIV, a specific inhibitor of Akt, completely reversed changes in TGF-β1–induced EMT marker expression in a concentration-dependent manner (Figure 7B). Consistent with this result, TβRI (T204D)-induced repression of E-cadherin promoter activity was significantly rescued by co-transfection with dominant negative forms of p85α or Akt (Figure 7C). ChIP and RT-PCR also revealed that TGF-β1–induced Smad3 binding to the Snail promoter (Figure 7D) and Snail expression at mRNA level (Figure 7E) were effectively weakened by treatment with AktIV or LY294002, a specific inhibitor of PI3K. In addition, treatment with AktIV or LY294002 resulted in remarkable attenuation in TGF-β1–induced cell migration (Figure 7F), and TGF-β1–mediated increase of MMP2 gene promoter activity was also significantly abolished by co-transfection with dominant negative forms of p85αΔiSH2-N or Akt1K179M (Figure 7G), indicating a crucial involvement of PI3K-Akt axis in metastatic TGF-β signaling pathway.
Figure 7.

PI3K/Akt1 pathway is crucial for TGF-β1–induced EMT, cell migration, and MMP2 activation and is inhibited by KF. (A) A549 cells were treated as in Figure 4A except for the TGF-β1 treatment for 90 minutes. (B) A549 cells were pretreated with DMSO or AktIV at the indicated concentrations for 30 minutes and then stimulated with 5 ng/ml of TGF-β1 for 48 hours. All cells were then subjected to Western blot analysis using anti–phospho-Akt, anti–E-cadherin, and anti–N-cadherin antibodies. β-Actin levels were monitored as a loading control for whole extracts. (C) A549 cells were transiently transfected with E-cadherin promoter-reporter (E-cadherin-Luc) combination with constitutively active TβRI (CA-TβRI, TβRI-T204D)-, dominant negative p85 (DN-p85, p85ΔiSH2-N)-, and dominant negative Akt1 (DN-Akt1, Akt1-K179M)-expressing plasmids and the corresponding empty vector (pcDNA). All quantitative data are the mean ± SD of three independent experiments. **P < .01. (D) A549 cells were pretreated with DMSO, 10 μM of LY294002, or 2.5 μM of AktIV for 30 minutes before treatment with 5 ng/ml of TGF-β1 for 24 hours and then subjected to ChIP assay using anti-Smad3 antibody. The purified DNA was analyzed as in Figure 3D. (E) A549 were pretreated with DMSO or 2.5 μM of AktIV for 30 minutes before treatment with 5 ng/ml of TGF-β1 for 3 hours, after which the expression of Snail mRNA was determined by RT-PCR analysis. β-Actin levels were monitored as a loading control for total RNA. (F) A549 cells (1 × 104) seeded on 8-mm porous Transwell chambers were treated as in D. Transmigrating cells were stained and counted as in Figure 2B. Quantitative data are shown as the mean ± SD of three independent experiments. **P < .01. (G) A549 cells were co-transfected as in C except for transfection with MMP2-Luc instead of E-cadherin-Luc and then stimulated with 5 ng/ml of TGF-β1 for 24 hours. All quantitative data are the mean ± SD of three independent experiments. **P < .01.
We further addressed whether Akt activation is necessary for TGF-β1–induced pSmad3L in A549 cells. Similar to the kaempferol effect, treatment with AktIV resulted in selective inhibition of TGF-β1–induced phosphorylation of Smad3 at Thr179 (Figure 8A). In contrast, overexpression of constitutively active form of Akt alone resulted in an increase of phosphorylation of Smad3 at Thr179 compared with expression of control counterpart (Figure 8B). Treatment with TGF-β1 resulted in increase of Akt binding with Smad3, and the binding reached a peak at 30 minutes and then declined to baseline at 60 minutes (Figure 8C). The TGF-β1–induced binding of Akt with Smad3 was completely abolished by pretreatment with kaempferol (Figure 8C). Given these findings that Akt controls phosphorylation of Smad3 at Thr179, we next assessed whether Akt-mediated phosphorylation of Smad3 at Thr179 would be sufficient to drive TGF-β1–induced repression of E-cadherin. Reporter gene assay revealed that TGF-β1–induced repression of E-cadherin promoter activity was further augmented in cells transfected with constitutively active forms of p110αK227E or Akt1E40K compared with that in cells transfected with empty vector. In contrast, in cells co-transfected with Smad3T179V and p110αK227E or Akt1E40K, TβRIT204D-induced repression of E-cadherin promoter activity was significantly rescued compared with that in cells co-transfected with control vector (Figure 8D), demonstrating Akt1 as a key mediator in TGF-β signaling pathway leading to E-cadherin repression through inducing the phosphorylation of Smad3 at Thr179.
