

Short abstract
The following work describes the development of a novel noninvasive transmucosal drug delivery system, the chitosan sponge matrix (CSM). It is composed of cationic chitosan (CS) nanoparticles (NPs) that encapsulate cisplatin (CDDP) embedded within a polymeric mucoadhesive CS matrix. CSM is designed to swell up when exposed to moisture, facilitating release of the NPs via diffusion across the matrix. CSM is intended to be administered topically and locally to mucosal tissues, with its initial indication being oral cancer (OC). Currently, intravenous (IV) administered CDDP is the gold standard chemotherapeutic agent used in the treatment of OC. However, its clinical use has been limited by its renal and hemotoxicity profile. We aim to locally administer CDDP via encapsulation in CS NPs and deliver them directly to the oral cavity with CSM. It is hypothesized that such a delivery device will greatly reduce any systemic toxicity and increase antitumor efficacy. This paper describes the methods for developing CSM and maintaining the integrity of CDDP NPs embedded in the CSM.
Keywords: oral cancer, chemotherapy, nanoparticles, platform, transmucosal, local, biodegradable, biocompatible, permeation, mucoadhesive, polymeric, chitosan, sponge, matrix, cisplatin, mucosa, topical, safe, drug, embedding, release
1. Introduction
There is a growing demand for safer, more effective routes of drug delivery. Two major methods, NP encapsulation and buccal administration, have been gathering a large amount of attention as alternatives to the more traditional oral and IV routes. Buccal delivery has been widely used in various forms, including gels [1], films [2], and tablets [3] to deliver vitamins [4], peptides [5], or antibiotics [6]. On the other hand, NP formulations have been used for targeted systemic delivery of agents [7]. CS, a natural cationic polysaccharide comprised of the copolymers N-acetyl glucosamine and glucosamine [8] has been widely utilized in such systems. It is produced via the deacetylation of chitin (Fig. 1), a structural polymer found in the shells of crustaceans [9] and widely considered to be among the most useful polymers in pharmaceutical research due to its abundance, biodegradability, and biocompatibility [10].
Fig. 1.
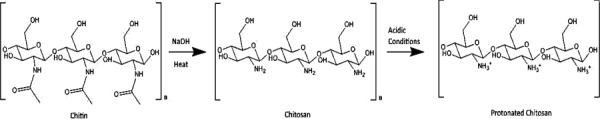
Molecular structures and formation conditions for chitin and CS. The acetyl groups (C2H3O) are removed from chitin to form CS. Note how the CS primary amine groups (NH2) become protonated in acidic conditions.
Localized buccal and NP delivery routes would be most suited in applications utilizing drugs with dose-limiting toxicities and severe side effects, such as chemotherapeutics. There is published literature regarding buccal and topical NP systems to administer chemotherapy agents such as 5-fluoruracil [11,12]. However, there are no reports of NPs physically incorporated within a buccal device. Consequently, this paper will discuss the development of a novel drug delivery system, the CSM (Fig. 2). CSM is composed of CS NPs encapsulating CDDP embedded within a polymeric CS matrix. CSM is intended for topical and local administration directly onto the tumor to reduce the toxic side effects of chemotherapy. Its initial indication is OC; however, it is a platform technology that can be used to treat other cancers such as melanoma.
Fig. 2.
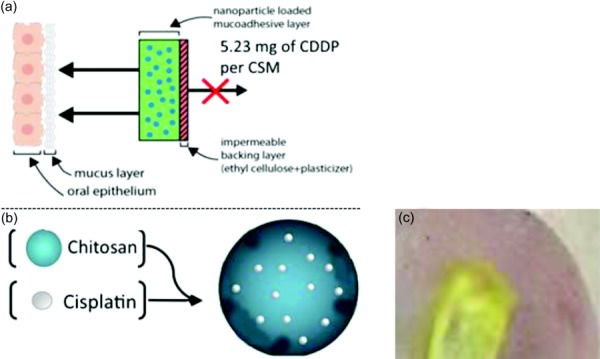
Schematic diagrams of the CSM. CSM can be molded to have several sizes, however the current size of 1.5 × 1.5 cm2 provides adequate coverage of a tumor that is 1 cm in diameter. The sponge is 2 mm in height. Note that the backing ethyl cellulose can be brushed on the back of CSM to create a unidirectional application. The CSM is designed to remain on the tumor for 1 hr and the remaining CSM with its non degradable ethylcellulose backing will be discarded (a). CS (CDDP NPs) (b). Ex vivo study: CSM is placed on the freshly harvested lamb's tongue for permeation studies. Transition of the CSM into a hydrogel after 5 mins of exposure to a wetting agent (c).
There is a significant unmet need in the treatment of OC. Unlike other cancers, the incidence of OC is on the rise in the U.S. primarily due to increasing rates of human papillomavirus-16 infection [13]. It is estimated that over 42,000 people in the U.S. and over 640,000 people worldwide were diagnosed with OC in 2014 [14]. Its five year relative survival rate remains at a mere 62.7% [14]. Despite this, there has been little progress in the development of innovative translational therapies for OC to both improve efficacy and reduce unwanted side effects. The current gold standard of care for OC is an IV CDDP infusion combined with radiotherapy and surgery [15]. Depending on the patient and condition, these infusions may be administered over a period ranging from weeks to months, with each treatment lasting several hours. For instance, patients with stage III locally advanced oral tumors will receive one 40–50 mg/m2 IV of CDDP daily for 6–7 weeks with concurrent radiation [16].
CDDP ((SP-4-2)-diamminedichloroplatinum (II)) is an alkylating agent that has been used in the treatment of a wide assortment of cancers [17–19]. First Food and Drug Administration (FDA) approved in 1978, CDDP remains the most effective choice of platinum-based chemotherapeutics. Its mechanism of action first involves CDDP entering the cell via both active and passive transport [20,21]. Upon entering the nucleus, CDDP loses its two chlorine atoms and binds to the nitrogen atoms of two consecutive purine bases on the deoxyribonucleic acid (DNA), forming CDDP-DNA cross-linked adducts. These adducts cause a conformational change in the DNA, allowing for the binding of high mobility group domain proteins. Once bound, high mobility group proteins destabilize the base pairs beyond reparation, halting DNA transcription and replication, ultimately inducing apoptosis [22]. Despite its proven efficacy when dosed properly, the dosing options of IV CDDP in patients are often limited to suboptimal treatment levels due to systemic toxicity and severe side effects. These side effects include nausea and vomiting, anemia, mucositis, ototoxicity, neurotoxicity, and nephrotoxicity [23,24]. Additionally, once in the bloodstream CDDP faces two significant issues: (a) CDDP is subject to chemical deactivation by substitution or elimination of amine groups [25], and (b) being a nontargeted molecule, only a fraction of CDDP actually reaches the tumor site. This can be attributed to the small size of CDDP molecules, which allows them to easily diffuse through a tumor's malformed and permeable vasculature [26] rather than remain local.
To address the aforementioned issues of IV CDDP, we have encapsulated CDDP within CS NPs and embedded them within CSM (Fig. 2) intended for topical and local application to OC tumors. Encapsulation of CDDP protects it from biological deactivation and promotes increased rates of cell uptake [27]. Additionally, using cationic CS as the polymer for both the NPs and CSM allows for electrostatic interactions between it and anionic domains of mucin proteins in the oral cavity. This mucoadhesive property is what allows the NPs, and subsequently CDDP, to remain local to the delivery sites.
