

Abstract
Understanding the functional consequences of genetic variation, and how it affects complex human disease and quantitative traits, remains a critical challenge for biomedicine. We present an analysis of RNA sequencing data from 1641 samples across 43 tissues from 175 individuals, generated as part of the pilot phase of the Genotype-Tissue Expression (GTEx) project. We describe the landscape of gene expression across tissues, catalog thousands of tissue-specific and shared regulatory expression quantitative trait loci (eQTL) variants, describe complex network relationships, and identify signals from genome-wide association studies explained by eQTLs. These findings provide a systematic understanding of the cellular and biological consequences of human genetic variation and of the heterogeneity of such effects among a diverse set of human tissues.
Over the past decade, there has been a marked increase in our understanding of the role of genetic variation in complex traits and human disease, especially via genome-wide association studies (GWAS) that have cataloged thousands of common genetic variants affecting human diseases and other traits (1–3). However, the molecular mechanisms by which this genetic variation predisposes individuals to disease are still poorly characterized, impeding the development of therapeutic interventions.
The majority of GWAS variants are noncoding, likely manifesting their effects via the regulation of gene expression (4, 5). Thus, characterization of the regulatory architecture of the human genome is essential, not only for understanding basic biology but also for interpreting GWAS loci. Expression quantitative trait locus (eQTL) analysis (6–8) is the most common approach used to dissect the effects of genetic variation on gene expression. However, comprehensive eQTL data from a range of human tissues are lacking, and eQTL databases are biased toward the most accessible tissues. Additionally, although many regulatory regions act in a tissue-specific manner (9, 10), it is unknown whether genetic variants in regulatory regions have tissue-specific effects as well. Complex diseases are often caused by the dysfunction of multiple tissues or cell types, such as pancreatic islets, adipose, and skeletal muscle for type 2 diabetes (11, 12), so it is not obvious a priori what the causal tissue(s) are for any given GWAS locus or disease. Hence, understanding the role of regulatory variants, and the tissues in which they act, is essential for the functional interpretation of GWAS loci and insights into disease etiology. The Genotype-Tissue Expression (GTEx) Project was designed to address this limitation by establishing a sample and data resource to enable studies of the relationship among genetic variation, gene expression, and other molecular phenotypes in multiple human tissues (13). To facilitate the collection of multiple different tissues per donor, the project obtains recently deceased donors through consented next-of-kin donation, from organ donation and rapid autopsy settings. The results described here were generated during the project’s pilot phase, prior to scaling up collection to 900 donors. All project data are made available at regular intervals to qualified researchers through dbGaP. Summary data are available on the GTEx Portal (http://gtexportal.org).
Study design
During the pilot, we recruited 237 postmortem donors, collecting an average of 28 tissue samples per donor spanning 54 distinct body sites (fig. S1 and tables S1 and S2). Blood-derived DNA samples were genotyped at approximately 4.3 million sites, with additional variants imputed using the 1000 Genomes phase I, resulting in ~6.8 million single-nucleotide polymorphisms (SNPs) with minor allele frequency (MAF) of ≥5% after quality control (tables S3 to S5) (14).
RNA was extracted from all tissues, but quality varied widely, with tissue site and sample specific ischemic time accounting for ~40% of the variance in RNA quality (fig. S2). To maximize statistical power, we prioritized RNA sequencing of samples from nine tissues that were most frequently collected and that routinely met minimum RNA quality criteria: adipose (subcutaneous), tibial artery, heart (left ventricle), lung, muscle (skeletal), tibial nerve, skin (Sun-exposed), thyroid, and whole blood (Table 1) (14).
Table 1. GTEx pilot samples.
Characteristics of the 1641 RNA-sequenced samples included in the pilot data freeze. The second and third columns show the tissue abbreviation and color assigned to each tissue (used throughout). The nine tissues prioritized for sequencing are indicated by red stars. Boxes highlight two regions of the brain that were sampled in duplicate. A region each from the cerebellum and cortex (BRNCHA and BRNCTXA) was sampled on site during initial tissue collection, and again after the brain was received by the brain bank (BRNCHB and BRNCTXB). Cell lines included are an EBV-transformed lymphoblastoid cell line from blood (LCL) and a cultured primary fibroblast cell line from fresh skin (FIBRBLS). RIN, RNA integrity number.
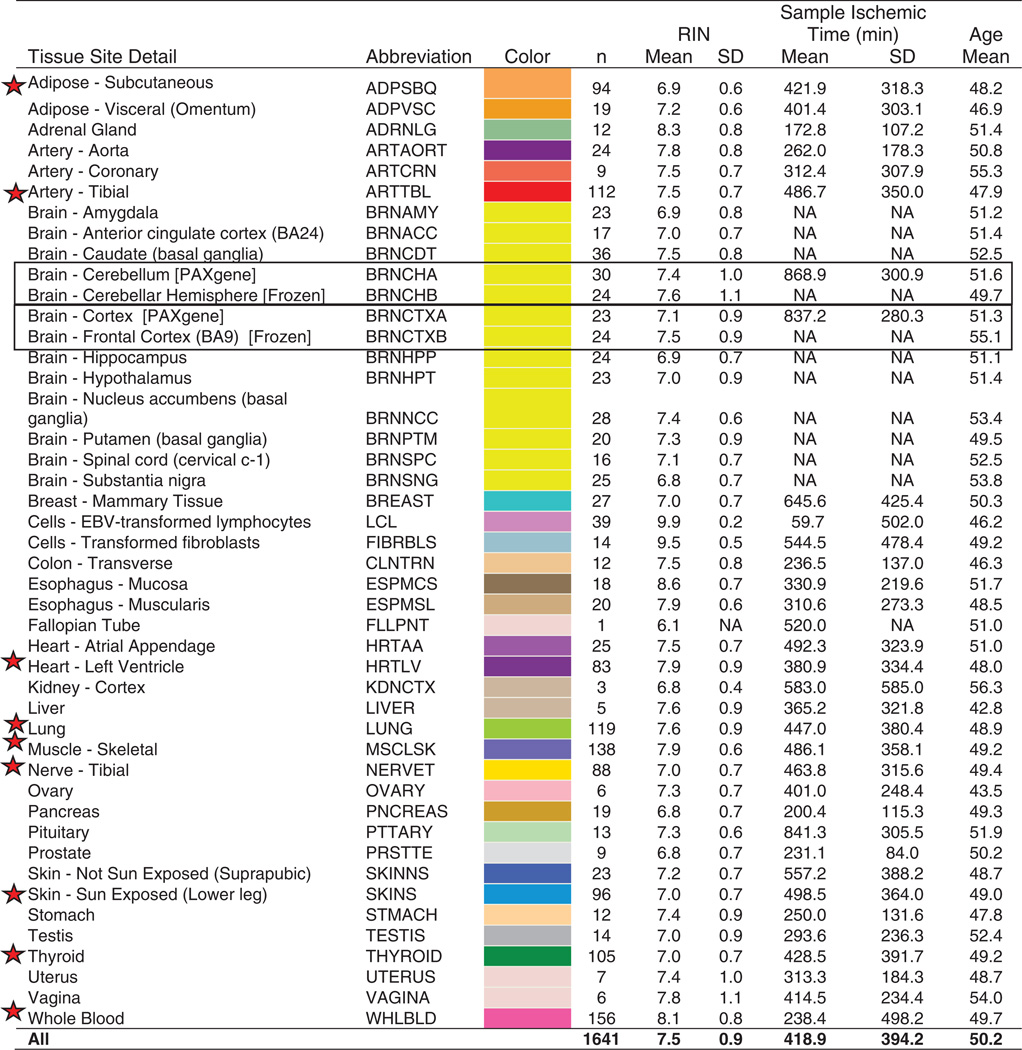 |
We performed 76–base pair (bp) paired-end mRNA sequencing on a total of 1749 samples, of which 1641 samples from 43 sites, and 175 donors, constituted our final “pilot data freeze” reported on here (14). Median sequencing depth was 82.1 million mapped reads per sample (fig. S3A). The final data freeze included samples from 43 body sites: 29 solid-organ tissues, 11 brain sub-regions (with two duplicated regions), a whole-blood sample, and two cell lines derived from donor blood [EBV-transformed lymphoblastoid cell lines (LCLs)] and skin samples (cultured fibroblasts) (Table 1 and tables S1 and S2). Median sample size for the nine high-priority tissues was 105; median sample size for the other 34 sampled sites was 18.5.
Gene expression across tissues
We examined the patterns of expression of 53,934 transcribed genes across tissues [on the basis of Gencode V12 annotations] (14, 15). The number of biotypes [protein-coding genes, pseudogenes, and long noncoding RNAs (lncRNAs)] that were transcribed above a minimal threshold [reads per kilobase per million mapped reads (RPKM) > 0.1] was similar for most tissues (average of 20,940) (fig. S3B). Testis was an outlier, with substantially more transcribed regions detected than other tissues (31,240 on average), many of which are lncRNAs. Whole blood was also an outlier, exhibiting the fewest detected transcribed regions (17,160 on average).