Figure 8.

Akt1 directly phosphorylates Smad3 at Thr179, and it mediates TGF-β1–induced E-cadherin repression. (A) A549 cells were pretreated with DMSO or AktIV at the indicated concentrations for 30 minutes and then stimulated with 5 ng/ml of TGF-β1 for 30 minutes. (B) A549 cells were transfected with vector for constitutively active HA-Akt1 (HA-CA-Akt1, HA-Akt1-E40K) or its corresponding empty vector, pcDNA, for 24 hours. All cells were then subjected to Western blot analysis using antibodies against phospho-Smad3 (Thr179), phospho-Smad3 (Ser208), phospho-Smad3 (Ser423/425), and HA. (C) A549 cells were pretreated with DMSO or 25 μM of KF for 30 minutes before treatment with 5 ng/ml of TGF-β1 for the indicated periods, and cell lysates were immunoprecipitated with anti-Smad3 antibody and immunoblotted as indicated. β-Actin levels were monitored as a loading control for whole extracts. (D) A549 cells were cotransfected with E-cadherin promoter-reporter construct (E-cadherin-Luc) in combination with constitutively active TβRI (CA-TβRI, TβRI-T204D)-, constitutively active p110α (CA-p110α, p110α-K227E)-, constitutively active Akt1 (CA-Akt1, Akt1-E40K)-, and Smad3-T179V (S3T179V)-expressing plasmids and the corresponding empty plasmid (pcDNA). All quantitative data are the mean ± SD of three independent experiments. **P < .01; ***P < .005.
Discussion
Although kaempferol has been extensively studied as a natural flavonoid that possesses chemopreventive effect on cancer, its antimetastatic effect has been less addressed. The enhancement of EMT in cancer cells is known to increase the risk of metastasis and closely associated with poor prognosis of many types of cancer. Fibroblast growth factor (FGF) signaling is important in promotion of EMT and invasion of cancer cells [34]. Kaempferol inhibits tyrosine phosphorylation of FGF receptor by FGF stimulation [35]. The study by Huang et al suggested that estrogen-related receptor (ERR) α promotes the migration and invasion of lung cancer A549 cells by inducing EMT [36]. Kaempferol was reported to inhibit cancer cell growth by antagonizing ERRα and ERRγ activities [37]. These findings strongly suggest that kaempferol may exert its antimetastatic activity through targeting key components of several signaling pathways involved in tumor metastasis. TGF-β1 potently induces EMT in epithelial tumor cells, conferring them migratory and invasive properties. Our data showed that kaempferol strongly inhibited TGF-β1–induced EMT, migration, and MMP-2 activation in human A549 lung cancer cells, which strongly support its potential as an antitumor agent that suppresses tumor metastasis.
Smad2 and Smad3 are key mediators for TGF-β signaling in both normal and pathological conditions. It has been established that Smad2 and Smad3 have distinct functions in TGF-β1–induced cellular responses. For example, Smad3 is critical to inducing TGF-β1–mediated growth inhibition [38,39], whereas Smad2 is uniquely essential for TGF-β signaling in CD4 T cell differentiation [40]. Our data demonstrated that Smad3, but not Smad2, was required for TGF-β1–induced E-cadherin repression and up-regulation of snail gene expression in A549 lung cancer cells. It was also notable that kaempferol significantly inhibited Smad3 binding to the promoter of snail gene and the induction of snail expression upon TGF-β1 stimulation, whereas it did not affect the canonical activation of Smad3 (C-terminal phosphorylation, complex formation with Smad4, and translocation into the nucleus) induced by TGF-β1. These results imply the possibility that Smad3 activity can be inhibited noncanonically by kaempferol before or after it enters the nucleus in response to TGF-β1.