The CSM is able to preserve NPs in a stable environment and promote passage across epithelial barriers. It is designed with an impermeable ethylcellulose backing to provide unidirectional drug delivery and prevent CDDP NP washout from saliva (Fig. 2). When hydrated with a wetting agent such as saline, CSM swells up and transforms to a mucoadhesive hydrogel, covering the tumor and allowing the NPs to diffuse across the porous matrix into the tumor tissue. Properties including NP size and surface charge can be controlled to allow for an even greater degree of targeting depending on the severity and size of the tumor. Several in vitro tests studying the release of CDDP from the NPs and NP cell uptake will be discussed in this paper. Based on what has been discussed above, it can be hypothesized that polymeric CS NPs can be embedded within CSM without damaging the structural integrity of the NPs or the sponge.
2. Materials and Methods
2.1. Materials and Equipment.
All the chemicals and equipment were obtained from commercial suppliers. The list of chemicals is shown in Table 1 and they were used as received. All water-based solutions were prepared with double deionized water.
Table 1.
List of chemical and materials
| Type | Molecular weight | Property | Manufacturer |
|---|---|---|---|
| CS chloride salt Cl113 (CS Cl113) | 50–150 kDa | Deacetylation degree 75–90% | Novamatrix, FMC BioPolymer |
| CS glutamate G113 (CS G113) | 50–150 kDa | Deacetylation degree 75–90% | Novamatrix, FMC BioPolymer |
| CDDP H6Cl2N2Pt | 300 g/mol | Chemotherapy agent | Strem Chemicals |
| Sodium TPP Na5P3O10 | 367.864 g/mol | Cross-linker used for ionic gelation | Sigma-Aldrich |
| Glacial acetic acid C2H4O2 | 60.05 g/mol | Solvent for CS | Sigma-Aldrich |
| MTS cytotoxicity assay | — | Cell viability assay | BioVision |
| FITC C21H11NO5S | 389.4 g/mol | Fluorescent labeling can be conjugated to CS | Life Technologies |
| ALX | 594 nm or 633 nm laser | Fluorescent labeling can be conjugated to CS | Life Technologies |
| Sodium hydroxide (NaOH) | 39.99 g/mol | Used for adjusting pH levels | Sigma-Aldrich |
2.1.1. Materials.
Hamster cheek pouch carcinoma cells (HCPC-1), human pharyngeal squamous cell carcinoma cells (FaDu), and buccal epithelial TR146 cells were used for the in vitro and in vivo studies.
2.1.2. Equipment.
Malvern ZetaSizer Nano ZS90 was used for measuring the size, polydispersity (PDI), and the polydistribution index. Malvern NanoSight was used for detecting NPs released from the CSM in human saliva. 30k Pall NanoSep centrifugal filters were used for filtration, separation, and purification of the NPs. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) was used for detecting any traces of CDDP.
2.2. Preparation of Cisplatin Nanoparticles (CDDP NPs).
Several optimization studies were performed by altering the mass ratio of CS to tripolyphosphate (TPP) and synthesis pH to determine a formulation that yielded NPs of desirable size and charge, with low PDI and high encapsulation efficiency. CDDP NPs were synthesized via the proven ionic gelation technique [28]. CDDP (1.5 mg/mL) was added to deionized (DI) water and the mixture was sonicated for 4 hrs until CDDP was completely dissolved. A 0.175% v/v acetic acid solution was prepared from 100% glacial acetic acid. CS Cl113 (1.0 mg/mL) was dissolved in this solution on a vortex for 10 mins. The CS solution was transferred to a beaker under magnetic stirring (600 rpm). TPP was then added to the CDDP solution (0.45 mg/mL) and vortexed for 1 min until completely dissolved. Immediately after, the CDDP-TPP solution was added dropwise at a flow rate of 1 mL/min, resulting in a final volume ratio of 1.1 mL CS:0.85 mL CDDP-TPP. The solutions were left to mix for 15 mins prior to characterization.
2.2.1. Preparation of Blank Nanoparticles.
Blank nanoparticles (BLK NPs) were synthesized employing ionic gelation [28]. TPP was dissolved in water (1.0 mg/mL) and filtered through a 0.22 μm syringe filter. A 0.175% v/v acetic acid solution was prepared from 100% glacial acetic acid. CS Cl113 (1.0 mg/mL) was dissolved in this solution on a vortex for 10 mins. The CS solution was transferred to a beaker under magnetic stirring (600 rpm). TPP solution was added dropwise from a pipette at a flow rate of 1 mL/min, resulting in a final volume ratio of 1.0 mL CS:0.25 mL TPP. The solutions were left to mix for 15 mins prior to characterization.
2.2.2. Preparation of Fluorescent Labeled CS NPs.
Fluorescein isothiocyanate salt-chitosan and Alexa Fluor 647-chitosan NPs (FITC-CS NPs and ALX-CS NPs) were synthesized in order to detect the NPs in permeation, release and cell uptake studies. Prior to NP preparation, CS Cl113 was hydrated in 0.175% v/v acetic acid (1.0 mg/mL). The solution was left to stir in a beaker for 1 hr at 500 rpm and the pH was adjusted to 6.0 with 0.1 M NaOH. FITC was dissolved in 100% ethanol (10 mg/mL). FITC solution was added to the CS Cl113 solution dropwise to achieve a final concentration of 0.04 mg/mL. 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) salt was added to the solution (0.84 mg/mL) and the mixture allowed to stir for 12 hrs and then dialyzed for two days (Fig. 3). The purified solution was frozen on dry ice and lyophilized for 72 hrs at 50 mTorr and −90 °C. The ionic gelation method was used to synthesize the FITC-CS NPs [28]. Lyophilized FITC-CS was rehydrated in 0.175% v/v acetic acid (1.0 mg/mL) and stirred overnight at 200 rpm. TPP was dissolved in water (1.0 mg/mL) and was added dropwise to the FITC-CS solution under magnetic stirring (600 rpm) at a flow rate of 1 mL/min. The final volume ratio of the solutions was 1.0 mL FITC-CS:0.5 mL TPP. The NP solution stirred for 15 mins prior to characterization. The same preparation was used for ALX-CS NPs, with a simple substitution of ALX for FITC.
Fig. 3.
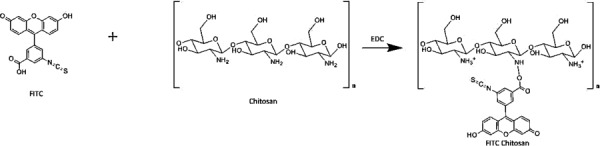
Molecular structures of FITC, CS, and FITC-CS. FITC-CS is synthesis is catalyzed by the addition of EDC. The primary amine of CS acts as a nucleophile to the carboxylic acid of FITC and forms a covalent bond between the two molecules. The secondary amine on FITC-CS is unable to become protonated and will remain neutral.
2.2.3. Characterization of CS NPs
2.2.3.1. Dynamic Light Scattering (DLS).
1.0 mL and 800 μl of each NP solution were placed in a separate polystyrene cuvette and zeta potential cell, respectively. Measurements were taken using a ZetaSizer Nano (Malvern) to determine the size, charge, and PDI of the NPs. Backscatter detection was employed at an angle of 173 deg.
2.2.3.2. Encapsulation Efficiency (EE).
The CDDP NP solution was transferred to a 30 k Pall NanoSep centrifugal filter and centrifuged for 8 mins at 1100 relative centrifugal force (RCF). The NPs encapsulating CDDP remained in the top compartment while free CDDP was in the bottom of the tube. The NPs were washed with 0.175% acetic acid to remove any CDDP stuck to the surface of the NPs and were subsequently centrifuged again (repeated 3×). The top and bottom compartments were analyzed for platinum (Pt) concentration with ICP-AES. Encapsulation efficiency was determined using the following equations:
| (1) |
| (2) |
| (3) |
| (4) |
2.2.4. In Vitro MTS (Colorimetric) Cytotoxicity Assay (CDDP NPs).
HCPC-1 and FaDu cells were each cultured in with Dulbecco's modified Eagle's medium and 10% fetal bovine serum until a minimum density of 2 × 105 cells/mL was reached. Cells were transferred to a 96-well microtiter plate. The well known cytotoxicity assay, MTS reagent was added according to the manufacturer's protocol. BLK NPs, CDDP NPs, and free CDDP were added to each well in varying concentrations and allowed to incubate for 48 hrs. Cell viability percentage was determined by the reagent's color intensity according to the manufacturer's guidelines. Color intensity was quantified by a plate reader.