Hierarchical clustering demonstrated that expression profiles accurately recapitulate tissue type, with blood samples forming the primary outgroup (Fig. 1A). The multiple brain regions cluster strongly together as a single unit, but among those the 11 individual sub-sampled regions are less distinct (Fig. 1A and fig. S4A). The most distinct brain region is the cerebellum (fig. S4A) (16), with preservation method having little impact on that signal (fig. S4B). The distribution of gene expression across tissues is described by Melé et al. (17), who show that tissue-specific transcription is typically dominated by a few genes that vary from tissue to tissue.
Fig. 1. Sample clustering based on gene expression and exon splicing profiles.

(A) Clustering performed on the basis of gene expression values for all genes from Gencode v12 annotation. Tissue type is the primary driver of expression differences, with the nonsolid tissues (blood and LCL cell lines) clustering separately from solid tissues. Hierarchical clustering was performed using as distance = 1 – Pearson correlation, and average method. (B) Sample clustering based on the “percent spliced in” (PSI) values for exons across samples. Tissue differentiation is less clearly a driver, and brain is now the main outgroup, driven largely by a cluster comprised of cerebellum and cortex samples.
We quantified splicing events (splice junctions, exons, transcripts) by estimating exon inclusion levels, measured as PSI (percent spliced in) scores (14, 18). Clustering samples by PSI scores also largely, although less clearly, recapitulates tissue type. Samples from the brain, not blood, form the primary outgroup (Fig. 1B), which is divided into two groups: A group of 227 samples (from the cerebellum and cortex) forms an independent subcluster (cluster 1), and a smaller group of 97 samples (cluster 2, dominated by the remaining subregions) clusters closer to samples from the rest of the tissues. This is consistent with isoform regulation playing a comparatively larger role in defining cellular specificity in the brain (18, 19). These analyses are extended in Melé et al. (17) to define tissue-specific splicing signatures and to investigate in depth the role of individual variation in splicing.
eQTL analyses: Single-tissue eQTL analysis
A primary goal of the GTEx project is to identify eQTLs for all genes for a range of human tissues. Past studies, hampered by the difficulties of obtaining human tissue samples, have typically examined no more than three tissues (8, 20). Although our pilot sample sizes are modest for eQTL discovery, the breadth of tissues provides an opportunity to assess differential eQTL discovery among tissues. Because of our small sample sizes, we primarily examined eQTLs that act in cis to the gene (cis-eQTLs; see box S1), as the expected effect size of trans-eQTLs (box S1) is too low to be efficiently detected at this time. We calculated cis-eQTLs separately for each of the nine tissues with sufficient sample sizes (>80 donors) for all SNPs within ±1 Mb of the transcriptional start site (TSS) of each gene (14). Significance correlations between genotypes and gene expression levels were determined by linear regression on quantile normalized gene-level expression values, after correction for known and inferred technical covariates (fig. S8) (14), using Matrix eQTL (21). To obtain gene-specific significance levels while correcting for testing multiple SNPs per gene, we computed permutation-adjusted P values for each gene for the most significant SNP per gene (14). We defined “eGenes” as genes with at least one SNP in cis significantly associated, at a false discovery rate (FDR) of ≤0.05, with expression differences of that gene (box S1) (14). A list of the significant SNP-gene pairs detected per eGene can be found on the GTEx Portal (http://gtexportal.org).
The number of eGenes ranged from 919 in heart to 2244 in thyroid, with a total of 6486 unique eGenes across the nine tissues (Fig. 2A). Rerunning the analysis on successively downsampled donor subsets from each tissue showed an approximately linear relationship between eGenes and sample size (slope of ~21 eGenes per sample; Fig. 2A). Interestingly, thyroid and nerve share a steeper slope with ~29 significant eGenes per sample, whereas muscle and blood share a shallower trajectory with ~15 eGenes per sample, which may reflect the lower transcript complexity observed for these two tissues (17). The number of eGenes identified showed no signs of plateauing at current sample sizes in any of the tissues.
Fig. 2. Number and sharing of significant ciseQTLs per tissue.

(A) Numbers of significant cis-eQTL genes (eGenes) per tissue according to single-tissue analysis. For each gene, the minimum nominal P value was used as the test statistic and an empirical P value was computed to correct for number of tests per gene, based on either permutation analysis of genotype sample labels applied to the full set of samples per tissue (◆) or Bonferroni correction, used for downsampling (line) to reduce computational burden (14). In the range of sample sizes tested, the number of identified eGenes increases linearly with sample size. (B) Dendrogram and heat map of pairwise eQTL sharing using the method of Nica et al. (22). Values are not symmetrical, since each entry in row i and column j is an estimate of π1 = Pr(eQTL in tissue i given an eQTL in tissue j). Blood has the lowest levels of eQTL sharing with other tissues while adipose shows higher levels of sharing. (C) Activity probabilities for both multitissue modeling approaches, applied to all nine tissues, indicate that the most likely configurations are for eQTLs that are active in only a few tissues or in many tissues. (D) For eQTLs in each tissue considered separately, analyzing multiple tissues jointly increases the number of discovered eQTL associations (FDR < 0.05), as assessed by the SNP-based multitissue model.
Consistent with previous work (20, 22), the majority of the significant cis-eQTLs clustered around the TSS of target genes in all nine tissues (fig. S9, A and B, and fig. S10). The eQTL signals also tended to show an upstream bias (fig. S10 and tables S6 and S7); an average of ~80% of significant eQTLs fell within ±100 kb around the TSS, and ~60% of all eQTLs were upstream despite testing a similar fraction of SNPs upstream and downstream of the TSS (tables S6 and S7) (23). A slight but distinct overrepresentation of nonsignificant eQTLs around the TSS (relative to other SNPs near the gene; red versus black line in figs. S9A and S11A) supports the existence of additional eGenes, which did not meet significance with current sample sizes.
To investigate the sensitivity and validity of our study, in particular because we used tissues from deceased donors, we compared the GTEx blood eQTLs to a previously reported eQTL study of whole blood samples in ~5300 individuals (7). Although many experimental and processing differences exist between these studies, a considerable fraction of GTEx eGenes (68%) were replicated in this study at FDR < 5% (14). Given the incomplete overlap in variants tested, we also compared our eQTLs against a smaller study of 911 blood samples taken from the Estonian Biobank, where we were able to apply a similar eQTL analysis pipeline to that used in GTEx and hence get better SNP coverage (fig. S11) (14). Notably, 98% of GTEx blood eQTLs at FDR < 5% showed consistent allelic direction with those eQTLs (P < 10−200, binomial test) (14).
Multitissue joint discovery of eQTLs: Tissue specificity and sharing of eQTLs
The specificity or sharing of eQTLs among different tissues and cell types is of considerable biological interest (8, 22, 24), yielding insights into differential genetic regulation among tissues. Furthermore, cross-referencing tissue-specific eQTLs with disease genetic associations could help identify tissues most relevant to disease biology. We used the GTEx pilot data to examine eQTL sharing across multiple tissues and leveraged the large tissue range to discover weak but constitutively active eQTLs.
We investigated patterns of eQTL sharing using 22,286 genes (with RPKM > 0.1 in at least 10 samples) for each of the nine tissues with both a simple non–model-based analysis of every pair of tissues (22) and more sophisticated Bayesian models for joint analysis of all nine tissues (24, 25). Analyses focused on a ±100-kb window surrounding the TSS for each gene, which is smaller than used for the single-tissue analysis, as this is where we observe the highest eQTL density.
The non–model-based pairwise analysis method identifies significant SNP-gene pairs in a first tissue, and then uses the distribution of the P values for these pairs in the second tissue to estimate π1, the proportion of non-null associations in the second tissue (22, 26). Estimated values of π1 ranged from 0.54 to 0.90 (Fig. 2B). For every pair of tissues, we observed a high level of sharing of eQTLs, and this pairwise sharing of eQTL among tissues roughly mirrored the tissue gene expression correlations (fig. S12).