Smad3 can be regulated noncanonically through phosphorylation of its linker region. The recent discovery of C-terminal domain phosphatase 1 as a specific phosphatase for Smad3 dephosphorylation in the linker region [41,42] highlights the need to consider Smad-linker phosphorylation as a reversible and tightly controlled TGF-β signaling event. Actually, the study by Matsuura et al. demonstrated that phosphorylation of Smad3 linker region by cyclin-dependent kinase inhibits its antiproliferative function, thus facilitating cell cycle progression [43]. Furthermore, increased phosphorylation of Smad3 linker region has been identified in many types of cancer cells and was proven to contribute to the TGF-β1–mediated signaling events to facilitate protumorigenic outcomes [44,45]. In line with these observations, our data demonstrated that TGF-β1 induced Smad3 linker phosphorylation, and Smad3 mutant lacking linker phosphorylation sites could reverse metastatic responses to TGF-β1 in A549 lung cancer cells. A remarkable finding is that the inhibitory effect of kaempferol on increased Smad3 linker phosphorylation by TGF-β1 treatment was specifically limited to Thr179 residue, but not to other linker sites, and it was sufficient to reverse TGF-β1–induced EMT and migration in A549 cells by expression of Smad3T179V alone. TGF-β1–induced Smad3 binding to the Snail promoter and subsequent repression of E-cadherin were also dependent on Smad3 phosphorylation at Thr-179. However, TGF-β1–induced p21WAF1 expression and cell cycle arrest in A549 cells were not affected by kaempferol treatment and overexpression of Smad3T179V (Supplementary Figure S1, A and B). These results newly characterized an important role of the Thr179 phosphorylated form of Smad3 in the metastatic TGF-β signaling in A549 lung cancer cells. However, we do not exclude the possibility that other Smad3 linker sites for phosphorylation could contribute to TGF-β1–induced metastatic responses.
Supplementary Figure S1.

Kaempferol (KF) and Smad3T179V do not affect TGF-β1–induced growth inhibition in A549 lung cancer cells. Effects of KF on TGF-β1–induced p21WAF1 expression (A) and growth inhibition (B). (C) FACS analysis of TGF-β1–induced cell cycle arrest in A549 cells infected with empty vector (pCAG), Smad3 wild type (S3WT), or Smad3T179V mutant (S3T179V) lentivirus.
A growing body of evidence has shown that the linker domain of Smad3 undergoes regulatory phosphorylation by various intracellular signaling kinases, including mitogen-activated protein kinase, cyclin-dependent kinase, glycogen synthase kinase 3-β, and protein kinase C, in normal and pathological conditions [46]. Because a previous study by Nguyen et al. showed that kaempferol activated ERK1/2 in A549 lung cancer cells [47], we investigated the involvement of ERK1/2 in phosphorylation of Smad3 linker region and EMT induced by TGF-β1. Pretreatment of A549 cells with U0126, a specific inhibitor of MEK1/2, completely inhibited phosphorylation of ERK1/2 but had no effects on phosphorylation of Smad3 linker region and EMT upon TGF-β1 stimulation (Supplementary Figure S2, A and B), indicating that ERK1/2 is not associated with these TGF-β1 responses in A549 cells. The PI3K/Akt pathway plays an important role in tumor development [48]. Recently, Lee et al. demonstrated that PI3K is a direct target molecule for the inhibitory effects of kaempferol on neoplastic cell transformation [25]. Our data demonstrated that TGF-β1–induced EMT and cell migration were dependent on the activation of the PI3K/Akt axis. Consistent with the effect of kaempferol on TGF-β1–induced Smad3 linker phosphorylation, Akt activation or inhibition showed prominent effects on Smad3 phosphorylation at Thr179 only. Moreover, Akt interacted with Smad3 in the presence of TGF-β1. These observations suggest the direct link between PI3-K/Akt axis and Smad3 linker phosphorylation at Thr179 in metastatic TGF-β signaling, thus extending our knowledge about the molecular mechanism of EMT and cell migration induced by TGF-β1 in cancer cells.