2.2.5. In Vitro Cell Uptake Study.
Cultured TR146 cells (human buccal squamous cell carcinoma) were separately incubated with ALX NPs (0.03 g/L) for periods of both 30 and 60 mins. Cells were analyzed using fluorescence-activated cell sorting (FACS) and percentage cell uptake was determined. Note: It was determined that utilizing both human oral buccal (TR146) and human oral pharyngeal (FaDu) cell lines would better represent he complex environment of the mouth.
2.2.6. CDDP NP Drug Release Study.
CDDP NPs were synthesized via ionic gelation as described above. The particles were then characterized with DLS for size and surface charge. NPs were washed with 0.175% acetic acid to remove any free CDDP on the surface. They were then purified via centrifugation (1100 RCF, 8 mins) in a 30k molecular weight cutoff (MWCO) PALL centrifuge filter. NPs were placed in a dialysis bag within a beaker of phosphate buffered saline (PBS) buffer (pH 7.2) to simulate salivary fluids. Samples were drawn at varying time points and analyzed for CDDP content using ICP-AES to calculate the release profile
| (5) |
2.3. Synthesis of CSM.
CS G113 was dissolved in a 1.0% v/v aqueous acetic acid solution (17.0 mg/mL) under magnetic stirring for 1 hr at 700 rpm. CDDP NP, BLK NP, or 1 mg/mL CS Cl113) solution was combined with the CS G113 solution in a ratio of 10.0 mL:1.0 mL. This was frozen in liquid nitrogen for 25 mins and lyophilized for two days.
2.3.1. Stability of NPs in CSM Solution.
Freshly synthesized NP solution (CDDP, ALX, FITC, and BLK) was combined with the CSM sponge solution in varying v/v aqueous ratios (1.1:0.85, 1.1:0.8, 1.1:0.5, 1.1:0.4, 1.1:0.45, and 1.1:0.30) of CDDP NP:CSM. The size, charge, and PDI of the NPs were measured using DLS.
2.3.2. Integrity of NPs in Lyophilized CSM.
CSMs were placed in human saliva and the released fluorescent NPs were visualized and quantified with NanoSight (Malvern). Note: Malvern NanoSight utilizes nanoparticle tracking analysis to characterize and analyze each NP individually. It allows for the detection of NPs even in solutions containing polymers and proteins.
2.3.3. Scanning Electron Microscopy (SEM).
CSMs were placed under a Denton-Vacuum Desk IV sputter coater and sprayed with a 10 nm thick layer of gold–palladium alloy. Samples were then mounted and imaged inside a JEOL 7401F field emission scanning electron microscope.
3. Results and Discussion
3.1. Synthesis of CS NPs.
Results of CDDP NP syntheses are described in Table 2. It was observed that increasing the CS solution concentration while keeping TPP constant (1 mg/mL) yielded CDDP NPs with a larger size and zeta potential (Figs. 4(a) and 4(b)). In contrast, as the TPP concentration was increased and CS kept constant (1 mg/mL) it lowered the surface charge of the CDDP NPs (Fig. 4(c)). There was no observable trend between NP size and the modulation of TPP concentration (Fig. 4(d)). Upon increasing the pH, the zeta potential and size of the NPs both increased (Figs. 4(e) and 4(f)). CDDP NPs displayed a high encapsulation efficiency of 78%. Two fluorescent variants of CS NPs displayed lower zeta potential and larger size and were functional when observed under their respective excitation wavelengths (495 nm and 647 nm).
Table 2.
The average properties observed for different types of CS NPs synthesized as described in text
| NP type | Avg. size (nm) (Std. Dev.) | Avg. charge (mV) (Std. Dev.) | Avg. PDI (Std. Dev.) |
|---|---|---|---|
| Blank NP | 122.97 (7.13) | +35.2 (3.45) | 0.29 (0.01) |
| CDDP NP | 70.46 (7.21) | + 31.4 (2.78) | 0.20 (0.02) |
| ALX NP | 185.27 (105.87) | +16.9 (3.76) | 0.34 (0.10) |
| FITC NP | 143.3 (25.67) | +19.3 (1.14) | 0.13 (0.03) |
Note: Values derived from DLS measurements. Each type of NP was synthesized a minimum of three times to ensure accuracy and reproducibility.
Fig. 4.

(a) Effect of increasing the TPP concentration on NP zeta potential. CS concentration was kept constant at 1.0 mg/mL (b) impact of increasing TPP concentration on NP size. CS concentration was kept constant at 1.0 mg/mL (c) results of increasing CS concentration on NP zeta potential. TPP concentration remained at 1.0 mg/mL (d) effect of increasing CS concentration on NP size. TPP concentration was fixed at 1.0 mg/mL (e) impact of increasing the CS solution pH used for NP synthesis with 0.1 M NaOH (f) results of increasing the CS solution pH used for NP synthesis with 0.1 M NaOH. When the TPP concentration was increased, there was a clear trend in the reduction of NP zeta potential, but no predictable impact was made on NP size. The zeta potential and size both increased when the CS concentration was heightened. The NPs produced with higher pH CS solutions showed both larger size and lower zeta potential.
We found that NP parameters such as size and charge were able to be modulated by altering the concentration of reagents and pH of the synthesis procedure. The functional portion of CS molecules is a primary amine group, which becomes protonated in acidic conditions (pKa = 6.5), endowing CS with cationic properties. We hypothesize that the larger size and zeta potential observed with increasing CS concentration can be attributed to the higher abundance of amine groups. The more positive charges present, the stronger the repulsive forces are between CS chains, increasing their intermolecular distance, thus the size of NPs. The reduced charge seen when increasing the concentration of TPP is due to the higher amount of TPP anions counteracting the cationic amine groups, effectively neutralizing their charge. Additionally, the observed negligible impact of TPP concentration on NP size suggests that the amount of cross-linking anions do not play a significant role in determining the size of the NPs. Increasing the pH of the CS solution prior to synthesis lowers the degree of amine protonation. This limits the number of potential cross-linking sites available for TPP anions to interact with while forming NPs. This lower cross-linking density leads to larger NPs, and the smaller amount of amine groups yields a lower overall positive charge. However, there is a slight deviation in the overall trends of increasing the CS concentration and TPP concentration. The spike in size at 1.25 mg/mL CS concentration (Fig. 4(d)) and 0.7 mg/mL TPP concentration (Fig. 4(b)) can be attributed to the close mass ratios of CS to TPP in both instances (5.00:1.00 and 5.71:1.00, respectively). This suggests a critical point is reached at these proportions where larger NPs are formed than usual and more studies will be conducted to investigate this phenomenon.
We hypothesize that the decreased zeta potential and increased size seen on both the FITC and ALX CS NPs are due to the fluorophore conjugation site being the amine group of CS. This reaction reduces the zeta potential because it transforms the primary amine into a secondary amine, eliminating its ability to become protonated and thus decreases its cationic potential. However, it should be noted that this reaction does not substitute every amine group, and the unreacted primary amines are what give the ALX/FITC-CS NPs a remaining degree of positive charge. We believe the increased size of the fluorescent NPs is due to both the reduction of TPP cross-linking sites and the increased steric hindrance of the large fluorescent molecule conjugates. (Fig. 3).