To assess patterns of sharing beyond tissue pairs, we applied two Bayesian methods that assess, for each SNP-gene pair, the evidence for each of the 512 (29) possible null/alternative eQTL configurations. The first method (24) is “gene-based” and assumes a single causal eQTL per gene. We extended the model to (i) support the fact that not all tissues were sampled across all GTEx donors; (ii) calculate a gene-level FDR without requiring permutations (27); and (iii) include a fine mapping approach across multiple tissues (28). We also used a “SNP-based” approach (25), which assesses association of each SNP-gene pair separately, working directly with the z-statistics for each SNP-gene pair within each tissue [see also (29)]. We summarized the estimates of tissue specificity using marginal posterior probabilities for the number of tissues in which a randomly selected gene (gene-based model) or SNP-gene pair (SNP-based model) is active (Fig. 2C). Both approaches show a U-shaped pattern, with high tissue specificity (activity in a single tissue) or tissue ubiquity (activity in all nine tissues) more common than profiles involving only a few tissues, despite many more possible combinatorial patterns for intermediate specificity. Notably, both methods indicate that more than 50% of all detected eQTLs are common to all nine tissues. Reassuringly, both Bayesian methods produce pairwise tissue sharing probabilities that show agreement with the non–model-based analysis (fig. S13). Figure S14 illustrates the value of the multitissue analysis for an example in which the tissue specificity of an eQTL supports NDRG4 as a candidate to influence QT interval in the heart (30). Despite superficial similarity between patterns of tissue-tissue gene expression and eQTL sharing, extensive comparisons of eQTL evidence to expression levels indicate that the tissue-specific patterns are only modestly correlated with average tissue expression levels (fig. S15).
We also examined effect size estimates for eQTLs that are shared between tissues. The vast majority of shared eQTLs show consistent effect directions in different tissues. However, some SNPs showed apparent opposite effect directions in different tissues (fig. S16). A number of examples that we investigated using multi-SNP multitissue analyses (28) appeared to be due to pairs of SNPs, in LD with one another, that were separate eQTLs in different tissues (fig. S17) rather than being a single eQTL with opposite effects.
Beyond the biological interest of eQTL sharing among tissues, the availability of multiple tissues can also increase the power to detect eQTLs active in multiple tissues by combining information across tissues (24). To investigate, we conducted a permutation analysis by holding the expression data fixed and permuting the genotypes. In this manner, we identified the significant eGenes per tissue (as in the single-tissue analysis above, but for a smaller window near the TSS), or jointly for any combination of tissues by considering the minimum P value across tissues under each permutation. With this approach, the number of eGenes identified for individual tissues was similar to the single-tissue eQTL analysis (Fig. 2A). Next, the minimum P value for each gene across all nine tissues was used to test for eQTL evidence, and subjected to gene-level false discovery control. A total of 7425 eGenes with FDR < 0.05 were identified, representing a factor of 3 increase relative to the maximum number of significant eGenes for any single tissue. The Bayesian models, which leverage the high proportion of tissue-common eQTLs, increase the power to detect eQTLs for an individual tissue by borrowing strength from the remaining tissues. Thus, of the original 22,286 expressed genes, 10,030 showed a significant eQTL (FDR < 0.05) with the gene-based Bayesian multitissue model (approximately 35% more than by joint permutation). Using the SNP-based Bayesian model, we selected four of the tissues with the largest sample sizes, and computed the marginal eQTL probability for each tissue alone after adding one to eight additional tissues in a fixed sequence. This resulted in a marked increase in the number of significant SNP-gene pairs (FDR < 0.05) (Fig. 2D). For downstream analyses using the multitissue results, a single SNP with highest posterior probability was chosen as representative across all tissues, recognizing that multiple causal SNPs would be difficult to resolve with the current sample size. Posterior probability profiles for both Bayesian models, as well as marginal posterior probabilities that an eQTL is active in each tissue, based on summing the probabilities across all configurations, are available for these genes (http://gtexportal.org).
Allele-specific expression analysis
To complement eQTL analysis, we used allele-specific expression (ASE) of genes to indirectly estimate the overall effect of cis-regulatory variants on the expression of nearby genes. Individuals that are heterozygous for a cis-regulatory variant may differentially express each of the two alleles of the affected gene. In these cases, assuming no other causes for allele-specific expression, imbalanced allelic expression of a gene can be used to infer the effect of cis regulation on the gene even without necessarily identifying the specific regulatory variant(s). In addition to the subtle cis-regulatory effects described here, ASE of premature stop variants is also informative of nonsense-mediated decay (31), and consistent monoallelic expression of specific genes can be used to detect epigenetic silencing by imprinting.
ASE can be estimated from the abundance of RNA-seq reads supporting each of the gene’s alleles, measured at heterozygous sites within the transcribed region. We identified a median of 6385 sites per sample (across all tissues and all individuals) that were both heterozygous for a transcript SNP in the imputed genotype data of the associated individual and well expressed in that sample (SNP covered by ≥ 30 RNA-seq reads) (fig. S18). Of these sites, a median of 390 sites per sample had significant ASE (binomial P < 0.005, or 144 sites with FDR 21%, after rigorous quality control of ASE sites and after downsampling to exactly 30 reads at each site to ensure equal power to detect ASE) (Fig. 3A and table S8A) (14). To assess whether different tissues vary in terms of the overall effect of cis-regulatory variation in them, we estimated the proportion of those sites with significant ASE in each of the tissues (combining data from all individuals) (14) and found that the fraction of significant ASE sites varied widely across tissues, with a range of 1.7 to 3.7% (median 2.3%). Brain regions appeared depleted for allelic effects (2.0% ASE in brain versus 2.7% in other tissues combined; P < 2 × 10−16, Fisher’s exact test) (Fig. 3A), consistent with the hypothesis that brain-specific genes exhibit reduced genetic diversity (32). Our results generally support our eQTL findings; in thyroid, where we observed a high number of eQTLs (Fig. 2A), we also see an increased amount of ASE (Fig. 3B). Blood and LCLs have a higher proportion of allelic effects (3.6% versus 2.3% in others; P < 2 × 10−16, Fisher’s exact test) (Fig. 3B), suggesting nongenetic sources of monoallelic expression possibly due to the more clonal nature of these cells (33).
Fig. 3. Quantification of regulatory diversity by ASE.

(A) Proportion of sites with significant ASE (P < 0.005) in each tissue (colored and labeled as in Table 1), with binomial confidence intervals. (B) Proportion of significant ASE sites for the nine tissues with eQTL data as a function of the proportion of eQTLs after regressing out the log of sample size. (C) Partitioning variation in allelic and total gene expression within and between individuals and tissues. We calculated pairwise Spearman rank correlations between all the samples using two metrics [(D) and (E)]. (D) Allelic ratios over sites (sampled to 30 reads each), which captures similarity in allelic effects that are a proxy for cis-regulatory variation. (E) Total read counts over the same sites, which captures similarity in total gene expression levels. The plots show the distributions of pairwise correlations for sample pairs that are from (1) different tissues and different individual, (2) different tissues within an individual, or (3) same tissues in different individuals. Gene expression levels are highly correlated within the same tissue (E3) (see Fig. 1A). However, allelic ratios show highest correlation among different tissues of the same individual that share the same genome (D2).
To compare between-sample and between-tissue sharing of ASE with the overall similarity of gene expression, we constructed two Spearman rank correlation matrices between all pairs of samples. In one, we correlated ASE ratios, capturing sharing of cis regulation; in the other, we correlated total coverage of the same sites, capturing similarity of overall gene expression levels, analyzing genes that are expressed in both tissues (both matrices used counts of reads covering heterozygous sites shared between the compared pair of samples) (14). The two matrices of tissue medians were highly correlated (Mantel test, r = 0.772, P < 0.0001; fig. S19), indicating that tissues with similar gene expression profiles also have a higher degree of sharing of genetic regulatory effects.
Interestingly, when partitioning the full sample-level matrix into correlations between samples from different tissues of the same individual, the same tissue across individuals, or different individuals and different tissues (Fig. 3C), we found more complex relationships between total expression and allelic ratios. As expected, expression levels are determined by tissue, and samples cluster by tissue (75.6% of variance; Fig. 3E and table S8B). However, allelic ratios show the opposite pattern (Fig. 3D), with higher correlation among tissues within the same individual (17.9% of variance) than among individuals for the same tissue (8.6%). These results indicate that ASE is primarily determined by the common genome among different tissues of the same individual (34). This suggests that the two dimensions of gene expression variability, gene expression levels and allelic ratios, are largely defined by independent factors.
ASE analysis can also be used as orthogonal confirmation of eQTL effects, on the basis of the expectation that individuals who are heterozygous for a cis-eQTL variant should manifest biased allelic ratios in the eQTL target gene. We performed this analysis at the genome-wide level across all the tissues in the GTEx data set, including the tissues with sample sizes that were too small for eQTL analysis. Examining the significant eQTLs identified by single-tissue analysis for the nine main tissues, we looked at their ASE effects separately in each of the 42 tissues, calculating the odds ratio of significant versus nonsignificant ASE for eQTL heterozygote versus homozygote individuals (e.g., for thyroid eQTLs we looked for ASE in all 42 tissues, then for blood eQTLs and so on) (figs. S20 and S21). In addition to replication of eQTL signals in the discovery tissue, we can estimate how relevant our eQTL findings from nine tissues are to a wide variety of other tissues (independent of sample size or allele frequency) and then assess which tissue is the best proxy for capturing regulatory effects in another tissues of interest.