Supplementary Figure S2.
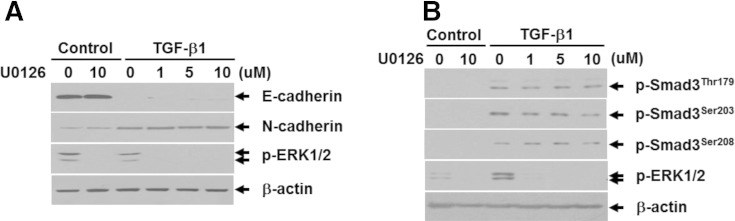
ERK1/2 is not involved in TGF-β1–induced EMT and phosphorylation of Smad3 linker region in A549 lung cancer cells. Effects of U0126 on TGF-β1–mediated repression of E-cadherin and induction of N-cadherin (A) and phosphorylation of Smad3 linker region (B).
In summary, this study provides the first evidence showing that the natural compound kaempferol can inhibit the TGF-β1–induced EMT and cell migration in human lung cancer cell by targeting phosphorylation of Smad3 linker region. The presenting data also highlight a loss of PI3K/Akt-mediated Smad3 phosphorylation at Thr179 as a key mechanism of inhibition by kaempferol of oncogenic TGF-β1 signaling (Figure 9) and suggest that the pharmacologic inhibition of Smad3 phosphorylation at Thr179 can be an effective strategy to impede lung cancer progression.
Figure 9.

A model illustrating the mechanism underlying inhibition of TGF-β1–induced EMT and cell migration by KF. In A549 lung cancer cells, TGF-β1 activates both canonical Smad pathway and noncanonical PI3K-Akt1 pathway. The activated Akt1 directly interacts with Smad3 and phosphorylated it at Thr179 in the linker region, which leads to induction of Snail, N-cadherin, and MMP2 expression as well as repression of E-cadherin that are necessary for induction of EMT and migration of cancer cells. Kaempferol inhibits TGF-β1–induced EMT and cell migration by suppressing its downstream Akt1 activity through binding with PI3K.
The following are the supplementary data related to this article.
Acknowledgements
We thank Fang Liu (The State University of New Jersey, USA) for providing phosphor-Smad3 (Thr179), phosphor-Smad3 (Ser203), phosphor-Smad3 (Ser208), and phosphor-Smad3 (Ser213) antibodies. This work was supported the National Research Foundation of Korea grant funded by the Korean Government (MEST) (2012R1A1A2044419 to B.C.K), and a grant of the Ministry of Science, ICT and Future Planning through the National Research Foundation of Korea (NRF-2014M3A9B5073918) and the National R&D Program for Cancer Control (1020420), Ministry for Health and Welfare, Republic of Korea.