The optimal mass ratio for blank/fluorescent NPs was 4.0 CS:1.0 TPP and 2.88 CS:1.0 TPP for CDDP-CS NPs. These were determined based on the proportions of CS to TPP that produced the smallest size, highest zeta potential, and lowest PDI for each formulation. Ultimately, the best combination for CDDP NPs yielded small NPs (<100 nm), a positive zeta potential (20–30 mV), and low PDI (<0.20). For the treatment of OC, these properties are ideal. A low PDI index indicates that the size distribution of the NPs is narrow, which is important for therapeutic efficacy. According to published literature, particles of this size range pass through the 50 cell layer thick oral epithelium [29] and reach the basement membrane where cancerous stem cells may reside [30]. The positive charge is critical to ensure that the NPs remain local and rapidly taken up by cells [27]. We observed that if the CDDP-TPP solution was not used immediately, its pH begins to decrease within 15 mins and such time gaps can alter the NP characteristics between batches. Based on our data: (1) The properties of CS-TPP NPs can be easily modulated by varying the mass ratio of CS to TPP and (2) CS concentration plays a greater role in altering the size and surface charge of NPs than TPP.
3.2. Stability of CDDP-TPP Solution.
Throughout the course of the NP synthesis procedure, the pH of all solutions was monitored for variation. We observed that the pH of the freshly synthesized CDDP solution pH was 4.25 (±0.07). This varied significantly from the pH of the DI water it was prepared in (5.54 ± 0.11, stored in an airtight container at 25 °C). Immediately after addition of TPP to the solution, the pH increased to 6.81 (±0.08) and proceeded to decrease linearly over the course of 9 hrs to 6.17 (±0.09) (Fig. 5(a)). The rate of this decrease was affected by the pH of the CDDP solution (adjusted with 0.1 M NaOH), with higher pH's corresponding to faster rates of pH depression (Fig. 5(b)). Additionally, it was observed that after 72 hrs at room temperature unprotected from light the CDDP-TPP solution formed a blue precipitate. This did not occur when it was stored in a light-blocking container.
Fig. 5.
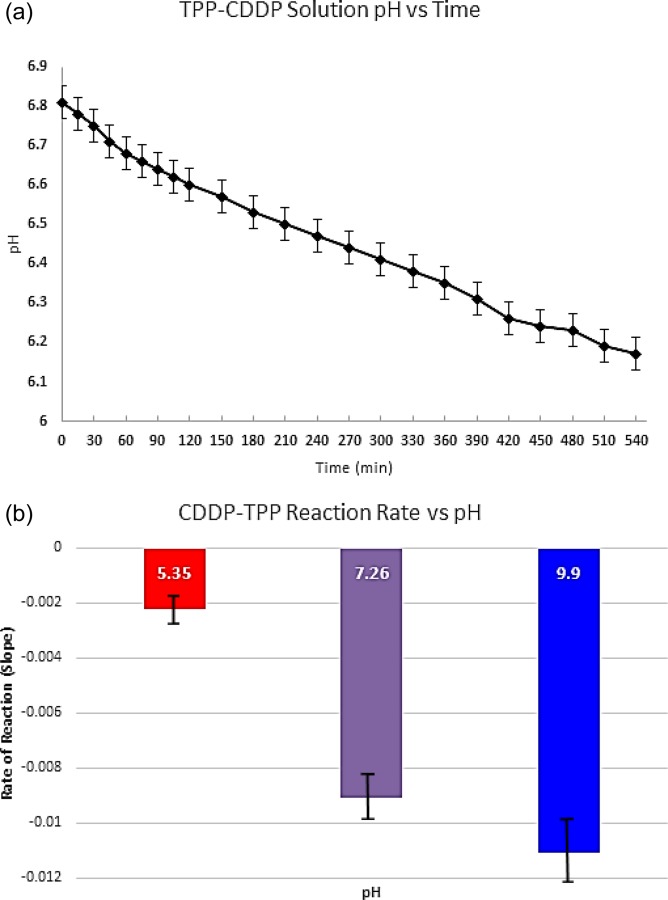
(a) pH of the CDDP-TPP solution measured at 15 mins intervals for the initial 2 hrs and at 30 mins intervals over the course of an additional 5 hrs and (b) rate of the hypothesized CDDP-TPP reaction. The rate was measured by plotting out the % decrease in pH over the first hour and analyzing the slopes of the lines for each condition. We believe that the greater pH depression rates can be correlated with a more rapid production of phosphoric acid.
We hypothesize that the decrease in pH is due to the hydrolysis of CDDP [31]. The chloride ions of CDDP are excellent leaving groups and are readily replaced by water ligands after dissolution [31]. This reaction results in the formation of positively charged mono or di aqueous CDDP species [31], and we hypothesize that free hydrochloric acid (HCl) is formed as a byproduct, lowering the pH of the solution. Hydrolysis of CDDP can be slowed by the addition of excess chloride ions to the system to create an equilibrium between free Cl− and Cl− on CDDP [32]. Even if CDDP becomes hydrolyzed, the mono and di aqueous Pt species are therapeutically potent [33,34] and can still be encapsulated in CS NPs for treatment. We plan on investigating the impacts of adding Cl− in the form of NaCl on the NP synthesis process and evaluating the presence of CDDP and its hydrolyzed species via high-performance liquid chromatography (HPLC).
In addition to the reactivity of CDDP with water, CDDP can also react with phosphate-based compounds such as TPP [35]. We believe one of the byproducts of these reactions is phosphoric acid (PO4 −), which is responsible for subsequent decline in pH seen when TPP is added to the CDDP solution. We also found these reactions to pH dependent (Figs. 5(a) and 5(b)). When CDDP-TPP solution pH was raised with 0.1 M NaOH, the rate of pH decline was more rapid compared to the unadjusted control. This suggests that a higher abundance of reactive deprotonated oxygens on TPP react with CDDP more quickly to produce of PO4 − in greater quantities. Because of these reactions, the NPs and CSM must be thoroughly characterized at each step of the synthesis process to identify and quantify the presence of CDDP-phosphato derivatives. Additionally, the blue precipitate observed is a dinuclear phosphate-platinum complex [35] formed by the bonding of two phosphate-platinum complexes. This formed more rapidly CDDP when TPP was exposed to light, inferring that light plays a key role in the catalytic formation of this product.
In conclusion, the mixing of TPP with the CDDP solution is a critical parameter to control during NP synthesis. Once made, the solution should be used immediately to minimize batch to batch variability caused by the changing pH and production of CDDP-TPP compounds. We will develop a method to identify and quantify the potential CDDP derivatives formed in our synthesis procedure and discuss the findings in our next publication. It is imperative that a translational product like CSM is thoroughly analyzed in order to meet the FDA's stringent characterization requirements for active drug substances.
3.3. In Vitro MTS Cytotoxicity Assay and Cell Uptake Study.
As shown in Fig. 6, HCPC-1 cells treated with CDDP NPs, free CDDP, and BLK NPs showed 23%, 38%, and 68% cell viability, respectively. Also shown is the viability of FaDu cells treated with CDDP NPs, free CDDP, and BLK NPs showed corresponding values of 21%, 49%, and 87%. Figure 7 illustrates that after 30 mins of incubation, 98% of TR-146 cells were positive for ALX CS NPs and in 60 mins, 95% of cells were positive for ALX CS NPs. No fluorescence was detected in the TR-146 control cells.
Fig. 6.