We found that the overall tissue specificity of the eQTL sets varies. For example, eQTLs identified in skeletal muscle were less active in other tissues. Some tissues, such as brain, were not well captured by any of the nine eQTL sets here. ASE analysis also allows quantification of genome-wide tissue sharing of cis-regulatory signals without relying on eQTL discovery, but instead by measuring how often a site with a significant ASE signal in one tissue is significant in another tissue of the same individual. The proportion of shared ASE effects between tissue pairs within an individual varies between 36% and 58% (mean 46%), with slightly increased sharing observed between closely related tissues (conditioning on each site being measurable in both of the tissues) (fig. S22A). This relatively high degree of sharing is consistent with the eQTL results described above. If we relax the constraint of requiring a gene to be expressed in both tissues, then the proportion of shared ASE effects is substantially lower (0.85 to 39%, mean 11%; fig. S22B). This finding represents the total probability of detecting a regulatory effect in another tissue, and highlights a high degree of apparent tissue specificity that derives from the fact that a gene expressed in one tissue is often simply not expressed in another. This is particularly pronounced in brain, where the large proportion of genes showing tissue-specific isoform expression in that organ (17) drives a lower degree of overall sharing. Whole blood and skeletal muscle are partial outliers, relative to other tissues, with lower sharing of eQTLs and lower average replication in other tissues (Fig. 2A, fig. S12, and figs. S20 to S22).
Although eQTL analysis and other association-based analyses are efficient for identifying common variants with phenotypic effects in populations, the genotype of those variants may not be a good indicator of the gene expression phenotype at an individual level. We investigated this using ASE analysis to estimate how consistent the allelic effect of single-tissue eQTL variants are across individuals that share the same genotype for the best associated eQTL variant per gene. We tested a subset of 606 eQTLs that had high read coverage and a large number of samples for transcript heterozygous sites in the eQTL discovery tissue. We identified 53 eQTLs with significant replication of the eQTL signal in ASE, as inferred by higher allelic imbalance in eQTL heterozygotes than in homozygotes (linear regression P < 0.05 after Bonferroni correction; Fig. 4, A and B). Further, for 22 of the 53 eQTL genes, individuals show variability in their allelic ratios that cannot be accounted for by the eQTL genotype or sampling error (Bonferroni corrected P < 0.05 from permutation of read counts; Fig. 4, C and D). These results highlight that common eQTLs do not explain all of the cis-regulatory effects in individuals and are inadequate predictors of allelic expression variance at the individual level. The higher variance in eQTL heterozygotes than in homozygotes (Mann-Whitney P = 2.13 × 10−7; Fig. 4B) suggests that part of this variance might originate from modification of the main eQTL effect by environmental or trans effects, or due to additional, independent regulatory variants (34).
Fig. 4. Cis-regulatory effects in individuals that are not explained by detected eQTLs.

(A) An eQTL showing individual homozygous (AA) for the eQTL SNP (left panel) or heterozygous (AG) (right panel). ASE is measured at the TC SNP. (B) An example of replication of an eQTL signal in ASE analysis in the NDRG4 gene, with eQTL heterozygotes showing higher ASE in the eQTL target gene than eQTL homozygotes (only a subset of individuals shown; linear regression P = 5.69 × 10−6). The error bars are from a binomial test for the allelic ratio. (C) For each eQTL gene where the eQTL signal was replicated in ASE (linear regression P < 0.05 after Bonferroni correction), the eQTL heterozygotes show higher variance in allelic ratio (Mann-Whitney P = 2.13 × 10−7). (D) Permuted P value for the variance between individuals, which is higher than expected in 22/53 genes (9 genes in homozygotes, 20 in heterozygotes).
Analysis of splicing QTLs
Beyond estimating overall levels of gene expression, RNA-seq data also allow for the quantification of expression levels of individual transcript isoforms, as well as components of these, such as exons, splice junctions, and untranslated regions. We refer to the quantitative variation of gene structure due to genetic variation as splicing QTLs (sQTLs) (Fig. 5A). To identify sQTLs, we used Altrans (35), a method that identifies SNPs that are associated with variation in the expression levels of exon junctions (sjQTLs; box S1) (14), and sQTLSeekeR (36), a method that identifies SNPs associated with the variation in the relative abundances of gene transcript isoforms (srQTLs; box S1) (14). Altrans identifies both novel and annotated splicing events, while sQTLSeekeR tests only annotated isoforms. Altrans, however, is restricted to changes in the usage of splice junctions, while sQTLSeekeR can in principle detect any variation in the relative abundance of transcript isoforms (fig. S23). Altrans was run using a ±1 Mb region around the TSS, while for sQTLseekeR, we tested within the body of the gene ±5 kb (14).
Fig. 5. Splicing QTLs.

(A) A splicing QTL that affects the relative usage of alternative splice isoforms for the tRNA methyltransferase 1 homolog gene (TRMT1). TRMT1 has three annotated isoforms, only two of which are abundant in skeletal muscle. The relative abundance of the two isoforms differs by genotype (number of individuals below each genotype), with heterozygotes showing an intermediate behavior. This SNP has not been detected as an eQTL. The right panel shows the exonic structure of the transcripts along with the location of the sQTL SNP (dotted line). (B) The relative proportions of the different types of splicing events detected by the two methods over the nine tested tissues (fig. S23). (C) Functional enrichment of sQTLs from Altrans and sQTLseekeR. For the top-ranked SNPs associated with a given splicing event, we computed the relative frequency with which they map to different biologically determined ENCODE functional domains.
We detected an average of ~1900 genes with Altrans, and ~250 with sQTLseekeR, with at least one cis-sQTL per tissue (FDR = 0.05; table S9). The greater genomic window tested around the TSS for Altrans, and the capacity to detect novel splicing events, explains the much larger number of sQTLs detected by Altrans than by sQTLseekeR (about 70% of Altrans sQTLs correspond to novel events, and only about 10% map within 5 kb of annotated genes). Despite the difference, Altrans and sQTLseekeR show strong complementarity, detecting variants that are associated with very different types of alternative splicing events. Most of the splice events detected by Altrans (80%) are exon-skipping events, while 60% of those detected by sQTLseekeR correspond to complex splice events, such as mutually exclusive exons (Fig. 5B and fig. S23). If we consider only sQTLs associated with exon-skipping events, the overlap in sQTLs identified by both methods is substantial (36% of sQTLseekeR are also found by Altrans, P = 0.004) (figs. S23 to S25). An example of an sQTL that we identified with potential biological relevance is shown in Fig. 5A (further examples are in fig. S26).
Significant sQTLs show a high degree of sharing among tissue pairs, with tissue-specific sQTLs accounting only for 7 to 21% of the total depending on the tissue (figs. S27 and S28). The highest degree of sharing is between heart left ventricle and whole blood, whereas the two tissues that share the fewest sQTLs are whole blood and Sun-exposed skin. In general, sQTLs identified in whole blood are shared at lower levels with other tissues, as was observed for eQTLs (Fig. 2A and fig. S12, A and B). Although we observe the same enrichment of sQTLs around the TSS as seen for eQTLs, the sQTLs that are shared across multiple tissues tend to be closer to the TSS than those that are tissue-specific (fig. S28). On average, 20% of sQTLs associated with changes in exon junction abundance by Altrans were also predicted to be eQTLs (π1 = 0.20, π1 = 0.14 to 0.27; table S10). An even larger fraction (48%) of sQTLs detected by sQTLseekeR associated with changes in relative abundances of gene transcript isoforms, were identified as eQTLs (π1 = 0.48, π1 = 0.13 to 0.70; table S10). This enrichment of eQTLs among sQTLs is larger than expected at FDR < 0.05, but a substantial fraction of sQTLs are unique and not detected by standard eQTL analysis.
Functional annotation of eQTLs in noncoding regions
Genetic variants affecting gene expression and splicing patterns have been shown to fall within regulatory elements, providing a potential molecular basis for their effects (37–39). To assess whether the eQTLs discovered across the nine tissues were enriched in regulatory regions, we used a set of regulatory annotations from the ENCODE project (10) and the Epigenomics Roadmap project (40), including regulator-bound locations, DNase I hypersensitivity sites, and maps of histone modifications for proximal and distal regulatory regions (14). For each tissue, we chose the top significant SNP per gene from the single-tissue eQTL analysis (14,431 eQTL SNPs). Discarding SNPs that were within annotated genes resulted in 4085 intergenic eQTL SNPs, which were compared to our regulatory annotations (14). Intergenic eQTL SNPs were enriched for transcription factor-bound sites, open chromatin, promoters, and enhancers (P = 4.3 × 10−18, 2.9 × 10−8, 1.7 × 10−19, and 0.003, respectively) relative to the density of these features within a 2.5-kb window of the tested eQTLs (fig. S29 and table S11). This enrichment was even more pronounced in a subset of 91 unambiguous intergenic eQTL SNPs (fig. S29). In contrast, we found sQTLs to be enriched within annotated splice junctions relative to other functional regions (Fig. 5C).
