

Abstract
Acetylcholine acts through nicotinic and muscarinic acetylcholine (ACh) receptors in ventral midbrain and striatal areas to influence dopamine (DA) transmission. This cholinergic control of DA transmission is important for processes such as attention and motivated behavior, and is manipulated by nicotine in tobacco products. Identifying and characterizing the key ACh receptors involved in cholinergic control of DA transmission could lead to small molecule therapeutics for treating disorders involving attention, addiction, Parkinson’s disease, and schizophrenia. α6-containing nicotinic acetylcholine receptors (nAChRs) are highly and specifically expressed in midbrain DA neurons, making them an attractive drug target. Here, we used genetic, pharmacological, behavioral, and biophysical approaches to study this nAChR subtype. For many experiments, we used mice expressing mutant α6 nAChRs (“α6L9S” mice) that increase the sensitivity of these receptors to agonists such as ACh and nicotine. Taking advantage of a simple behavioral phenotype exhibited by α6L9S mice, we compared the ability of full versus partial α6* nAChR agonists to activate α6* nAChRs in vivo. Using local infusions of both agonists and antagonists into brain, we demonstrate that neurons and nAChRs in the midbrain are sufficient to account for this behavioral response. To complement these behavioral studies, we studied the ability of in vivo α6* nAChR activation to support plasticity changes in midbrain DA neurons that are relevant to behavioral sensitization and addiction. By coupling local infusion of drugs and brain slice patch clamp electrophysiology, we show that activating α6* nAChRs in midbrain DA areas is sufficient to enhance glutamatergic transmission in VTA DA neurons. Together, these results from in vivo studies strongly suggest that α6* nAChRs expressed by VTA DA neurons are positioned to strongly influence both DA-mediated behaviors and the induction of synaptic plasticity by nicotine.
Keywords: nicotine, addiction, dopamine, glutamate, locomotor, plasticity
1
The brain dopamine (DA) pathway is a crucial neurotransmitter system involved in a myriad of processes, including reward/reinforcement, attention, cognition, and voluntary movement. DA neurons in the ventral tegmental area (VTA) that project to nucleus accumbens (NAc) make up the mesolimbic DA pathway, whereas DA neurons in the substantia nigra pars compacta (SNc) that project to dorsal striatum comprise the nigrostriatal DA pathway. Disrupted DA transmission causes the cardinal features of Parkinson’s disease as well as addiction to drugs of abuse, and is clearly implicated in disorders such as schizophrenia, bipolar disorder, mood disorders, and attention deficit hyperactivity disorder. Cholinergic signaling strongly regulates the DA system in animal models and human disease. For example, in Parkinson’s disease, enhanced cholinergic signaling often plays a role in the pathological circuit changes that give rise to the motor features of the disease (Pisani et al., 2007). Brain areas rich in DA-producing cell bodies and axon terminals receive dense cholinergic innervation, and numerous proteins involved in cholinergic transmission are expressed in these areas (Woolf and Butcher, 1981, Bolam et al., 1984, Woolf and Butcher, 1986). These include acetylcholinesterase, choline acetyltransferase, muscarinic acetylcholine receptors, and nicotinic acetylcholine receptors (nAChRs). Recently, nAChRs have emerged as crucial regulators of DA transmission (Rice and Cragg, 2004, Zhang and Sulzer, 2004, Exley and Cragg, 2008, Cachope et al., 2012, Threlfell et al., 2012), and many groups are actively searching for novel compounds designed to specifically manipulate nAChRs in the DA system.
nAChRs in the central nervous system are either homomeric α7 receptors or heteromeric receptors comprised of multiple α and β subunits. α4β2* (* indicates a receptor containing the indicated subunits but may contain other unknown subunits) nAChRs exhibit widespread expression in brain (Nashmi et al., 2007, Shih et al., 2014), along with high affinity for nicotine and acetylcholine (Salminen et al., 2004, Salminen et al., 2007, Grady et al., 2010). α4β2* nAChRs are both necessary and/or sufficient for most aspects of nicotine addiction in mouse models, including reward, self-administration, tolerance, behavioral and cellular sensitization, and enhanced firing of VTA DA neurons (Tapper et al., 2004, Maskos et al., 2005, Mameli-Engvall et al., 2006, Tapper et al., 2007, Pons et al., 2008, Exley et al., 2011). α4β2* nAChRs are expressed by most neurons of the midbrain DA system (Azam et al., 2002, Nashmi et al., 2007). α6β2* nAChRs are the other major nAChR subtype in the mesolimbic DA system. These receptors are also strongly expressed in some VTA and SNc neurons (Azam et al., 2002, Mackey et al., 2012), and they are necessary for nicotine self-administration (Pons et al., 2008). GABAergic neurons in substantia nigra and VTA do not appear to express α6 subunits (Drenan et al., 2008a), but DA neurons express multiple α6β2* subtypes: α4α6β2β3, α4α6β2, α6β2β3, and α6β2 (referred to hereafter as “α6*”) (Salminen et al., 2004, Salminen et al., 2007, Drenan et al., 2010). Due to their restricted expression pattern in only a few cell types in brain (Mackey et al., 2012, Shih et al., 2014), along with their high sensitivity to nicotine and ACh (Salminen et al., 2007), manipulation of α6* nAChR activity with drugs may offer a more selective approach to treating disorders involving DA transmission (Quik and McIntosh, 2006). Unfortunately, very few α6-selective drugs have been developed due to the complexity of α6* nAChR stoichiometry and the challenges associated with in vitro expression and characterization of the various α6* subtypes (Drenan et al., 2008b, Drenan and Lester, 2012).