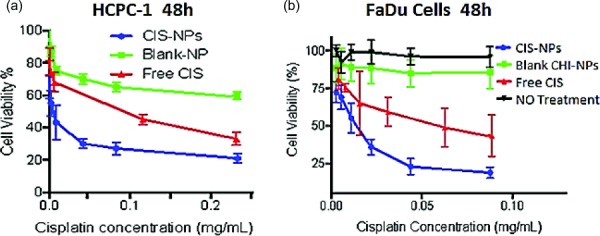
The percentage cell viability observed when free CDDP, BLK NPs, and CDDP NPs were incubated with HCPC-1 cells (a) and FaDu cells (b) over the course of 48 hrs at 37 °C and 5% CO2. Both cells lines showed the lowest viability when exposed to the CDDP NPs, followed by free CDDP and BLK NPs, respectively.
Fig. 7.
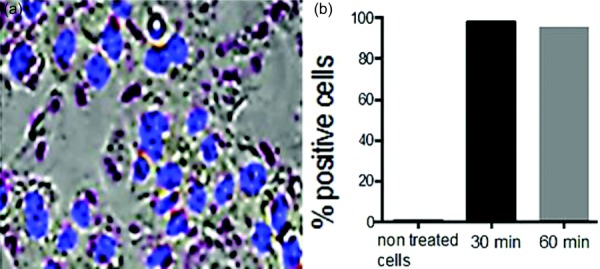
(a) NP cell uptake microscopy image after 1 hr incubation of OC TR-146 cells with ALX-CS NPs and DAPI stained cell nuclei shown and (b) evaluation of cell uptake by flow cytometry, percentage of positive cells after 30 and 60 mins incubation with NPs at a concentration of 0.03 g/L. NPs were taken up by more than 95% of the cells in both cases.
We postulate that the variability seen in the cell uptake percentage versus time is due to the slight degradation of NPs in the cell culture medium; however, a difference of only 3% is negligible and can also be attributed to the natural variability of an in vitro experiment with live cells. The lower viability observed in the CDDP NP group is due to the NPs having a high rate of cell uptake (Fig. 7) via direct membrane translocation (DMT) [27]. This energy-autonomous mechanism is facilitated by the cationic surface charge of NPs due to protonated amine groups. Although DMT is not fully understood, it is known that this path provides a rapid rate of uptake without harming the membrane or causing cytotoxicity [27]. Higher uptake efficiency contributes to a proficient destruction of cancerous cells and minimizes the potential for mutations to develop via cell proliferation, creating genomic heterogeneity [36]. The greater efficacy of CDDP NPs observed in these experiments compared to free CDDP is due to the encapsulation within NPs, which provides swift uptake via DMT and protection from chemical deactivation. It is important to note that the decreased cell viability seen for BLK NPs can be attributed to the natural anticancer properties of CS [37]. CS or CS NPs exhibit virtually no cytotoxicity in noncancerous cell lines [38].
3.4. CDDP NP Release Study.
In order to be therapeutically efficacious, CDDP NPs must release their payload in a manner which destroys cancer cells as rapidly as possible. Figure 8 illustrates the bolus release profile observed with the CDDP NPs in pH 7.2 PBS. Eighty percentage of CDDP was released within the first 5 hrs, with 100% release seen at 50 hrs. Release of CDDP is catalyzed by the swelling of the NPs at pH 7.2. A neutral pH causes the deprotonation of amine groups on CS and their subsequent loss of a net positive charge ( → NH2). This loss inhibits the electrostatic interactions with TPP and lowers the NPs cross-linking density, effectively breaking their structure. This lack of electrostatic interaction increases the size of the NPs (less TPP interactions to tightly bind the CS polymers together). This increases the size and intermolecular distance between CS chains and provides a route for CDDP diffusion out of the NPs. The rate of release can be modulated by modifying parameters of both the NP synthesis and postsynthesis conditions. For example, the concentration of TPP can be increased to heighten the cross-linking density of the CDDP NPs and slow the release rate [39]. Additionally, the molecular weight of CS plays a critical role in the release profile of CS NPs [39]. In vitro testing provides a good model for CDDP release; however, there are many more variables that exist in vivo capable of impacting the release profile, such as the presence of lysozyme which cleaves the β-1,4 glycosidic bonds in between CS monomers [40]. The bolus release profile we observed is critical for our application of treating oral squamous cell carcinoma. A large amount of drug released immediately will destroy cancer cells quickly, reducing the risk of CDDP resistance developing [33]. Additionally, the release profile of the NPs can be controlled by adjusting the pH of the surrounding medium [41] and cross-linking density. In addition to the in vitro and ex vivo studies, properties of the CSM were studied in hamster in vivo experiments. OC tumors were induced in hamsters' cheek pouch and CSM were applied topically to the tumor for 1 hr. The details of these studies are described in the upcoming publications but it should be noted that CSM showed improved tumor shrinkage over CDDP injections. Pharmacokinetic studies indicated minor traces of CDDP in other organs and minimal side effects were observed in the CSM group. The rapid tumor shrinkage by CSM in hamsters further supports the in vitro data of CSM efficacy in destroying cancer cells while remaining local resulting in safer treatment. In addition, they indicate that CSM is able to release the CDDP NPs and the NPs can permeate deep into the tumor. These in vivo studies have also shown adequate mucoadhesion.
Fig. 8.
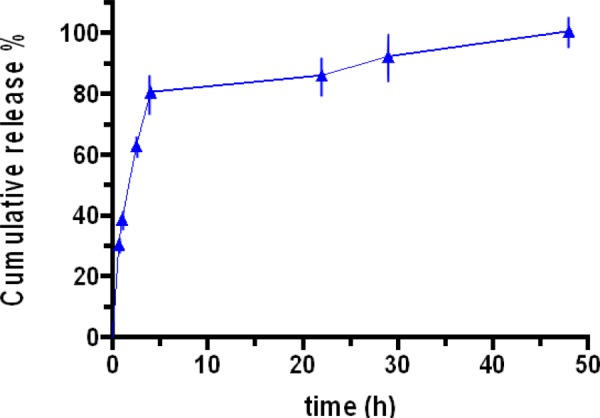
Release profile of CDDP NPs (2.88:1.00) at pH 7.2 in PBS. Percentage release was determined using Eqs. (1)–(4) using Pt concentrations quantified with ICP-AES.
3.5. Synthesis of CSM and SEM.
CSMs were successfully synthesized by the described methods. They were bright white in color and fluffy in texture (Fig. 9), yet able to be flexed or compressed without breaking. NPs were intact within CSM when visualized under SEM (Fig. 10(a)) based on the similar textures observed between isolated lyophilized CDDP NPs and the CSM. The NP control (lyophilized CDDP NPs, Fig. 10(a)) and CSM embedded with CDDP NP (Fig. 10(b)) showed multitextured porous surfaces with many physical inconsistencies. In contrast, the CSM control (lyophilized CS, Fig. 10(c)) displayed a uniform homogenous texture with no notches or grooves. We postulate that this texture is present in the NP samples because the CS in that case is cross-linked, which modifies its natural structure. We postulate that the degree of porosity and perhaps amount of CS NPs impacts the release rate and texture of CSM. This porous structure is critical to its functionality, as an increased number of pores allows for a high water swelling capacity and rate of NP release. Contrastingly, the lyophilized CS had a very uniform, tight structure and virtually no pores, which would inhibit the uptake of water and rate of NP release.
Fig. 9.

Photograph of CSM containing CDDP NPs. Note the smooth surface texture and pure white color. 1.5 cm × 1.5 cm × 2 mm.
Fig. 10.