We also asked whether the specific SNP-gene links (“genetic links”) predicted by our eQTL analysis were supported by enhancer-gene links based on functional correlation (“functional links”) (14). We found that open chromatin regions containing an eQTL SNP show higher correlation to the corresponding gene, relative to open chromatin regions without an eQTL at similar distances and linked to genes also containing an independent eQTL (r = 0.037 versus 0.030, paired Mann-Whitney two-sided P = 4.3 × 10−11, SE = 0.00114, n = 32,168 eQTL-SNP pairs).
This demonstrates the value of eQTL information to establish functional links between regulatory elements and genes in the genome, an analysis that will increase in power as additional tissues and subjects are added to the data set.
Gene network inference within tissues using cross-individual expression variation
To provide a view of coordinated gene regulation arising from both cis and trans genetic effects and nongenetic sources, we inferred gene-gene coexpression networks within tissues. Studying each of the nine tissues in isolation and examining variation of gene expression across individuals, we linked pairs of genes that show correlated expression (top 1% of pairs by Pearson correlation) (14), revealing similar patterns of coexpression across tissue pairs (Fig. 6A). The median π1 statistic (fraction of true positive results) (26), estimating the total fraction of coexpressed gene pairs identified in one tissue that are replicated in a second tissue, is 0.44, ranging from 0.30 to 0.58 (Fig. 6A). Furthermore, the specific tissue pairs with higher overlap of coexpressed genes also have a greater overlap of cis-eQTLs than other tissue pairs, indicating a similar pattern of tissue relatedness underlying both results (P < 0.05 for correlation between similarity matrices of Figs. 2B and 6A). Thus, although coexpression networks primarily capture non-cis mechanisms, including trans regulation and environmental factors, the overall level of sharing and the specific patterns of tissue relatedness agree with the patterns observed from cis-eQTLs, which suggests that regulatory mechanisms beyond cis effects may be shared across tissues.
Fig. 6. Coexpression networks within tissues and individuals.

(A) Similarity of coexpression networks discovered in each tissue separately (rows) and replicated across all other tissues (columns), on the basis of the correlation in gene-pair expression levels across all individuals for a given tissue, as quantified by the π1 statistic. The tissues in this heat map are ordered as in Fig. 2B. (B) Coexpression modules learned within adipose tissue on the basis of weighted gene coexpression network analysis (WGCNA). The heat map shows the similarity in gene expression patterns (across individuals) for each pair of genes expressed within adipose tissue (red = high correlation, blue = low correlation). Non-gray colors highlight separate modules. (C and D) Genes in the same adipose coexpressed module [(C), rows] show enrichment for similar gene ontology (GO) categories (columns) and are co-bound by the same transcription factors (TF) [(D), columns] in their transcription start site (blue = Benjamini-Hochberg corrected P < 0.01). Dendrogram (top) denotes TF-to-TF similarity in module targeting. (E) Average expression level (red = high, blue = low) in each tissue (rows) across 117 expression modules (columns). Modules highlighted include Mod6, showing highest expression in whole blood and cortex; Mod95, showing highest expression in noncortex brain; and Mod101, showing brainwide expression. (F) Expression pattern of 175 individuals (columns) across 45 tissues (rows) for the ZFP57 gene encoding a KRAB domain transcription factor. Colored entries denote expression levels (heat map). White entries denote missing expression measurements for an individual in a given tissue. (G) Probability of membership of each individual (columns) in each expression module (rows) for the three most significant modules [highlighted in (E)]. (H) Genotype of the three top modQTL SNPs (rows) across individuals (columns) shows correlation with module membership probability.
We also used a weighted gene coexpression network analysis (WGCNA) approach (41) to construct coexpression networks and extract gene modules for each tissue (Fig. 6B). The clustering of coexpressed genes into modules allows us to identify active biological processes across tissues (Fig. 6C). Modules enriched for common Gene Ontology (GO) biological processes were observed in all nine tissues (e.g., cell cycle, protein transport), while other biological processes were only seen in specific tissues (e.g., fatty-acyl-CoA metabolic process enrichment seen only in adipose, Benjamini-Hochberg corrected P = 2.6 × 10−7) (14). In addition to functional annotation, the identified coexpression networks also enabled us to search for potential transcriptional regulators of these modules (Fig. 6D). We found enrichment of transcription factor binding in promoter regions of genes in the same module using ENCODE chromatin immunoprecipitation (ChIP)–seq data (42), suggesting that some modules could be regulated by a large number of transcription factors (fig. S30A). Finally, we compared coexpression modules learned in each tissue individually to those learned in other tissues, based on common gene membership and correlation of first principal components of expression variation across individuals (fig. S30B). Many modules showed correlation between principal components to other modules across tissues but lacked common genes, demonstrating that similarity in patterns of variation is sometimes only visible at the module level.
Gene network inference within individuals using cross-tissue expression variation
The availability of multiple tissues per individual further allowed us to define modules of co-regulated genes within each individual, by correlating gene expression across tissues for the same individual (14). Merging modules across individuals produced 117 modules (fig. S6E), containing between 25 and 414 genes. Each of these modules corresponds to a multitissue expression pattern for a gene, enabling us to study changes in the regulatory program of genes that affect multiple tissues at a time.
We used these modules to identify instances of coordinated variation in multitissue expression patterns across individuals. For each gene, we estimated the proximity of each individual’s expression pattern to the median expression pattern of each module, corresponding to a “module membership score” (Fig. 6F and fig. S31A). We then calculated a module membership score for each individual (Fig. 6G) (14). Cases where depletion in membership of one module was accompanied by a corresponding increase in membership of a different module are called “module switching events.” The majority of genes showed conserved multitissue gene expression patterns among individuals, remaining in the same module or switching between modules with similar expression patterns (correlation distance < 0.5). However, we identified 3965 genes (21%) that show switching between dissimilar modules (correlation distance > 0.5), which may have important biological consequences (fig. S31B). Using module membership scores as a quantitative trait, we searched for neighboring SNPs that are correlated with these module membership scores (Fig. 6H), which we call module-switching QTLs (modQTLs). Searching a window of 1Mbp around each gene in cis, we found a total of 2102 modQTLs associated with statistically significant switching (FDR < 0.05) between dissimilar gene modules (correlation distance > 0.5), suggesting that genetic variation for those genes leads to changes in multitissue regulatory programs. For example, the ZFP57 gene shows three distinct patterns of multitissue expression across individuals, and three nearby SNPs are associated with these changes, suggesting a genetic basis for these multitissue differences (Fig. 6H).
We compared the 2102 modQTLs with eQTLs discovered by both the single-tissue and multitissue eQTL analyses. At all correlation levels of stringency, the modQTLs capture 53% of tissue-specific eQTLs (calculated as the percentage of lead SNPs in LD r2 > 0.8 with lead modQTLs) and 60% of multitissue eQTLs, and these two together account for only 42% of modQTL SNPs. Hence, 58% of modQTLs are not discovered by other methods, which suggests that the approach has the potential to reveal novel genetic effects on the modulation of genes across tissues (fig. S31C).
Personal transcriptomics and implications for human disease
The in-depth analysis of multitissue transcriptomes enables both an understanding of the population-level properties of the transcriptome as well as individual level properties inferred from analyses of single transcriptomes or the transcriptomes of multiple tissues from a single individual. As is the case for personalized genomics, this individual-level transcriptome analysis is likely to become a crucial addition to the personalized assessment of an individual’s biology and likely disease status.
Impact of individual gene-disrupting variants on splicing and expression
Assessing the functional impact of DNA sequence variants identified in whole-genome and exome sequencing studies remains a major challenge. Variants predicted to result in the truncation of proteins (splice, stop-gain SNVs, or frame-shift indels) may have large effects on biochemical function, but are also highly enriched for annotation artifacts (43). Errors in predicting the true functional impact of these and other variants can substantially reduce discovery power in common complex diseases and, more important, can affect disease diagnosis in clinical settings (44).
The GTEx multitissue expression data provide an opportunity to assess the real impact of protein-truncating variants (PTVs) on the human transcriptome [see also (31)]. We combined exome sequencing and RNA-seq data from 173 GTEx individuals to assess the global properties of predicted high-confidence PTVs (14). PTVs are enriched in alternatively spliced exons, with just 38.4% of high-confidence PTVs having annotation support across all reported transcripts (Fig. 7A), and only 51 to 55% supported by the major transcript of at least one tissue (for all tissues with at least 10 samples). These numbers highlight the need for careful transcript-specific assessment of functional annotation for all classes of variation. Furthermore, if we require that a fixed percentage of the dominant isoforms across all sequenced tissues support this annotation, we find that the percentage of predicted PTVs with annotation support of PTV decreases as we increase the threshold for the proportion of tissues with major transcript isoform support for PTV prediction (Fig. 7B). This highlights the need for empirically derived reference transcript sets across a broad array of tissues to enhance clinical interpretation for personal genome sequencing and disease studies. An example with clinical ramifications is shown in fig. S32.