To address these obstacles, we developed and have studied transgenic mice expressing mutant, hypersensitive α6 nAChR subunits. In these mice (referred to as “α6L9S”), a Leu to Ser mutation at the “9 prime” (9′) position in the second transmembrane domain of the α6 subunit renders α6* nAChR populations hypersensitive to acetylcholine and nicotine (Drenan et al., 2008a). Previously, we used these mice to elucidate several aspects of α6* nAChR neurobiology. In behavioral experiments, we demonstrated that increased activity of α6* nAChRs leads to locomotor activation. Using slice electrophysiology, we found that α6* nAChR activity stimulates action potential firing in VTA DA neurons (Drenan et al., 2008a). A neurochemical analysis revealed that α6L9S mice exhibit enhanced synthesis and release of DA in NAc and dorsal striatum (Drenan et al., 2008a, Drenan et al., 2010, Wang et al., 2013). We also demonstrated that α4 subunits are required for most of these effects, highlighting the importance of α4α6* nAChRs (Drenan et al., 2010). More recently, we used these mice to show that selective activation of α6* nAChRs in DA neurons is sufficient to enhance glutamatergic synaptic plasticity, which is a key molecular change occurring during the nicotine dependence/addiction process (Engle et al., 2013, Engle et al., 2015). Despite these advances, important questions remain to be answered to further validate the α6L9S model for future drug discovery studies on mesolimbic DA system α6* nAChRs. Although the VTA to NAc pathway is presumed to be the key brain area that mediates most or all of these effects, we have not directly shown this. In the present study, we used behavioral, systems, and biophysical approaches to determine whether the VTA is required for selective α6* nAChR activation to support locomotor activity and induction of synaptic plasticity.
2. Experimental Procedures
2.1. Animals
Adult α6L9S mice and non-transgenic (nonTG) littermates were used in the current study. All mice were maintained on a standard 12 hour light/dark cycle at 22°C with food and water ad libitum. Mice were weaned on postnatal day 21 and subsequently group-housed with same-sex littermates. Genotype analysis by polymerase chain reaction (PCR) from tail biopsies was completed as previously described (Drenan et al., 2010). Care of all animals was carried out in agreement with the National Institutes of Health Office of Laboratory Animal Welfare as well as a protocol approved by the Institutional Animal Care and Use Committee at Purdue University. α6L9S mice were generated as previously published (Drenan et al., 2008a). Briefly, a Leu9′ to Ser mutation was introduced into a bacterial artificial chromosome (BAC) at the α6 nAChR subunit gene, Chrna6. Mutant BAC DNA was then injected into FVB/N embryos and the embryos were implanted into pseudopregnant Swiss-Webster surrogates. The insertion site in the mouse genome is unknown. Founder animals were identified and back-crossed to C57BL/6 mice for 12 or more generations. As a result, 90–95% of the genome of the α6L9S strain is expected to be C57BL/6 although FVB/N allelic DNA close to the insertion site is likely to remain. The L9S mutation leaves α6* nAChRs 10- to 100- fold more sensitive to ligands such as nicotine and acetylcholine compared to non-α6* nAChRs, depending upon the assay (Drenan et al., 2008a, Drenan et al., 2010, Cohen et al., 2012). Previous studies have confirmed that α6* nAChRs are not overexpressed, nor expressed in ectopic brain regions in α6L9S mice (Drenan et al., 2008a, Drenan et al., 2010).
2.2. Drugs and Chemicals
(−) Nicotine hydrogen tartrate salt was from Glentham Life Sciences (Wiltshire, United Kingdom). All nicotine doses are reported as freebase. α-conotoxin MII (αCtxMII) was synthesized as described in previously published reports (Azam et al., 2010). ABT-089 powder was provided by Abbvie. Varenicline, SCH23390, and all other chemicals without a specified supplier were from Sigma (St. Louis, MO).
2.3. Locomotor Activity
Horizontal locomotor activity in α6L9S mice and nonTG littermates was measured using a Panasonic WV-CP294 camera and converted into distance traveled using TopScan LITE software (CleverSys; Reston, VA). Prior to behavioral testing, all mice were handled, scruffed, and given a saline injection (i.p.) once per day for a minimum of three days. Groups of 6–8 mice were placed in a fresh home cage and baseline locomotor activity was recorded in one min intervals for 8 min. Mice were removed from the cage and injected with either a test compound (nicotine, varenicline, etc.) or saline and returned to the cage within 20 seconds. Locomotor activity was then recorded in one min intervals for > 30 min following the injection. For dose-response studies, mice were administered saline then each successive dose of test compound at 3–4 day intervals.