Footnotes
This work was supported the National Research Foundation of Korea grant funded by the Korean Government (MEST) (2012R1A1A2044419 to B.C.K), and a grant of the Ministry of Science, ICT and Future Planning through the National Research Foundation of Korea (NRF-2014M3A9B5073918) and the National R&D Program for Cancer Control (1020420), Ministry for Health and Welfare, Republic of Korea.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;27:2192–2206. doi: 10.1101/gad.225334.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Voulgari A, Pintzas A. Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim Biophys Acta. 2009;1796:75–90. doi: 10.1016/j.bbcan.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Bierie B, Moses HL. TGF-β and cancer. Cytokine Growth Factor Rev. 2006;17:29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A. 2003;100:8621–8623. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boudreau HE, Casterline BW, Rada B, Korzeniowska A, Leto TL. Nox4 involvement in TGF-beta and SMAD3-driven induction of the epithelial-to-mesenchymal transition and migration of breast epithelial cells. Free Radic Biol Med. 2012;53:1489–1499. doi: 10.1016/j.freeradbiomed.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts AB, Tian F, Byfield SD, Stuelten C, Ooshima A, Saika S, Flanders KC. Smad3 is key to TGF-beta-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor Rev. 2006;17:19–27. doi: 10.1016/j.cytogfr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Saika S, Kono-Saika S, Ohnishi Y, Sato M, Muragaki Y, Ooshima A, Flanders KC, Yoo J, Anzano M, Liu CY. Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. Am J Pathol. 2004;164:651–653. doi: 10.1016/S0002-9440(10)63153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuxe J, Vincent T, Garcia de Herreros A. Transcriptional crosstalk between TGFβ and stem cell pathways in tumor cell invasion: role of EMT promoting Smad conplexes. Cell Cycle. 2010;9:2363–2374. doi: 10.4161/cc.9.12.12050. [DOI] [PubMed] [Google Scholar]
- 10.Hua F, Mu R, Liu J, Xue J, Wang Z, Lin H, Yang H, Chen X, Hu Z. TRB3 interacts with SMAD3 promoting tumor cell migration and invasion. J Cell Sci. 2011;124:3235–3246. doi: 10.1242/jcs.082875. [DOI] [PubMed] [Google Scholar]
- 11.Xue J, Lin X, Chiu WT, Chen YH, Yu G, Liu M, Feng XH, Sawaya R, Medema RH, Hung MC. Sustained activation of SMAD3/SMAD4 by FOXM1 promotes TGF-β-dependent cancer metastasis. J Clin Invest. 2014;124:564–579. doi: 10.1172/JCI71104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahan SM, Cha JY, Kim J, Kim D, Trang HT, Kim YM, Cho YH, Park D, Hong S. Smad3 regulates E-cadherin via miRNA-200 pathway. Oncogene. 2012;31:3051–3059. doi: 10.1038/onc.2011.484. [DOI] [PubMed] [Google Scholar]
- 13.Miyazono K, Suzuki H, Imamura T. Regulation of TGF-β signaling and its roles in progression of tumors. Cancer Sci. 2003;94:230–234. doi: 10.1111/j.1349-7006.2003.tb01425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi Y, Massagué J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 15.Engel ME, McDonnell MA, Law BK, Moses HL. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem. 1999;274:37413–37420. doi: 10.1074/jbc.274.52.37413. [DOI] [PubMed] [Google Scholar]
- 16.Kamaraju AK, Roberts AB. Role of Rho/ROCK and p38 MAPK kinase pathways in transforming growth factor-beta-mediated Smad-dependent growth inhibition of human breast carcinoma cells in vivo. J Biol Chem. 2005;280:1024–1036. doi: 10.1074/jbc.M403960200. [DOI] [PubMed] [Google Scholar]