Fig. 7. Integration of transcriptome data improves annotation of putative protein truncating variants (PTVs).

(A) The majority of annotated PTV variants are partial PTV, meaning that only a fraction of the RNA-seq transcripts support PTV annotation. (B) For all the predicted PTV variants, we ask what percentage of variants maintain a PTV annotation if we require that a fixed percentage of the dominant isoforms across all sequenced tissues support a PTV prediction; 70% of PTV variants are relevant if the threshold is 10%, whereas only 40% of PTV variants are relevant if the threshold is 100%.
GWAS and eQTLs
The ultimate goal of the GTEx project is to provide a framework for biological interpretation of disease-related variants. To evaluate the relevance of the discovered eQTLs in disease mapping studies, we tested the eQTLs identified in each tissue for association with disease using the Wellcome Trust Case Control Consortium (WTCCC) studies of seven complex disorders (45) (see Fig. 8A). Using eQTLs identified in whole blood, we found an enrichment for top associations with autoimmune diseases (shown as a leftward shift from the null distribution in the Q-Q plot in Fig. 8A), for Crohn’s disease, rheumatoid arthritis, and type 1 diabetes among eQTLs (P < 2.2 × 10−16 for each disease), consistent with the utility of blood and lymphoblastoid cell lines (LCLs) in trait mapping for autoimmune disorders (46, 47). In contrast to the autoimmune disorders, we found no enrichment for association (no shift in P value distribution) with bipolar disorder or type 2 diabetes among the blood eQTLs (P > 0.05) (Fig. 8A). This tissue specificity of autoimmune enrichment results suggests they are not due to the confounding effects that result from similar underlying genomic properties between eQTL and disease association regions. More generally, we observed trait-specific levels of enrichment for the WTCCC disease traits among the nine different single-tissue eQTL sets.
Fig. 8. Tissue-dependent GWAS eQTL enrichment Q-Q plots.

(A) eQTLs are enriched for trait associations with an important class of complex diseases. eQTLs discovered in whole blood (plotted in red) show significant enrichment for SNPs associated with autoimmune disorders from the WTCCC study (type 1 diabetes, Crohn’s disease, and rheumatoid arthritis) relative to null expectation (shown in gray) defined by non-eQTLs. (B) Enrichment of eQTLs for disease associations is tissue-dependent. Single-tissue eQTL annotation can be used to increase power to detect associations with hypertension, a disease for which the WTCCC study failed to yield significant associations. Notably, eQTLs discovered in adipose are enriched relative to muscle, lung, thyroid, skin, heart, and tibial artery (P < 0.05, Kolmogorov-Smirnov test) for known SNP associations with the hypertension.
Remarkably, the use of eQTLs increased power to detect associations with hypertension (Fig. 8B). In particular, eQTLs in subcutaneous adipose were significantly enriched for multiple associations with hypertension relative to muscle, lung, thyroid, skin, heart, and tibial artery (P < 0.05, Kolmogorov-Smirnov test) (Fig. 8B). This is particularly noteworthy because the WTCCC GWAS of this disease did not yield any genome-wide significant associations, which suggests that larger sample sizes were required to identify highly significant SNP associations in the absence of functional data. Because the majority of GWAS-identified variants (~95%) lie in noncoding regions of the genome, we determined which genome-wide significant trait associations (P < 5 × 10−8) reported to date are in LD with at least one GTEx-identified eQTL. We merged NCBI’s Phenotype-Genotype Integrator (PheGenI) (48) and the NHGRI GWAS catalog (49), yielding 10,129 genome-wide significant SNP associations with nearly 630 distinct complex traits. In total, 5195 “independent” SNPs were identified after LD pruning at r2 ≥ 0.8 and counting SNPs only once (14). Of these, 308 (~6%) are in strong LD(r2 ≥ 0.8), with a “best eQTL per gene” (at FDR < 0.05) from either the single-tissue or multitissue eQTL discovery analysis (table S12) in at least one of the nine tissues tested. For two-thirds of these cases (211 SNPs), no putative deleterious coding variants (nonsynonymous or splice variants) in the target gene product lie in LD (r2 ≥ 0.8) with the GWAS SNP; this finding suggests that regulatory effects may underlie the causal mechanism, although additional work is needed to prove causality. GWAS SNPs in LD with an eQTL show a factor of 2 higher representation in coding regions relative to all GWAS SNPs (11% versus 4.6%; table S13). Notably, about one-third of the eQTLs in LD with GWAS SNPs were detected only with methods that leverage the multitissue nature of GTEx data. Increasing both sample sizes and the range of tissues will likely increase the number of detected GWAS-eQTL loci.
Annotating a GWAS SNP with an eQTL can help to highlight candidate causal genes within a locus (i.e., the eQTL target gene). We found that proximity-based and eQTL-based gene assignments for GWAS SNPs were often discordant (47). A surprising proportion of trait-associated SNPs in LD (r2 ≥ 0.80) with a GTEx eQTL showed disagreement between the strongest eQTL-derived target gene and the genes that were physically proximal to the GWAS SNP (table S14). Of 190 GWAS loci (P < 5 × 10−8) where the lead SNP is an eQTL from the single-tissue analysis (FDR < 0.05) with only a single target gene, in 65 cases (~34%) this eQTL target gene differs from any of the genes that were closest to the SNPs in LD. These results were also observed when we restricted the target genes to protein-coding genes, when we pruned the GWAS SNPs for each trait examined (r2 ≥ 0.80), and when we used the eQTLs identified from the multitissue joint eQTL analysis.
In addition to prioritizing causal genes in GWAS loci, an eQTL catalog from multiple human tissues can highlight the relevant tissue(s) of action, evaluate the tissue specificity of GWAS loci, and characterize pleiotropic associations. We demonstrate the value of multitissue data to explore and resolve these issues for the GWAS intronic SNP, rs633185, located in ARHGAP42 (Fig. 9). This GWAS SNP is in high LD (r2 = 0.93) with the best cis-eQTL (rs604723) targeting ARHGAP42, and the best cis-eQTL for a neighboring gene, TMEM133 in tibial artery. Evaluating eQTL significance in all nine tissues shows that although the eQTL’s significance is indeed strongest in tibial artery, several other tissues may merit consideration in exploring the causal function of this locus, such as subcutaneous adipose and skin (Fig. 9, B and C, and fig. S33). This supports the need to explore the genetic basis of disease in the fuller context of a wide range of human tissues. The GTEx eQTLs may also be useful in highlighting the role of noncoding genes in disease risk and other complex traits (fig. S34). We found that ~13% of candidate genes proposed by GTEx eQTLs, and in LD to genome-wide significant GWAS SNPs, are noncoding genes (table S15).
Fig. 9. A blood pressure-associated SNP is a significant eQTL in tibial artery, for ARHGAP42 and TMEM133.

(A) The GWAS SNP, rs633185 in the intron of ARHGAP42, is associated with systolic blood pressure (P = 1.2 × 10−17) and diastolic blood pressure (P = 2 × 10−15). This GWAS SNP is in tight LD (r2 = 0.93) with the most significant eQTL for ARHGAP42 in tibial artery, rs604723 (P = 1 × 10−8), and is the most significant eQTL for TMEM133 in tibial artery (P = 2.7 × 10−8). Tibial artery was the only significant tissue at FDR < 0.05 according to the single-tissue eQTL discovery method. (B) Average posterior probability of the most significant cis-eQTL, rs607562 for ARHGAP42 at FDR < 0.05 from the multitissue eQTL methods. (C) Similar plot for TMEM133. The most significant cis-eQTL for TMEM133 from the multitissue methods at FDR < 0.05 is the GWAS SNP, rs633185, in tibial artery.
Conclusions
We have described a large in-depth data set of multitissue human gene expression. We assessed the variability of the transcriptome among individuals in a large number of tissues at a resolution that provides unique insights in to the diversity and regulation of gene expression among tissues. This analysis provides a unified view of genetic effects on gene expression across a broad range of tissue types, most of which have not been studied for eQTLs previously. We look forward to scaling up the resource to create a data set that will transform our understanding of how genetic variability influences different tissues and biological systems and ultimately complex diseases.