2.4. Bilateral Cannulation of VTA and NAc
α6L9S mice and nonTG littermates were anesthetized with a ketamine/xylazine cocktail (100 mg/kg ketamine, 10 mg/kg xylazine, injected i.p.). The surgical area was shaved and cleansed via three applications of alternating iodide ointment and 70% ethanol. Mice were then placed into a stereotaxic frame (Kopf; Tujunga, CA) and a small incision was made to expose the skull. The skull was leveled in the coronal and sagittal planes using the coordinates for bregma and lambda as landmarks. Bilateral holes were drilled in the skull according to adjusted coordinates from the third edition of the Franklin and Paxinos mouse brain atlas (for VTA: M/L: ±0.5 mm from bregma, A/P: −3.2 mm from bregma; for NAc: M/L: ±0.5 mm from bregma, A/P: +1.62 mm from bregma). The A/P coordinate was adjusted for each animal to accommodate individual variations in size; the distance between bregma and lambda was measured for each mouse and divided by the published distance in this species (4.21 mm), and this ratio was then multiplied by the proper A/P coordinate from the atlas to determine the proper A/P coordinates for each animal. Guide cannulae 3.0 mm in length (Plastics One; Roanoke, VA) along with a dummy cannula (also 3.0 mm in length; Plastics One) were slowly lowered into position and secured using Geristore cement (Den-Mat; Lompoc, CA). Animals remained in the stereotaxic apparatus until the cement fully dried. Once removed from the stereotaxic apparatus, a dust cap was screwed onto the dorsal portion of the guide cannula to keep the dummy cannula in place and to prevent contamination of the guide cannula. Following surgery, mice were given ketoprofen (5 mg/kg, s.c.) and allowed to recover on a heating pad under close observation until ambulatory. Mice were single-housed following cannulation surgery and were allowed to recover for at least 5 days prior to the start of behavioral testing.
2.5. Intracranial Infusions and Locomotor Activity
α6L9S mice or nonTG littermates received intra-VTA infusions of nicotine (1.7 nmol) or vehicle (sterile saline) approximately 10 minutes prior to the initiation of each locomotor activity session. To determine the role of α6* nAChRs in nicotine-mediated locomotion, some α6L9S mice received intra-VTA co-infusions of nicotine (1.7 nmol) and the α6* nAChR antagonist αCtxMII (10 pmol) before the locomotor monitoring session. Infusions were carried out using a dual syringe pump connected to internal cannulae (extending 1.5 mm beyond guide cannulae; Plastics One) via two identical Hamilton syringes and PE50 tubing (Plastics One). Mice were anesthetized using isoflurane (5% for initiation of anesthesia; 1.8% for maintenance). While maintained on isoflurane anesthesia, the dust cap and dummy cannula were removed and the internal cannula was fully inserted into the guide cannula. Drugs were infused at a rate of 0.1 μL/min for 5 minutes for a total volume of 0.5 μL. The internal cannula was left in place for an additional 5 minutes to prevent backflow into the guide cannula. Following the infusion, the internal cannula was removed and the dummy cannula and dust cap were replaced. The animal was immediately removed from the isoflurane and allowed to recover in the home cage for 10 minutes prior to each locomotor activity session. Following recovery from the isoflurane anesthesia, mice were injected with 0.02 mg/kg nicotine (i.p.) or saline, immediately placed into a fresh home cage and locomotor activity was then measured as described above for 30 minutes. A separate group of α6L9S mice received intra-NAc infusions of either SCH23390 (150 ng) or vehicle (sterile saline) before i.p. saline or nicotine using the procedures detailed above prior to locomotor testing for 30 minutes. Following the last locomotor activity session, mice were perfused and brains were retained for histological analysis/confirmation of cannulae placement.
2.6. Nicotine Injections for Electrophysiology
A set of experiments involved nicotine exposure in a novel environment prior to brain slice preparation for electrophysiology studies. Prior to receiving nicotine injections mice were habituated to being handled. For three consecutive days, at the same time each day, mice were picked up and held but not scruffed. On the fourth day, mice were scruffed and given a mock injection. On the fifth day (the day before the i.p. nicotine injection) mice were scruffed and received an i.p. saline injection. One the day of the experiment mice were given an i.p. nicotine (0.03 or 0.17 mg/kg) or vehicle (saline) injection and placed into a novel environment. A subset of mice were cannulated and given 7–10 days to recover before beginning the handling procedure. These mice were given an infusion of vehicle (saline) or αCtxMII immediately prior to the nicotine injection.
2.7. Brain Slice Preparation
Sixty minutes after the nicotine injection, mice were anesthetized with an injection of sodium pentobarbital (100 mg/kg, i.p.) prior to cardiac perfusion. Brain slices were prepared as previously described (Engle et al., 2012). Mice were perfused with oxygenated (95% O2/5% CO2) 4°C N-methyl-D-glucamine (NMDG)-based recovery solution containing (in mM): 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 Na+ ascorbate, 2 thiourea, 3 Na+ pyruvate, 10 MgSO4·7H2O, and 0.5 CaCl2·2H2O. The osmolarity was adjusted to 300–310 mOsm with sucrose and the pH to 7.3–7.4 with 10 N HCl. Brains were dissected and put in oxygenated 4°C recovery solution for 1 minute before cutting 200 μm coronal brain slices through the VTA on a vibrating microslicer. Slices were placed in oxygenated 33°C recovery solution for 12 minutes and then kept in room temperature holding solution for an hour or more until used for electrophysiology. Holding solution contained (in mM): 92 NaCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 Na+ ascorbate, 2 thiourea, 3 Na+ pyruvate, 2 MgSO4·7H2O, and 2 CaCl2·2H2O. The osmolality and pH were adjusted to 300–310 mOsm and 7.2–7.4.