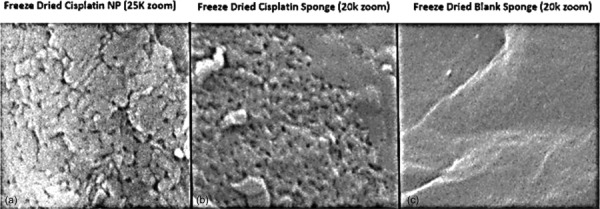
SEM images of lyophilized free CDDP NPs (a), CDDP NPs embedded within CSM (b), and an image of CS sponge synthesized without any NPs (c). The morphology of the free CDDP NPs and CDDP NPs in CSM are nearly identical, with both exhibiting a granulated texture. In contrast, the sponge composed of only CS showed a flat, smooth surface texture with no abnormal structures. This suggests that the NPs remain intact within CSM.
3.6. Stability of NPs in CSM.
It is critical to confirm that after being subjected to the harsh conditions of being frozen at −196 °C and lyophilized for three days, the NPs embedded within CSM remain both structurally intact and still maintain their functionality. We were able to confirm that viable NPs were released from CSM into human saliva after being subjected to three days of lyophilization using NanoSight (Malvern) to quantify the number and size of NPs (Fig. 11). This method of detection was chosen over DLS because we found that upon dissolving CSM for DLS measurement, there was background signal created from the matrix polymers that interfered with NP values. This signal was not present in NanoSight. We observed an increased size of the NPs released from the CSM compared to when they were freshly synthesized (Fig. 11). The measured pH of the human saliva sample was 7.02 compared to the 3.41 of the NP solution prior to lyophilization. The higher pH caused the CDDP NPs to increase in size due to the mechanisms previously described. CDDP NPs are designed to swell in biological conditions to allow for drug release, thus this experiment confirmed both their integrity and functionality when embedded with CSM and subsequently released into a simulated biological environment.
Fig. 11.
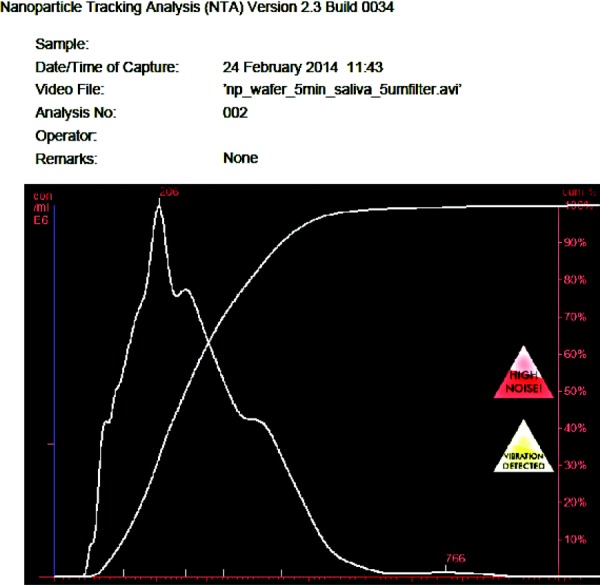
The concentration and % size increase in FITC NPs released from CSM in artificial human saliva (pH 7.0) taken using NanoSight (Malvern). This data confirm the presence of intact NPs within CSM. The increased size can be attributed to the higher pH of the saliva compared to the NP synthesis conditions.
4. Conclusions
We found that the properties of CS NPs can be modulated by varying parameters of the synthesis conditions, such as CS concentration, TPP concentration, and the pH of CS solution. By controlling these variables, CS NPs can be engineered with a particular size, charge, and release profile to suit different in vivo applications. The timely preparation of the CDDP-TPP solution during NP synthesis is a critical step and should always be used to prevent any variability between batches due to changing pH. Some of the key characteristics for success in this application are stability, integrity of NPs after freezing, and release profile. These are important because the NPs within CSM must remain intact and stable throughout the freezing process to ensure the CDDP remains encapsulated. Additionally, both the release rate of CDDP from the CS NPs and NPs from CSM play a vital role in its efficacy.
The ultimate end goal of this paper was to synthesize a transmucosal buccal drug delivery system containing CDDP NPs for the local, topical treatment of OC. We have developed a polymeric CS sponge matrix (CSM) capable of CDDP NPs, which was confirmed in a series of in vitro experiments. CSMs were successfully synthesized and tested for their functionality and presence of NPs, and their structural integrity was verified with both SEM and NanoSight. This innovation represents a significant development in the field of oncology, allowing for topical and local chemotherapy. Our next publication will discuss the in-depth physical and chemical characterization of CSM as well as optimization of the CS NP synthesis procedure.
Acknowledgment
We would like to acknowledge MIT's Koch Cancer Institute and its core facility department for their research resources and technical support. We would also like to acknowledge the financial assistance of MIT Deshpande Center, Massachusetts Life Sciences Center, and the generosity of Aaron Foundation. Special thanks to the National Institutes of Health and National Science Foundation, and the National Cancer Institute's Nanotechnology Characterization Laboratory for their support. Thanks to Ms. Ellen Milano for guidance on analytical method development, Dr. Stephen Lippard's Lab at MIT for technical support, and to Dr. Earl Ada for SEM services at the University of Massachusetts in Lowell. Erkin Aydin was supported by The Scientific and Technological Research Council of Turkey (TUBITAK) through 2219 Postdoctoral Research Scholarship Program.
Contributor Information
Manijeh Goldberg, Massachusetts Institute of Technology, Cambridge, MA 02139; David H. Koch Institute for Integrative Cancer Research, Department of Chemical Engineering, Cambridge, MA 02142; University of Massachusetts Lowell, Department of Mechanical Engineering, Lowell, MA 01854; Privo Technologies, LLC, Cambridge, MA 02141.
Aaron Manzi, Massachusetts Institute of Technology, Cambridge, MA 02139; David H. Koch Institute for Integrative Cancer Research, Department of Chemical Engineering, Langer Lab, Cambridge, MA 02142; Privo Technologies, LLC, Cambridge, MA 02141.
Erkin Aydin, Massachusetts Institute of Technology, Cambridge, MA 02139; David H. Koch Institute for Integrative Cancer Research, Department of Chemical Engineering, Langer Lab, Cambridge, MA 02142; Abdullah Gul University, Department of Mechanical Engineering, Kocasinan, Turkey.
Gurtej Singh, Massachusetts Institute of Technology, Cambridge, MA 02139; David H. Koch Institute for Integrative Cancer Research, Department of Chemical Engineering, Langer Lab, Cambridge, MA 02142.
Payam Khoshkenar, Massachusetts Institute of Technology, Cambridge, MA 02139; David H. Koch Institute for Integrative Cancer Research, Department of Chemical Engineering, Langer Lab, Cambridge, MA 02142; Privo Technologies, LLC, Cambridge, MA 02141.
Amritpreet Birdi, Massachusetts Institute of Technology, Cambridge, MA 02139; David H. Koch Institute for Integrative Cancer Research, Department of Chemical Engineering, Langer Lab, Cambridge, MA 02142; Privo Technologies, LLC, Cambridge, MA 02141.
Brandon LaPorte, Massachusetts Institute of Technology, Cambridge, MA 02139; David H. Koch Institute for Integrative Cancer Research, Department of Chemical Engineering, Langer Lab, Cambridge, MA 02142; Privo Technologies, LLC, Cambridge, MA 02141.
Alejandro Krauskopf, Massachusetts Institute of Technology, Cambridge, MA 02139.
Geralle Powell, Wellesley College, Department of Biology, Wellesley, MA 02481.
Julie Chen, University of Massachusetts Lowell, Department of Mechanical Engineering, Lowell, MA 01854.
Robert Langer, Massachusetts Institute of Technology, Cambridge, MA 02139; David H. Koch Institute for Integrative Cancer Research, Department of Chemical Engineering, Langer Lab, Cambridge, MA 02142.