Supplementary Material
ACKNOWLEDGMENTS
We thank the donors and their families for their generous gifts of organ donation for transplantation and tissue donations for the GTEx research study; the Genomics Platform at the Broad Institute for data generation; J. Nedzel, K. Huang, and K. Hadley for work on the GTEx Portal; L. Gaffney for help with figures; and members of the Analysis Working Group for scientific editing and feedback. Supported by the Common Fund of the Office of the Director, U.S. National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, NIA, NIAID, and NINDS through NIH contracts HHSN261200800001E (Leidos Prime contract with NCI), 10XS170 (NDRI), 10XS171 (Roswell Park Cancer Institute), 10X172 (Science Care Inc.), 12ST1039 (IDOX), 10ST1035 (Van Andel Institute), and HHSN268201000029C (Broad Institute) and through NIH grants R01 DA006227-17 (Univ. of Miami Brain Bank), R01 MH090941 (Univ. of Geneva), R01 MH090951 and R01 MH090937 (Univ. of Chicago), R01 MH090936 (Univ. of North Carolina–Chapel Hill), R01 MH090948 (Harvard Univ.), R01 GM104371 (Massachusetts General Hospital), and R01AG046170, R01CA163772, and U01AI111598-01 (Icahn School of Medicine, Mount Sinai). Additional support: European Research Council, Swiss National Science Foundation, and Louis-Jeantet Foundation (E.T.D.); Wellcome Trust grant 098381 (M.I.M.); Clarendon Scholarship, a NDM Studentship, and a Univ. of Oxford Green Templeton College Award (M.A.R.). J.K.P. is compensated for his work on the scientific advisory board for 23 and Me and computational advisory board for DNA Nexus; W.W. is an employee and shareholder of Novartis Inc.; A. Battle is a shareholder of Google, Inc.; J. Fleming is executive director of the American Medical and Research Association; and J.B.V. and R. Little serve on the board of NDRI.
The GTEx Consortium
Authorship of this paper should be cited as “GTEx Consortium”; participants are arranged by area of contribution and then by institution. Analysis Working Group LDACC: Kristin G. Ardlie,1 David S. DeLuca,1 Ayellet V. Segrè,1 Timothy J. Sullivan,1 Taylor R. Young,1 Ellen T. Gelfand,1 Casandra A. Trowbridge,1 Julian B. Maller,1,2 Taru Tukiainen,1,2 Monkol Lek,1,2 Lucas D. Ward,1,3 Pouya Kheradpour,1,3 Benjamin Iriarte,3 Yan Meng,1 Cameron D. Palmer,1,4 Tõnu Esko,1,4,5 Wendy Winckler,1 Joel N. Hirschhorn,1,4 Manolis Kellis,1,3 Daniel G. MacArthur,1,2 Gad Getz1,6; UNC/NCSU: Andrey A. Shabalin,7 Gen Li,8 Yi-Hui Zhou,9 Andrew B. Nobel,8 Ivan Rusyn,10,11 Fred A. Wright9; Univ. of Geneva: Tuuli Lappalainen,12,13,14,15,16,17 Pedro G. Ferreira,12,13,14 Halit Ongen,12,13,14 Manuel A. Rivas,18 Alexis Battle,19,20 Sara Mostafavi,19 Jean Monlong,21,22,23 Michael Sammeth,21,22,24 Marta Melé,21,22,25 Ferran Reverter,21,26 Jakob M. Goldmann,21,27 Daphne Koller,19 Roderic Guigó,21,22,28 Mark I. McCarthy,18,29,30 Emmanouil T. Dermitzakis12,13,14; Univ. of Chicago: Eric R. Gamazon,31,32 Hae Kyung Im,31 Anuar Konkashbaev,31,32 Dan L. Nicolae,31 Nancy J. Cox,31,32 Timothée Flutre,33,34 Xiaoquan Wen,35 Matthew Stephens,33,36 Jonathan K. Pritchard33,37,38; Harvard: Zhidong Tu,39,40 Bin Zhang,39,40 Tao Huang,39,40 Quan Long,39,40 Luan Lin,39,40 Jialiang Yang,39,40 Jun Zhu,39,40 Jun Liu41 Biospecimen and data collection, processing, quality control, storage, and pathological review caHUB Biospecimen Source Sites, National Disease Research Interchange (NDRI): Amanda Brown,42 Bernadette Mestichelli,42 Denee Tidwell,42 Edmund Lo,42 Michael Salvatore,42 Saboor Shad,42 Jeffrey A. Thomas,42 John T. Lonsdale42; Roswell Park: Michael T. Moser,43 Bryan M. Gillard,43 Ellen Karasik,43 Kimberly Ramsey,43 Christopher Choi,43 Barbara A. Foster43; Science Care Inc.: John Syron,44 Johnell Fleming,44 Harold Magazine,44; Gift of Life Donor Program: Rick Hasz45; LifeNet Health: Gary D. Walters46; UNYTS: Jason P. Bridge,47 Mark Miklos,47 Susan Sullivan47 caHUB ELSI study VCU: Laura K. Barker,48 Heather M. Traino,48,49 Maghboeba Mosavel,48 Laura A. Siminoff48,49 caHUB comprehensive biospecimen resource Van Andel Research Institute: Dana R. Valley,50 Daniel C. Rohrer,50 Scott D. Jewell50 caHUB pathology resource center NCI: Philip A. Branton51; Leidos Biomedical Research Inc.: Leslie H. Sobin,52 Mary Barcus52 caHUB comprehensive data resource Leidos Biomedical Research Inc.: Liqun Qi,52 Jeffrey McLean,52 Pushpa Hariharan,52 Ki Sung Um,52 Shenpei Wu,52 David Tabor,52 Charles Shive52 caHUB operations management Leidos Biomedical Research Inc.: Anna M. Smith,52 Stephen A. Buia,52 Anita H. Undale,52 Karna L. Robinson,52 Nancy Roche,52 Kimberly M. Valentino,52 Angela Britton,52 Robin Burges,52 Debra Bradbury,52 Kenneth W. Hambright,52 John Seleski,53 Greg E. Korzeniewski52; Sapient Government Services: Kenyon Erickson54 Brain bank operations University of Miami: Yvonne Marcus,55 Jorge Tejada,55 Mehran Taherian,55 Chunrong Lu,55 Margaret Basile,55 Deborah C. Mash55 Program management NHGRI: Simona Volpi,56 Jeffery P. Struewing,56 Gary F. Temple,56 Joy Boyer,57 Deborah Colantuoni56; NIMH: Roger Little,58 Susan Koester59; NCI: Latarsha J. Carithers,51 Helen M. Moore,51 Ping Guan,51 Carolyn Compton,51 Sherilyn J. Sawyer,51 Joanne P. Demchok,60 Jimmie B. Vaught,51 Chana A. Rabiner,51 Nicole C. Lockhart51,57 Writing committee Kristin G. Ardlie,1 Gad Getz,1,6 Fred A. Wright,9 Manolis Kellis,1,3 Simona Volpi,56 Emmanouil T. Dermitzakis12,13,14
1Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA. 2Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA 02114, USA. 3MIT Computer Science and Artificial Intelligence Laboratory, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. 4Center for Basic and Translational Obesity Research and Division of Endocrinology, Boston Children’s Hospital, Boston, MA 02115, USA. 5Estonian Genome Center, University of Tartu, Tartu, Estonia. 6Cancer Center and Department of Pathology, Massachusetts General Hospital, Boston, MA 02114, USA. 7Center for Biomarker Research and Personalized Medicine, Virginia Commonwealth University, Richmond, VA 23298, USA. 8Department of Statistics and Operations Research and Department of Biostatistics, University of North Carolina, Chapel Hill, NC 27599, USA. 9Bioinformatics Research Center and Departments of Statistics and Biological Sciences, North Carolina State University, Raleigh, NC 27695, USA. 10Department of Environmental Sciences and Engineering, University of North Carolina, Chapel Hill, NC 27599. 11Department of Veterinary Integrative Biosciences, Texas A&M University, College Station, TX 77843, USA. 12Department of Genetic Medicine and Development, University of Geneva Medical School, 1211 Geneva, Switzerland. 13Institute for Genetics and Genomics in Geneva (iG3), University of Geneva, 1211 Geneva, Switzerland. 14Swiss Institute of Bioinformatics, 1211 Geneva, Switzerland. 15Department of Genetics, Stanford University, Stanford, CA 94305, USA. 16New York Genome Center, New York, NY 10011, USA. 17Department of Systems Biology, Columbia University Medical Center, New York, NY 10032, USA. 18Wellcome Trust Centre for Human Genetics Research, Nuffield Department of Clinical Medicine, University of Oxford, Oxford OX3 7BN, UK. 19Department of Computer Science, Stanford University, Stanford, CA 94305, USA. 20Department of Computer Science, Johns Hopkins University, Baltimore, MD 21218, USA. 21Centre for Genomic Regulation (CRG), 08003 Barcelona, Catalonia, Spain. 22Universitat Pompeu Fabra, 08003 Barcelona, Catalonia, Spain. 23Human Genetics Department, McGill University, Montréal, Quebec H3A 0G1, Canada. 24National Institute for Scientific Computing, Petropolis 25651-075, Rio de Janeiro, Brazil. 25Department of Stem Cell and Regenerative Biology, Harvard University, Cambridge, MA 02138, USA. 26Universitat de Barcelona, 08028 Barcelona, Spain. 27Radboud University Nijmegen, Netherlands. 28Institut Hospital del Mar d’Investigacions Mèdiques (IMIM), 08003 Barcelona, Spain. 29Oxford Centre for Diabetes, Endocrinology and Metabolism, University of Oxford, Churchill Hospital, Oxford OX3 7LJ, UK. 30Oxford NIHR Biomedical Research Centre, Churchill Hospital, Oxford OX3 7LJ, UK. 31Section of Genetic Medicine, Department of Medicine and Department of Human Genetics, University of Chicago, Chicago, IL 60637, USA. 32Division of Genetic Medicine, Department of Medicine, Vanderbilt University, Nashville, TN 37232, USA. 33Department of Human Genetics, University of Chicago, Chicago, IL 60637, USA. 34INRA, Department of Plant Biology and Breeding, UMR 1334, AGAP, Montpellier, 34060, France. 35Department of Biostatistics, University of Michigan, Ann Arbor, MI 48109, USA. 36Department of Statistics, University of Chicago, Chicago, IL 60637, USA. 37Department of Genetics and Biology, Stanford University, Stanford, CA 94305, USA. 38Howard Hughes Medical Institute, Chicago, IL, USA. 39Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA. 40Icahn Institute of Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA. 41Department of Statistics, Harvard University, Cambridge, MA 02138. 