2.8. Patch Clamp Electrophysiology
Patch pipettes were prepared on a P-97 programmable micropipette puller (Sutter Instruments; Novato, CA) from borosilicate glass capillary tubes. The tip resistance was 4.5 to 8.0 MΩ when filled with an internal solution containing (in mM): 117 CsCH3SO3, 20 HEPES, 0.4 EGTA, 2.8 NaCl, 5 TEA-Cl, 2.5 MgATP, 0.1 spermine, and 0.25 MgGTP. The osmolarity of the internal solution was adjusted to 290 mOsm with sucrose and the pH was adjusted to 7.25 with Tris base. A VTA-containing brain slice was placed in a recording chamber and superfused at a rate of 1.5–2.0 mL/min with oxygenated 32°C recording solution containing (in mM): 124 NaCl, 2.5 KCl, 1.2 NaH2PO4, 24 NaHCO3, 12.5 glucose, 2 MgSO4·7H2O, and 2 CaCl2·2H2O. The osmolality and pH were to 300–310 mOsm and 7.2–7.4. Picrotoxin (75 mM) and tetrodotoxin (0.5 μM) were also included in the recording solution to isolate the effect of α-amino-3-hydroxy-5-methyl-isoxazolepropionic acid (AMPA) to the recorded neuron. A Nikon FN-1 upright microscope using infrared or visible differential interference contrast optics was used to visualize neurons. VTA neurons were identified as previously described (Engle et al., 2013) and found at coordinates of approximately −3.5 mm from bregma, 4.0–4.5 mm ventral to the surface, and 0.5–1.0 mm from the midline. Whole-cell recordings were acquired using the following instruments/software from Molecular Devices Corp. (Sunnyvale, CA): an Axopatch 200B amplifier, a 16-bit Digidata 1440 A/D converter, and pCLAMP 10.3 software.
Cells were held at −60 mV and AMPA receptor (AMPAR) function was analyzed as previously described (Engle et al., 2012, Engle et al., 2013, Engle et al., 2015). A micropipette filled with AMPA (100 μM) was loaded onto a single-dimension piezoelectric translator attached to a micromanipulator. pClamp 10 was used to run a protocol that triggered a Picospritzer III to puff-apply AMPA to the recorded neuron with a 250 ms, 12 psi pressure ejection. The protocol triggered the pipette to move 20–40 μm away from the recorded neuron, trigger the pressure ejection, and then retract the pipette. Movement of the neuron, stability of the seal, and the rise time of the response were all assessed to determine whether the response was optimal. These metrics were previously described (Engle et al., 2013).
2.9. Statistics
Statistical tests were done using GraphPad Prism 6 software. To evaluate statistical significance, data sets were analyzed for normality with a D’Agostino & Pearson omnibus normality test. For all experiments conducted, some experimental groups exhibited parametric distribution while others did not. Therefore, non-parametric distribution was assumed for all data sets, and Mann-Whitney U-tests were performed. P values for statistically significant results are presented in the figure legends.
3. Results
VTA DA and GABA neurons express moderate to high levels of nAChRs. VTA DA neurons produce α4α6*, α6(non-α4)*, and α4(non-α6)* nAChRs (Salminen et al., 2004, Salminen et al., 2007). In contrast, VTA GABA neurons appear to only express α4(non-α6)* nAChRs (Drenan et al., 2008a, Powers et al., 2013). This differential nAChR expression pattern (Fig. 1A) suggests that drugs targeting α4α6* and/or α6(non-α4)* nAChRs may offer a unique way to manipulate the mesolimbic DA system in various human disorders. To isolate the action of α6* nAChRs in DA neurons, we studied “α6L9S” mice. These mice express mutant, hypersensitive α6* nAChRs in midbrain DA neurons. This allows low concentrations of nicotine, acetylcholine, or other α6* nAChR agonists to be used as highly selective ligands for α6* nAChRs (Fig. 1B).
Figure 1. α6 expression and rationale for the chosen model.
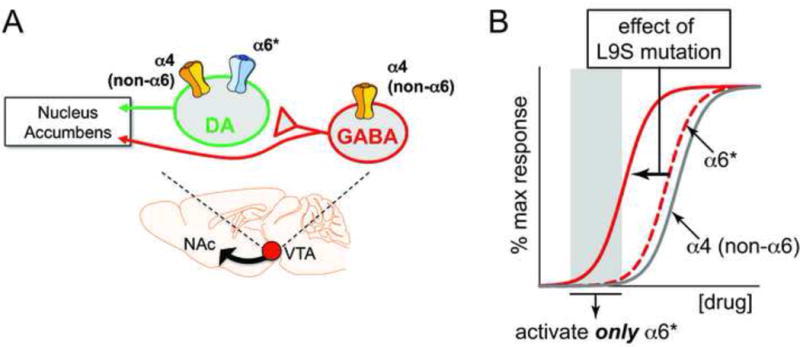
A) Schematic of VTA neuron connectivity. Whereas α4 subunits are expressed in both DA and GABA neurons in VTA, α6 subunits are specifically expressed in DA neurons but excluded from GABA neurons.