References
- [1]. Ceschel, G. C. , Maffei, P. , Sforzini, A. , Borgia, S. L. , Yasin, A. , and Ronchi, C. , 2002, “In Vitro Permeation Through Porcine Buccal Mucosa of Caffeic Acid Phenetyl Ester (CAPE) From a Topical Mucoadhesive Gel Containing Propolis,” Fitoterapia, 73, pp. S44–S52.10.1016/S0367-326X(02)00190-9 [DOI] [PubMed] [Google Scholar]
- [2]. Mura, P. , Corti, G. , Cirri, M. , Maestrelli, F. , Mennini, N. , and Bragagni, M. , 2010, “Development of Mucoadhesive Films for Buccal Administration of Flufenamic Acid: Effect of Cyclodextrin Complexation,” J. Pharm. Sci., 99(7), pp. 3019–3029.10.1002/jps.22068 [DOI] [PubMed] [Google Scholar]
- [3]. Ceschel, G. C. , Maffei, P. , Borgia, S. L. , and Ronchi, C. , 2001, “Design and Evaluation of Buccal Adhesive Hydrocortisone Acetate (HCA) Tablets,” Drug Delivery, 8(3), pp. 161–171 10.1080/107175401316906937. [DOI] [PubMed] [Google Scholar]
- [4]. Odukoya, O. , Hawach, F. , and Shklar, G. , 1985, “Retardation of Experimental Oral Cancer by Topical Vitamin E,” Nutr. Cancer, 6(2), pp. 98–104.10.1080/01635588509513813 [DOI] [PubMed] [Google Scholar]
- [5]. Senel, S. , Kremer, M. , Nagy, K. , and Squier, C. , 2001, “Delivery of Bioactive Peptides and Proteins Across Oral (Buccal) Mucosa,” Curr. Pharm. Biotechnol., 2(2), pp. 175–186.10.2174/1389201013378734 [DOI] [PubMed] [Google Scholar]
- [6]. Van Roey, J. , Haxaire, M. , Kamya, M. , Lwanga, I. , and Katabira, E. , 2004, “Comparative Efficacy of Topical Therapy With a Slow-Release Mucoadhesive Buccal Tablet Containing Miconazole Nitrate Versus Systemic Therapy With Ketoconazole in HIV-Positive Patients With Oropharyngeal Candidiasis,” JAIDS J. Acquired Immune Defic. Syndr., 35(2), pp. 144–150.10.1097/00126334-200402010-00007 [DOI] [PubMed] [Google Scholar]
- [7]. Wong, H. L. , Bendayan, R. , Rauth, A. M. , Li, Y. , and Wu, X. Y. , 2007, “Chemotherapy With Anticancer Drugs Encapsulated in Solid Lipid Nanoparticles,” Adv. Drug Delivery Rev., 59(6), pp. 491–504.10.1016/j.addr.2007.04.008 [DOI] [PubMed] [Google Scholar]
- [8]. Illum, L. , 1998, “Chitosan and Its Use as a Pharmaceutical Excipient,” Pharm. Res., 15(9), pp. 1326–1331.10.1023/A:1011929016601 [DOI] [PubMed] [Google Scholar]
- [9]. Santiago de Alvarenga, E. , 2011, “Characterization and Properties of Chitosan,” Material Science Biotechnology of Bipolymers, pp. 91–108 10.5772/17020. [Google Scholar]
- [10]. Garcia-Fuentes, M. , and Alonso, M. J. , 2012, “Chitosan-Based Drug Nanocarriers: Where Do We Stand?,” J. Controlled Release, 161(2), pp. 496–504.10.1016/j.jconrel.2012.03.017 [DOI] [PubMed] [Google Scholar]
- [11]. Simeonova, M. , Velichkova, R. , Ivanova, G. , Enchev, V. , and Abrahams, I. , 2003, “Poly (Butylcyanoacrylate) Nanoparticles for Topical Delivery of 5-Fluorouracil,” Int. J. Pharm., 263(1), pp. 133–140.10.1016/S0378-5173(03)00373-9 [DOI] [PubMed] [Google Scholar]
- [12]. Giannola, L. I. , De Caro, V. , Giandalia, G. , Siragusa, M. G. , Paderni, C. , Campisi, G. , and Florena, A. M. , 2010, “5-Fluorouracil Buccal Tablets for Locoregional Chemotherapy of Oral Squamous Cell Carcinoma: Formulation, Drug Release and Histological Effects on Reconstituted Human Oral Epithelium and Porcine Buccal Mucosa,” Curr. Drug Delivery, 7(2), pp. 109–117.10.2174/156720110791011792 [DOI] [PubMed] [Google Scholar]
- [13]. Westra, W. H. , 2009, “The Changing Face of Head and Neck Cancer in the 21st Century: The Impact of HPV on the Epidemiology and Pathology of Oral Cancer,” Head Neck Pathol., 3(1), pp. 78–81.10.1007/s12105-009-0100-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14]. National Cancer Institute, 2015, Cancer of the Oral Cavity and Pharynx, Surveillance, Epidemiology, and End Results Program Turning Cancer Data Into Discovery, http://seer.cancer.gov/statfacts/html/oralcav.html, Last accessed January 2, 2015. [Google Scholar]
- [15]. Rischin, D. , 2005, “Induction Chemotherapy in Head and Neck Cancer,” Squamous Cell Head and Neck Cancer, Humana Press, New York, pp. 165–170 10.1007/978-1-59259-938-7_11. [Google Scholar]
- [16]. Bachaud, J. M. , Cohen-Jonathan, E. , Alzieu, C. , David, J. M. , Serrano, E. , and Daly-Schveitzer, N. , 1996, “Combined Postoperative Radiotherapy and Weekly Cisplatin Infusion for Locally Advanced Head and Neck Carcinoma: Final Report of a Randomized Trial,” Int. J. Radiat. Oncol. Biol. Phys., 36(5), pp. 999–1004.10.1016/S0360-3016(96)00430-0 [DOI] [PubMed] [Google Scholar]
- [17]. Ihde, D. C. , Mulshine, J. L. , Kramer, B. S. , Steinberg, S. M. , Linnoila, R. I. , Gazdar, A. F. , Edison, M. , Phelps, R. M. , Lesar, M. , and Phares, J. C. , 1994, “Prospective Randomized Comparison of High-Dose and Standard-Dose Etoposide and Cisplatin Chemotherapy in Patients With Extensive-Stage Small-Cell Lung Cancer,” J. Clin. Oncol., 12(10), pp. 2022–2034 10.1093/jnci/djg017. [DOI] [PubMed] [Google Scholar]
- [18]. Studer, U. E. , Bacchi, M. , Biedermann, C. , Jaeger, P. , Kraft, R. , Mazzucchelli, L. , Markwalder, R. , Senn, E. , and Sonntag, R. W. , 1994, “Adjuvant Cisplatin Chemotherapy Following Cystectomy for Bladder Cancer: Results of a Prospective Randomized Trial,” J. Urol., 152(1), pp. 81–84 10.1016/j.eururo.2013.08.033. [DOI] [PubMed] [Google Scholar]
- [19]. Pearcey, R. , Brundage, M. , Drouin, P. , Jeffrey, J. , Johnston, D. , Lukka, H. , MacLean, G. , Souhami, L. , Stuart, G. , and Tu, D. , 2002, “Phase III Trial Comparing Radical Radiotherapy With and Without Cisplatin Chemotherapy in Patients With Advanced Squamous Cell Cancer of the Cervix,” J. Clin. Oncol., 20(4), pp. 966–972.10.1200/JCO.20.4.966 [DOI] [PubMed] [Google Scholar]