- 17.Liu F. Smad3 phosphorylation by cyclin-dependent kinases. Cytokine Growth Factor Rev. 2006;17:9–17. doi: 10.1016/j.cytogfr.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 18.Millet C, Yamashita M, Heller M, Yu LR, Veenstra TD, Zhang YE. A negative feedback control of transforming growth factor-beta signaling by glycogen synthase kinase 3-mediated Smad3 linker phosphorylation at Ser-204. J Biol Chem. 2009;284:19808–19816. doi: 10.1074/jbc.M109.016667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mori S, Matsuzaki K, Yoshida K, Furukawa F, Tahashi Y, Yamagata H, Sekimoto G, Seki T, Matsui H, Nishizawa M. TGF-β and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker region. Oncogene. 2004;23:7416–7429. doi: 10.1038/sj.onc.1207981. [DOI] [PubMed] [Google Scholar]
- 20.Murata M, Matsuzaki K, Yoshida K, Sekimoto G, Tahashi Y, Mori S, Uemura Y, Sakaida N, Fujisawa J, Seki T. Hepatatis B virus X protein shifts human hepatic transforming growth factor (TGF)-beta from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology. 2009;49:1203–1217. doi: 10.1002/hep.22765. [DOI] [PubMed] [Google Scholar]
- 21.Kretzschmar M, Doody J, Timokhina I, Massagué J. A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi J, Park SJ, Jo EJ, Lee HY, Hong S, Kim SJ, Kim BC. Hydrogen peroxide inhibits transforming growth factor-β1-induced cell cycle arrest by promoting Smad3 linker phosphorylation through activation of Akt-ERK1/2-linked signaling pathway. Biochem Biophys Res Commun. 2013;435:634–639. doi: 10.1016/j.bbrc.2013.05.035. [DOI] [PubMed] [Google Scholar]
- 23.Calderón-Montaño JM, Burgos-Morón E, Pérez-Guerrero C, López-Lázaro M. A review on the dietary flavonoid kaempferol. Mini Rev Med Chem. 2011;11:298–344. doi: 10.2174/138955711795305335. [DOI] [PubMed] [Google Scholar]
- 24.Cho YY, Yao K, Kim HG, Kang BS, Zheng D, Bode AM, Dong Z. Ribosomal S6 kinase 2 is a key regulator in tumor promoter induced cell transformation. Cancer Res. 2007;67:8104–8112. doi: 10.1158/0008-5472.CAN-06-4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee KM, Lee DE, Seo SK, Hwang MK, Heo YS, Lee KW, Lee HJ. Phosphatidylinositol 3-kinase, a novel target molecule for the inhibitory effects of kaempferol on neoplastic cell transformation. Carcinogenesis. 2010;31:1338–1343. doi: 10.1093/carcin/bgq102. [DOI] [PubMed] [Google Scholar]
- 26.Smith JA, Maloney DJ, Clark DE, Xu Y, Hecht SM, Lannigan DA. Influence of rhamnose substituents on the potency of SL0101, an inhibitor of the Ser/Thr kinase, RSK. Bioorg Med Chem. 2006;14:6034–6042. doi: 10.1016/j.bmc.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 27.Xie F, Su M, Qiu W, Zhang M, Guo Z, Su B, Liu J, Li X, Zhou L. Kaempferol promotes apoptosis in human bladder cancer cells by inducing the tumor suppressor, PTEN. Int J Mol Sci. 2013;14:21215–21226. doi: 10.3390/ijms141121215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo H, Rankin GO, Liu L, Daddysman MK, Jiang BH, Chen YC. Kaempferol inhibits angiogenesis and VEGF expression through both HIF dependent and independent pathways in human ovarian cancer cells. Nutr Cancer. 2009;61:554–563. doi: 10.1080/01635580802666281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin CW, Chen PN, Chen MK, Yang WE, Tang CH, Yang SF, Hsieh YS. Kaempferol reduces matrix metalloproteinase-2 expression by down-regulating ERK1/2 and the activator protein-1 signaling pathways in oral cancer cells. PLoS One. 2013;8:e80883. doi: 10.1371/journal.pone.0080883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:575156–575159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 31.Kim AN, Jeon WK, Lim KH, Lee HY, Kim WJ, Kim BC. Fyn mediates transforming growth factor-beta1–induced down-regulation of E-cadherin in human A549 lung cancer cells. Biochem Biophys Res Commun. 2011;407:181–184. doi: 10.1016/j.bbrc.2011.02.134. [DOI] [PubMed] [Google Scholar]
- 32.Duffy MJ, Maguire TM, Hill A, McDermott E, O’Higgins N. Metalloproteinases: role in breast carcinogenesis, invasion and metastasis. Breast Cancer Res. 2000;2:252–257. doi: 10.1186/bcr65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Runyan CE, Schnaper HW, Poncelet AC. The phosphatidylinositol 3-kinase/Akt pathway enhances Smad3-stimulated mesangial cell collagen I expression in response to transforming growth factor-beta1. J Biol Chem. 2004;279:2632–2639. doi: 10.1074/jbc.M310412200. [DOI] [PubMed] [Google Scholar]