B) In α6L9S mice, a Leu to Ser mutation was introduced at the 9′ position within transmembrane domain 2 of the α6 nAChR subunit protein. This mutation increases the sensitivity of the population of channels containing the mutant α6 subunit, allowing ligands such as ACh and nicotine to selectively activate these channels when low concentrations are used. The theoretical concentration response curve for most responses involving α6-containing receptors is left-shifted.
3.1. Locomotor Activity in α6L9S Mice
Previously, we used a rudimentary beam-break locomotor activity apparatus to study locomotion in α6L9S mice. We reported that α6L9S mice exhibit profound psychomotor activation when low doses of nicotine are administered systemically (Drenan et al., 2008a, Drenan et al., 2010, Grady et al., 2010, Cohen et al., 2012). Because nonTG littermate mice do not respond to these low nicotine doses with any change in locomotor activity, we suggested that these behavioral responses were due specifically to α6* nAChR activation in midbrain DA neurons. In this study, we have expanded upon those prior/initial studies. To study locomotor activity in α6L9S mice, we employed video tracking software designed to monitor/measure horizontal distance traveled. This system utilizes an overhead video camera that records mouse horizontal movement, allowing the software to create a spatial map of mouse ambulatory activity (Fig. 2A). To confirm our previous results, we measured nicotine-stimulated locomotor activity using this system. Mice were placed in a new home cage and allowed to ambulate for 8 min. Then, mice were quickly removed, injected, and returned to the monitoring cage within 20 sec. Locomotor activity was measured for an additional 30 min. As expected, nicotine (i.p.) produced a dose-dependent increase in locomotor activity in α6L9S mice (Fig. 2B). Nicotine-elicited locomotor activation was robust but brief, often declining to baseline within 30 min or less. Distance-traveled during min 9–15 was significantly increased by 0.02 mg/kg and 0.05 mg/kg nicotine in α6L9S mice compared to nonTG mice (Fig. 2C).
Figure 2. Locomotor activation in α6L9S mice by nicotine.
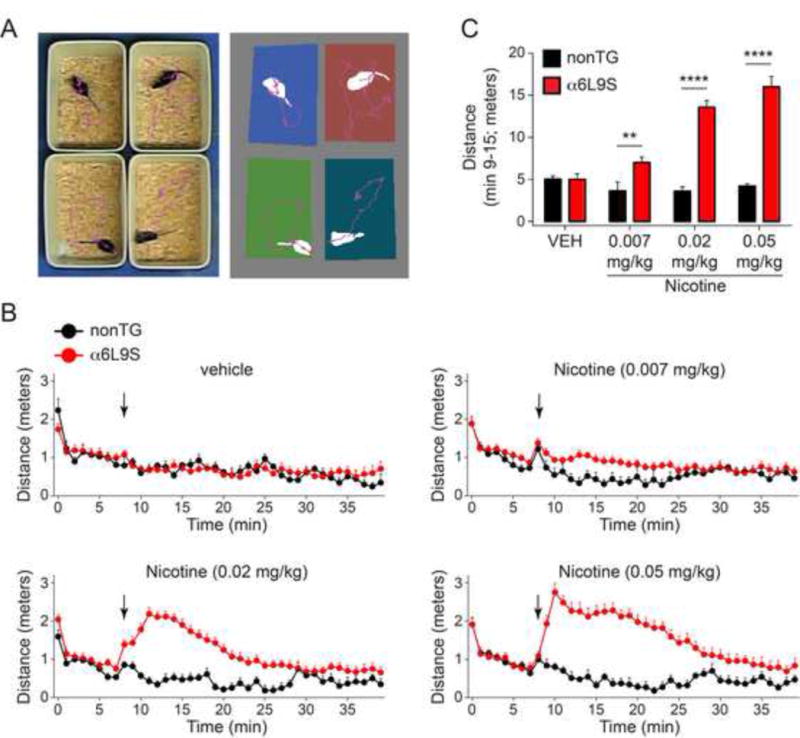
A) Locomotor activity measurements. α6L9S (n=19) and nonTG (n=5) littermate mice are injected i.p. with the indicated drug, followed by measurement of distance traveled via video tracking software while animals are ambulating in a novel home cage.
B) Locomotor activation in α6L9S mice by nicotine. α6L9S or nonTG littermate mice were placed in a fresh home cage and locomotor activity was recorded. After 8 min to establish baseline locomotor activity, mice were injected with the indicated dose of nicotine (0.05, 0.02, or 0.007 mg/kg; i.p.) and immediately returned to the cage for an additional 30 min of monitoring.