42National Disease Research Interchange, Philadelphia, PA 19103, USA. 43Roswell Park Cancer Institute, Buffalo, NY 14263, USA. 44Science Care Inc., Phoenix, AZ, USA. 45Gift of Life Donor Program, Philadelphia, PA 19103, USA. 46LifeNet Health, Virginia Beach, VA 23453, USA. 47UNYTS, Buffalo, NY 14203, USA. 48Virginia Commonwealth University, Richmond, VA 23298, USA. 49Department of Public Health, Temple University, Philadelphia, PA 19122, USA. 50Van Andel Research Institute, Grand Rapids, MI 49503, USA. 51Biorepositories and Biospecimen Research Branch, National Cancer Institute, Bethesda, MD 20892, USA. 52Biospecimen Research Group, Clinical Research Directorate, Leidos Biomedical Research, Inc., Rockville, MD 20852, USA. 53iDoxSolutions Inc., Bethesda, MD 20814, USA. 54Sapient Government Services, Arlington, VA 22201, USA. 55Brain Endowment Bank, Department of Neurology, Miller School of Medicine, University of Miami, Miami, FL 33136, USA. 56Division of Genomic Medicine, National Human Genome Research Institute, Bethesda, MD 20892, USA. 57Division of Genomics and Society, National Human Genome Research Institute, Bethesda, MD 20892, USA. 58Office of Science Policy, Planning, and Communications, National Institute of Mental Health, Bethesda, MD 20892, USA. 59Division of Neuroscience and Basic Behavioral Science, National Institute of Mental Health, Bethesda, MD 20892, USA. 60Cancer Diagnosis Program, National Cancer Institute, Bethesda, MD 20892, USA.
Footnotes
The primary and processed data used to generate the analyses presented here are available in the following locations: all primary sequence and clinical data files, and any other protected data, are deposited in and available from dbGaP (www.ncbi.nlm.nih.gov/gap) (phs000424.v3.p1, except for whole-exome sequencing data, which are part of phs000424.v5.p1); derived analysis files are available on the GTEx Portal (www.gtexportal.org). Biospecimens remaining from the study may be requested for research studies. The sample request form, biospecimen access policy, and material transfer agreement (MTA) are available on the GTEx Portal (www.gtexportal.org/home/samplesPage).
Author contributions: The GTEx Consortium contributed collectively to this study. Biospecimens were provided by the Biospecimen source sites and the brain bank operations, and processed by the LDACC and comprehensive biospecimen resource. Pathological review of specimens was conducted by the pathology resource center, and all donor data entry and review by the comprehensive data resource. Data generation was undertaken by the Laboratory, Data Analysis and Coordinating Center (LDACC), and analyses were performed by all members of the Analysis Working Group. Project activities were coordinated by caHUB operations and overseen by the NHGRI, NIMH, and NCI project teams. We acknowledge the following investigators of the Analysis Working Group who contributed substantially to analyses presented here. Transcriptome variation: D.S.D., P.G.F. Single-tissue eQTL analysis: A.A.S., D.S.D., A.V.S. Multitissue eQTL analysis: T.F., X.W., A.A.S., G.L. Allele-specific expression: T.L. Splice QTLs: J.M., H.O. Functional annotation: L.D.W., P.K. Network Analyses: A. Battle, S.M., Z.T., T.H., B.I. Protein-truncating analyses: M.A.R., M.L. GWAS analysis: E.R.G., A.V.S., L.D.W.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/348/6235/648/suppl/DC1
Materials and Methods
Box S1
Figs. S1 to S34
Tables S1 to S15
References (50–86)
REFERENCES AND NOTES
- 1.Welter D, et al. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Visscher PM, Brown MA, McCarthy MI, Yang J. Am. J. Hum. Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stranger BE, Stahl EA, Raj T. Genetics. 2011;187:367–383. doi: 10.1534/genetics.110.120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ward LD, Kellis M. Nat. Biotechnol. 2012;30:1095–1106. doi: 10.1038/nbt.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maurano MT, et al. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morley M, et al. Nature. 2004;430:743–747. doi: 10.1038/nature02797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Westra HJ, et al. Nat. Genet. 2013;45:1238–1243. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grundberg E, et al. Nat. Genet. 2012;44:1084–1089. doi: 10.1038/ng.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ernst J, et al. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.ENCODE Project Consortium. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Voight BF, et al. Nat. Genet. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Small KS, et al. Nat. Genet. 2011;43:561–564. doi: 10.1038/ng.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.GTEx Consortium. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.See supplementary materials on Science Online.
- 15.Harrow J, et al. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang HJ, et al. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melé M, et al. Science. 2015;348:660–665. doi: 10.1126/science.aaa0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barbosa-Morais NL, et al. Science. 2012;338:1587–1593. doi: 10.1126/science.1230612. [DOI] [PubMed] [Google Scholar]
- 19.Yeo G, Holste D, Kreiman G, Burge CB. Genome Biol. 2004;5:R74. doi: 10.1186/gb-2004-5-10-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dimas AS, et al. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shabalin AA. Bioinformatics. 2012;28:1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nica AC, et al. PLOS Genet. 2011;7:e1002003. doi: 10.1371/journal.pgen.1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Veyrieras JB, et al. PLOS Genet. 2008;4:e1000214. doi: 10.1371/journal.pgen.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flutre T, Wen X, Pritchard J, Stephens M. PLOS Genet. 2013;9:e1003486. doi: 10.1371/journal.pgen.1003486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li G, Shabalin AA, Rusyn I, Wright FA, Nobel AB. 2013 http://arxiv.org/abs/1311.2948. [Google Scholar]
- 26.Storey JD, Tibshirani R. Proc. Natl. Acad. Sci. U.S.A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wen X. 2013 http://arxiv.org/abs/1311.3981. [Google Scholar]
- 28.Wen X. Biometrics. 2014;70:73–83. doi: 10.1111/biom.12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sul JH, Han B, Ye C, Choi T, Eskin E. PLOS Genet. 2013;9:e1003491. doi: 10.1371/journal.pgen.1003491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newton-Cheh C, et al. Nat. Genet. 2009;41:399–406. doi: 10.1038/ng.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rivas MA, et al. Science. 2015;348:666–669. doi: 10.1126/science.1261877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khurana E, et al. Science. 2013;342:1235587. doi: 10.1126/science.1235587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chess A. Nat. Rev. Genet. 2012;13:421–428. doi: 10.1038/nrg3239. [DOI] [PubMed] [Google Scholar]
- 34.Buil A, et al. Nat. Genet. 2015;47:88–91. doi: 10.1038/ng.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ongen H, Dermitzakis ET. 2015 http://biorxiv.org/content/early/2015/01/22/014126. [Google Scholar]
- 36.Monlong J, Calvo M, Ferreira PG, Guigó R. Nat. Commun. 2014;5:4698. doi: 10.1038/ncomms5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ward LD, Kellis M. Nucleic Acids Res. 2012;40:D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaffney DJ, et al. Genome Biol. 2012;13:R7. doi: 10.1186/gb-2012-13-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Battle A, et al. Genome Res. 2014;24:14–24. doi: 10.1101/gr.155192.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernstein BE, et al. Nat. Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang B, et al. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerstein MB, et al. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MacArthur DG, et al. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dewey FE, et al. JAMA. 2014;311:1035–1045. doi: 10.1001/jama.2014.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wellcome Trust Case Control Consortium. Nature. 2007;447:661–678. [Google Scholar]
- 46.Nicolae DL, et al. PLOS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nica AC, et al. PLOS Genet. 2010;6:e1000895. doi: 10.1371/journal.pgen.1000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramos EM, et al. Eur. J. Hum. Genet. 2014;22:144–147. doi: 10.1038/ejhg.2013.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hindorff LA, et al. Proc. Natl. Acad. Sci. U.S.A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.