- 34.Zhao D, Lu Y, Yang C, Zhou X, Xu Z. Activation of FGF receptor signaling promotes invasion of non-small-cell lung cancer. Tumor Biol. 2015;36:3637–3642. doi: 10.1007/s13277-014-3001-y. [DOI] [PubMed] [Google Scholar]
- 35.Lee CJ, Lee MH, Cho YY. Fibroblast and epidermal growth factors utilize different signaling pathways to induce anchorage-independent cell transformation in JB6 Cl41 mouse skin epidermal cells. J Cancer Prev. 2014;19:199–208. doi: 10.15430/JCP.2014.19.3.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang JW, Guan BZ, Yin LH, Liu FN, Hu B, Zheng QY, Li FL, Zhong YX, Chen Y. Effects of estrogen-related receptor alpha (ERRα) on proliferation and metastasis of human lung cancer A549 cells. J Huazhong Univ Sci Technolog Med Sci. 2014;34:875–881. doi: 10.1007/s11596-014-1367-0. [DOI] [PubMed] [Google Scholar]
- 37.Wang H, Gao M, Wang J. Kaempferol inhibits cancer cell growth by antagonizing estrogen-related receptor α and γ activities. Cell Biol Int. 2013;37:1190–1196. doi: 10.1002/cbin.10152. [DOI] [PubMed] [Google Scholar]
- 38.Ashcroft GS, Yang X, Glick AB, Wenstein M, Letterio JL, Mizel DE, Anzano M, Greenwell-Wild T, Wahl SM, Deng C. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol. 1999;1:260–266. doi: 10.1038/12971. [DOI] [PubMed] [Google Scholar]
- 39.Kretschmer A, Moepert K, Dames S, Stemberger M, Kaufmann J, Klippel A. Differential regulation of TGF-beta signaling through Smad2, Smad3 and Smad4. Oncogene. 2003;22:6748–6763. doi: 10.1038/sj.onc.1206791. [DOI] [PubMed] [Google Scholar]
- 40.Malhotra N, Robertson E, Kang J. SMAD2 is essential for TGF beta-mediated Th17 cell generation. J Biol Chem. 2010;285:29044–29048. doi: 10.1074/jbc.C110.156745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wrighton KH, Willis D, Long J, Liu F, Lin X, Feng XH. Small C-terminal domain phosphatases dephosphorylate the regulatory linker regions of Smad2 and Smad3 to enhance transforming growth factor-beta signaling. J Biol Chem. 2006;281:38365–38375. doi: 10.1074/jbc.M607246200. [DOI] [PubMed] [Google Scholar]
- 42.Sapkota G, Knockaert M, Alarcón C, Montalvo E, Brivanlou AH, Massagué J. Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-beta pathways. J Biol Chem. 2006;281:40412–40419. doi: 10.1074/jbc.M610172200. [DOI] [PubMed] [Google Scholar]
- 43.Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430:226–231. doi: 10.1038/nature02650. [DOI] [PubMed] [Google Scholar]
- 44.Bae E, Sato M, Kim RJ, Kwak MK, Naka K, Gim J, Kadota M, Tang B, Flanders KC, Kim TA. Definition of smad3 phosphorylation events that affect malignant and metastatic behaviors in breast cancer cells. Cancer Res. 2014;74:6139–6149. doi: 10.1158/0008-5472.CAN-14-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagata H, Hatano E, Tada M, Murata M, Kitamura K, Asechi H, Narita M, Yanagida A, Tamaki N, Yagi S. Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesis to tumor-suppression in rat hepatocellular carcinoma. Hepatology. 2009;49:1944–1953. doi: 10.1002/hep.22860. [DOI] [PubMed] [Google Scholar]
- 46.Matsuzaki K. Smad phosphor-isoforms direct context-dependent TGF-β signaling. Cytokine Growth Factor Rev. 2013;24:385–399. doi: 10.1016/j.cytogfr.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen TT, Tran E, Ong CK, Lee SK, Do PT, Huynh TT, Nguyen TH, Lee JJ, Tan Y, Ong CS. Kaempferol-induced growth inhibition and apoptosis in A549 lung cancer cells is mediated by activation of MEK-MAPK. J Cell Physiol. 2003;197:110–121. doi: 10.1002/jcp.10340. [DOI] [PubMed] [Google Scholar]
- 48.Zhang L, Zhou F, ten Dijke P. Signaling interplay between transforming growth factor-β receptor and PI3K/AKT pathways in cancer. Trends Biochem Sci. 2013;38:612–620. doi: 10.1016/j.tibs.2013.10.001. [DOI] [PubMed] [Google Scholar]