C) Quantification of nicotine-elicited locomotor activity in α6L9S (n=19) and nonTG (n=5) mice. Total distance traveled between and including minutes 9 to 15 from the experiment described in B was summed for each mouse and averaged across all mice in each group. Mean distance traveled from minute 9 to 15 is plotted for each nicotine dose and each strain (α6L9S and nonTG). Mann-Whitney U-test: **p<0.01 (actual: p=0.0089), ****p<0.0001
Next, we conducted several experiments to test the hypothesis that nicotine-elicited locomotor activation in α6L9S mice is mediated by nAChRs in the ventral midbrain. Groups of mice were implanted with bilateral guide cannulae above the VTA. On the day of the experiment, mice were anesthetized with isoflurane. The first goal was to test the ability of intra-VTA αCtxMII to block locomotor activation elicited by systemic nicotine in α6L9S mice. The specificity of αCtxMII is 5.6-fold for α6* nAChRs vs. α3*, and has low antagonist activity at other nAChR subtypes (Cartier et al., 1996, McIntosh et al., 2004). α3* nAChR subunits are expressed at low levels in VTA DA neurons (Champtiaux et al., 2002, Shih et al., 2014). Thus, in the DA system, and in conjunction with the sensitizing α6L9S manipulation, αCtxMII is sufficient to block the activity of α6* nAChRs in the α6L9S model (Drenan et al., 2008a). Using an internal cannula, we infused αCtxMII (10 pmol) or vehicle into the VTA. At t = 0 min after mice recovered from isoflurane anesthesia and were ambulatory, 0.02 mg/kg nicotine (i.p.) was administered and distance traveled was measured. Vehicle-infused α6L9S mice responded normally to systemic nicotine, but locomotor activation was blocked in mice infused with αCtxMII (Fig. 3A). Control saline injections after the same intra-VTA infusion regimen demonstrated the specificity of 0.02 mg/kg nicotine in α6L9S mice (Fig. 3B). Quantification of distance traveled during the 30-minute experiment demonstrated a significant difference in nicotine-injected (but not saline-injected) mice infused with vehicle and αCtxMII (Fig. 3C). Cannulae placement and injection sites in VTA was verified post-hoc (Fig. 3D).
Figure 3. VTA αCtxMII-sensitive nAChRs mediate locomotor activation in α6L9S mice.
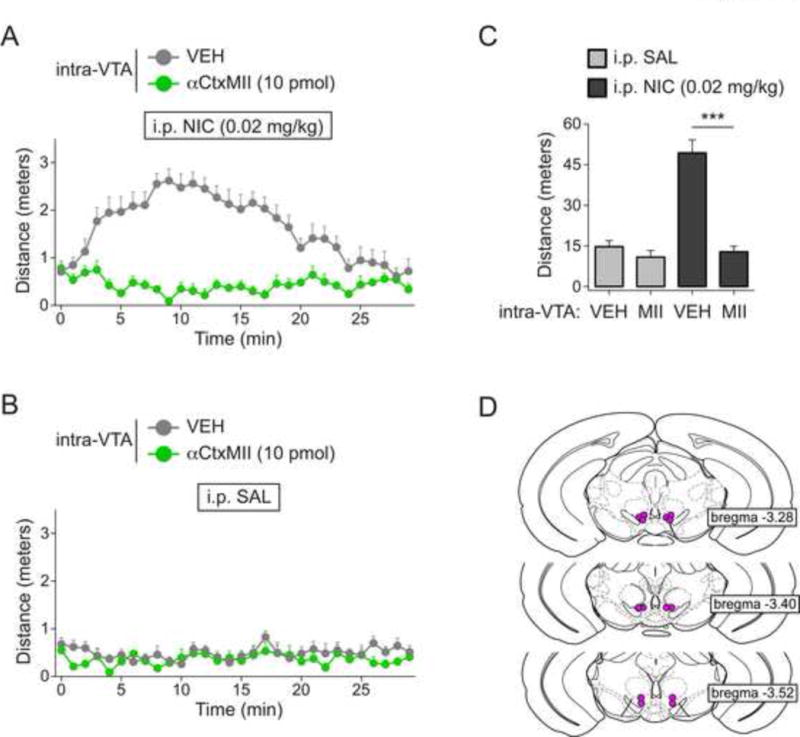
A) Locomotor activity in α6L9S mice infused into the VTA with vehicle (n=7) or αCtxMII (10 pmol; n=8) prior to systemic nicotine (0.02 mg/kg) injection. α6L9S mice were infused as indicated, followed by injection (i.p.) with nicotine prior to locomotor activity monitoring for 30 min.
B) Locomotor activity in α6L9S mice infused into the VTA with vehicle (n=7) or αCtxMII (10 pmol; n=7) prior to systemic saline injection. α6L9S mice were infused as indicated, followed by injection (i.p.) with saline prior to locomotor activity monitoring for 30 min.
C) Quantification of distance traveled for α6L9S mice injected with saline or nicotine following infusion of either vehicle or αCtxMII. Mann-Whitney U-test: ***p<0.001 (actual: p=0.0003)
D) Cannula placement in VTA for infusions of VEH/αCtxMII shown in (A) and (B)
To further study the mechanism by which systemic nicotine activates locomotion in α6L9S mice, α6L9S mice were cannulated to allow delivery of drugs into the nucleus accumbens shell (NAcs). NAcs is a key site of action for α6* nAChRs in the response to nicotine (Brunzell et al., 2009). We tested the hypothesis that dopamine signaling is required for nicotine-elicited locomotor activity in α6L9S mice by infusing mice with the dopamine D1 receptor antagonist SCH23390 (150 ng) prior to a systemic nicotine (0.02 mg/kg, i.p.) challenge. Consistent with a role for dopamine D1 receptor-mediated signaling in α6L9S locomotor activation, SCH23390 infusion into NAcs substantially reduced locomotor activation following systemic nicotine injection (Fig. 4A). Systemic saline control injections revealed a small but significant effect of SCH23390 alone on locomotor activity (Fig. 4B and C). Quantification of distance traveled during the 30-minute experiment demonstrated a significant difference in NIC-injected mice infused with vehicle vs. SCH23390 (Fig. 4C). Cannulae placement and injection sites in NAcs was verified post-hoc (Fig. 4D).