- [20]. Blair, B. G. , Larson, C. A. , Adams, P. L. , Abada, P. B. , Pesce, C. E. , Safaei, R. , and Howell, S. B. , 2011, “Copper Transporter 2 Regulates Endocytosis and Controls Tumor Growth and Sensitivity to Cisplatin In Vivo,” Mol. Pharmacol., 79(1), pp. 157–166.10.1124/mol.110.068411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21]. Sharp, S. Y. , Rogers, P. M. , and Kelland, L. R. , 1995, “Transport of Cisplatin and Bis-Acetato-Ammine-Dichlorocyclohexylamine Platinum (IV)(JM216) in Human Ovarian Carcinoma Cell Lines: Identification of a Plasma Membrane Protein Associated With Cisplatin Resistance,” Clin. Cancer Res., 1(9), pp. 981–989 10.3892/or.2014.3257. [PubMed] [Google Scholar]
- [22]. Fuertes, M. A. , Castilla, J. , Alonso, C. , and Perez, J. M. , 2003, “Cisplatin Biochemical Mechanism of Action: From Cytotoxicity to Induction of Cell Death Through Interconnections Between Apoptotic and Necrotic Pathways,” Curr. Med. Chem., 10(3), pp. 257–266.10.2174/0929867033368484 [DOI] [PubMed] [Google Scholar]
- [23]. Hamers, F. P. T. , Gispen, W. H. , and Neijt, J. P. , 1991, “Neurotoxic Side-Effects of Cisplatin,” Eur. J. Cancer Clin. Oncol., 27(3), pp. 372–376.10.1016/0277-5379(91)90549-S [DOI] [PubMed] [Google Scholar]
- [24]. Von Hoff, D. D. , Schilsky, R. , Reichert, C. M. , Reddick, R. L. , Rozencweig, M. , Young, R. C. , and Muggia, F. M. , 1978, “Toxic Effects of Cis-Dichlorodiammineplatinum (II) in Man,” Cancer Treat. Rep., 63(9–10) pp. 1527–1531 10.1007/BF00254137742. [PubMed] [Google Scholar]
- [25]. Lau, J. K. , and Deubel, D. V. , 2005, “Loss of Amine From Platinum (II) Complexes: Implications for Cisplatin Inactivation, Storage, and Resistance,” Chem.- Eur. J., 11(9), pp. 2849–2855.10.1002/chem.200401053 [DOI] [PubMed] [Google Scholar]
- [26]. Jain, R. K. , 2001, “Normalizing Tumor Vasculature With Anti-Angiogenic Therapy: A New Paradigm for Combination Therapy,” Nat. Med., 7(9), pp. 987–989.10.1038/nm0901-987 [DOI] [PubMed] [Google Scholar]
- [27]. Lin, J. , and Alexander-Katz, A. , 2013, “Cell Membranes Open ‘Doors' for Cationic Nanoparticles/Biomolecules: Insights Into Uptake Kinetics,” ACS Nano., 7(12), pp. 10799–10808.10.1021/nn4040553 [DOI] [PubMed] [Google Scholar]
- [28]. Fernandez-Urrusuno, R. , Calvo, P. , Remunan-Lopez, C. , Vila-Jato, L. , and Alonso, M. , 1999, “Enhancement of Nasal Absorption of Insulin Using Chitosan Nanoparticles,” Pharm. Res., 16(10), pp. 1576–1581.10.1023/A:1018908705446 [DOI] [PubMed] [Google Scholar]
- [29]. Shojaei, A. H. , 1998, “Buccal Mucosa as a Route for Systemic Drug Delivery: A Review,” J. Pharm. Pharm. Sci., 1(1), pp. 15–30 10.5138/ijdd.v3i4.426. [PubMed] [Google Scholar]
- [30]. Nakamura, T. , and Kinoshita, S. , 2003, “Ocular Surface Reconstruction Using Cultivated Mucosal Epithelial Stem Cells,” Cornea, 22(7), pp. S75–S80.10.1097/00003226-200310001-00011 [DOI] [PubMed] [Google Scholar]
- [31]. Berners-Price, S. J. , Frenkiel, T. A. , Frey, U. , Ranford, J. , and Sadler, P. J. , 1992, “Hydrolysis Products of Cisplatin: pKa Determinations Via [1H, 15N] NMR Spectroscopy,” J. Chem. Soc., Chem. Commun., 1992(10), pp. 789–791.10.1039/c39920000789 [Google Scholar]
- [32]. Corden, B. J. , 1987, “Reaction of Platinum (II) Antitumor Agents With Sulfhydral Compounds and the Implications for Nephrotoxicity,” Inorg. Chim. Acta, 137(1), pp. 125–130.10.1016/S0020-1693(00)87131-5 [Google Scholar]
- [33]. Galluzzi, L. , Senovilla, L. , Vitale, I. , Michels, J. , Martins, I. , Kepp, O. , Castedo, M. , and Kroemer, G. , 2012, “Molecular Mechanisms of Cisplatin Resistance,” Oncogene, 31(15), pp. 1869–1883.10.1038/onc.2011.384 [DOI] [PubMed] [Google Scholar]
- [34]. Long, D. F. , and Repta, A. J. , 1981, “Review Article Cisplatin: Chemistry, Distribution and Biotransformation,” Biopharm. Drug Dispos., 2(1), pp. 1–16.10.1002/bdd.2510020102 [DOI] [PubMed] [Google Scholar]
- [35]. Bose, R. N. , Cornelius, R. D. , and Viola, R. E. , 1985, “Kinetics and Mechanisms of Platinum (II)-Promoted Hydrolysis of Inorganic Polyphosphates,” Inorg. Chem., 24(24), pp. 3989–3996.10.1021/ic00218a006 [Google Scholar]
- [36]. Shafee, N. , Smith, C. R. , Wei, S. , Kim, Y. , Mills, G. B. , Hortobagyi, G. N. , Stanbridge, E. J. , and Lee, E. Y.-H. P. , 2008, “Cancer Stem Cells Contribute to Cisplatin Resistance in Brca1/p53–Mediated Mouse Mammary Tumors,” Cancer Res., 68(9), pp. 3243–3250.10.1158/0008-5472.CAN-07-5480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37]. Dass, C. R. , and Choong, P. F. M. , 2008, “The Use of Chitosan Formulations in Cancer Therapy,” J. Microencapsulation, 25(4), pp. 275–279.10.1080/02652040801970461 [DOI] [PubMed] [Google Scholar]
- [38]. Huang, M. , Khor, E. , and Lim, L.-Y. , 2004, “Uptake and Cytotoxicity of Chitosan Molecules and Nanoparticles: Effects of Molecular Weight and Degree of Deacetylation,” Pharm. Res., 21(2), pp. 344–353.10.1023/B:PHAM.0000016249.52831.a5 [DOI] [PubMed] [Google Scholar]
- [39]. Agnihotri, S. A. , Mallikarjuna, N. N. , and Aminabhavi, T. M. , 2004, “Recent Advances on Chitosan-Based Micro-and Nanoparticles in Drug Delivery,” J. Controlled Release, 100(1), pp. 5–28.10.1016/j.jconrel.2004.08.010 [DOI] [PubMed] [Google Scholar]
- [40]. Freier, T. , Koh, H. S. , Kazazian, K. , and Shoichet, M. S. , 2005, “Controlling Cell Adhesion and Degradation of Chitosan Films by N-Acetylation,” Biomaterials, 26(29), pp. 5872–5878.10.1016/j.biomaterials.2005.02.033 [DOI] [PubMed] [Google Scholar]
- [41]. Pan, Y. , Li, Y. J. , Zhao, H. Y. , Zheng, J. M. , Xu, H. , Wei, G. , Hao, J. S. , and Cui, F. D. , 2002, “Bioadhesive Polysaccharide in Protein Delivery System: Chitosan Nanoparticles Improve the Intestinal Absorption of Insulin In Vivo,” Int. J. Pharm., 249(1), pp. 139–147.10.1016/S0378-5173(02)00486-6 [DOI] [PubMed] [Google Scholar]