Figure 4. Locomotor activation in α6L9S mice involves D1 dopamine receptors in nucleus accumbens shell.
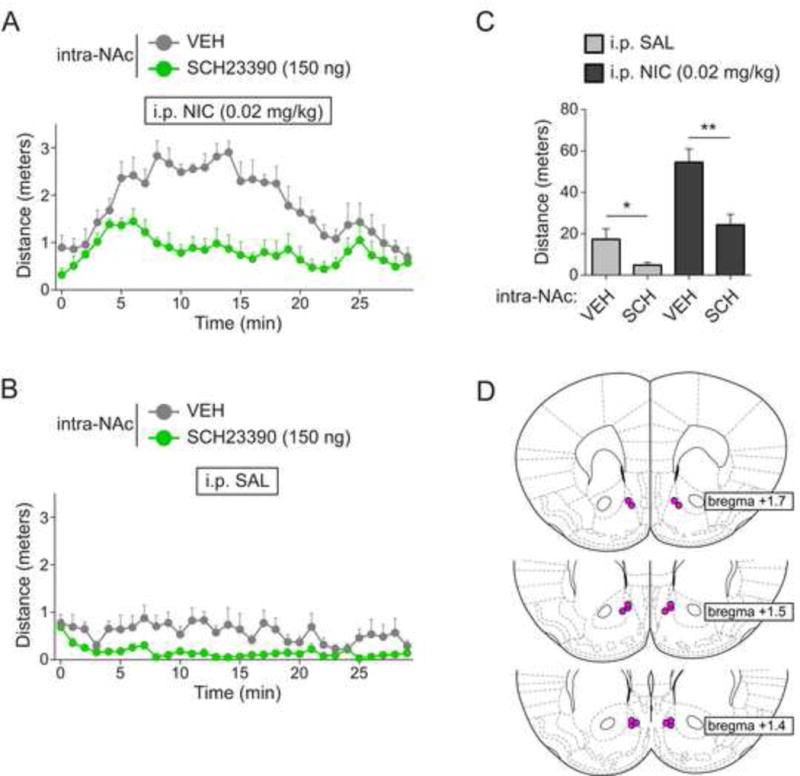
A) Locomotor activity in α6L9S mice infused into the NAc with vehicle (n=7) or SCH23390 (150 ng; n=8) prior to systemic nicotine (0.02 mg/kg) injection. α6L9S mice were infused as indicated, followed by injection (i.p.) with nicotine prior to locomotor activity monitoring for 30 min.
B) Locomotor activity in α6L9S mice infused into the NAc with vehicle (n=7) or SCH23390 (150 ng; n=7) prior to systemic saline injection. α6L9S mice were infused as indicated, followed by injection (i.p.) with saline prior to locomotor activity monitoring for 30 min.
C) Quantification of distance traveled for α6L9S mice injected with saline or nicotine following infusion of either vehicle or SCH23390. Mann-Whitney U-test: *p<0.05 (actual: p=0.0262), **p<0.01 (actual: p=0.0037)
D) Cannula placement in NAc for infusions of VEH/SCH23390 shown in (A) and (B)
Next, we tested the ability of intra-VTA nicotine to recapitulate the effect of systemic nicotine in α6L9S mice. We infused vehicle or nicotine (1.7 nmol) into the VTA. Immediately after isoflurane was removed and mice resumed ambulatory activity, we measured locomotor activity for 30 min. Whereas nonTG mice did not respond appreciably to 1.7 nmol nicotine relative to vehicle (Fig. 5A and E), α6L9S mice responded with increased locomotor activity when 1.7 nmol nicotine was infused into the VTA (Fig. 5C and E). Infusion sites were confirmed with histological analysis after the experiment (nonTG: Fig. 5B; α6L9S: Fig. 5D). To test the hypothesis that α6* nAChRs mediate the effect of nicotine in this experiment, α6L9S mice were co-infused with 1.7 nmol nicotine and αCtxMII, an antagonist of α6* nAChRs. In contrast to nicotine, co-infusion of nicotine with αCtxMII did not result in locomotor activation in α6L9S mice (Fig. 5C and E). Together, these results indicate that activation of α6* nAChRs in VTA neurons is sufficient to stimulate locomotor activity in mice.
Figure 5. Locomotor activation in α6L9S mice requires α6* nAChRs in VTA.
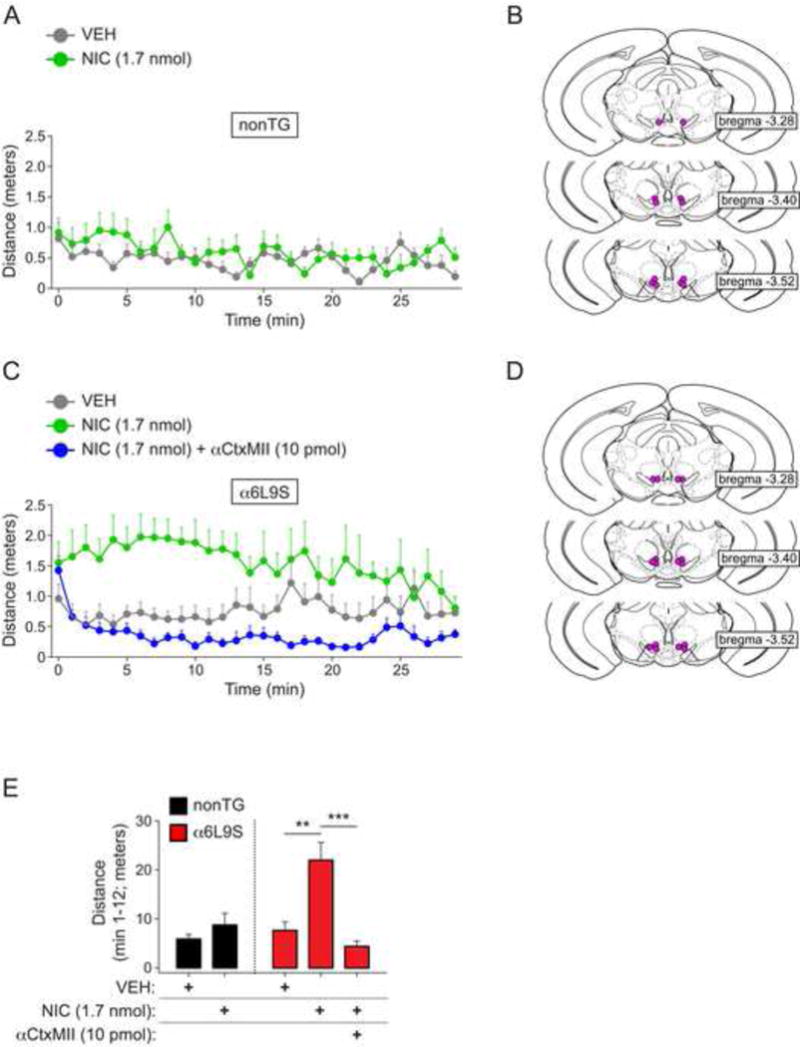
A) Low-dose nicotine infusion into VTA of nonTG mice does not stimulate locomotor activity. Vehicle (VEH; n=6) or 1.7 nmol of nicotine (NIC 1.7 nmol; n=6) was infused into the VTA of isoflurane-anesthetized nonTG mice. Following infusion, mice were immediately removed from isoflurane and locomotor activity was measured for 30 min.
B) Cannula placement in VTA for infusions of VEH/NIC in VTA of nonTG mice. Approximate site of nicotine delivery is marked with a purple dot.
C) Nicotine infusion into VTA of α6L9S mice stimulates locomotor activity, and co-infusion of nicotine with αCtxMII blocks locomotor activation. Vehicle (VEH; n=8), 1.7 nmol of nicotine (NIC 1.7 nmol; n=6), or 1.7 nmol of nicotine plus 10 pmol of αCtxMII (NIC 1.7 nmol + MII 10 pmol; n=8) were infused into the VTA of isoflurane-anesthetized mice. Following infusion, mice were immediately removed form isoflurane and locomotor activity was measured for 30 min.
D) Cannula placement in VTA for infusions of VEH/NIC/NIC+αCtxMII in VTA of α6L9S mice. Approximate site of nicotine delivery is marked with a purple dot.
E) Quantification of locomotor activity. Distance traveled following infusion of the indicated drug(s) into nonTG (A; n=6) and α6L9S (C; n=6–8) mice was recorded for 30 min, and mean distance traveled from minute 1 to 12 for each group is plotted. Mann-Whitney U-test: **p<0.01 (actual: p=0.0027), ***p<0.001 (actual: p=0.0007)
Having confirmed that locomotor activation in α6L9S mice is mediated by α6* nAChRs in VTA, we tested the ability of several nicotinic ligands to support a similar behavioral response. Varenicline (Fig. 6A), an FDA-approved drug for smoking cessation, is a high-affinity partial agonist at α4β2 nAChRs (Coe et al., 2005, Rollema et al., 2007). More recently, it has also been shown to exhibit similar partial agonism at α6* nAChRs in the midbrain DA system (Grady et al., 2010, Bordia et al., 2012). To further study α6* nAChRs using α6L9S locomotor activation, we tested the ability of varenicline to stimulate locomotor activity in α6L9S mice. As with nicotine, mice injected (i.p.) with low doses of varenicline exhibited a dose-dependent increase in locomotor activity compared to nonTG littermate control mice (Fig. 6D). Using distance-traveled during min 9–39, varenicline produced a significant increase in locomotor activity at 0.01 mg/kg and 0.03 mg/kg (Fig. 6F).
Figure 6. Locomotor activation in α6L9S mice by nAChR partial agonists varenicline and ABT-089.
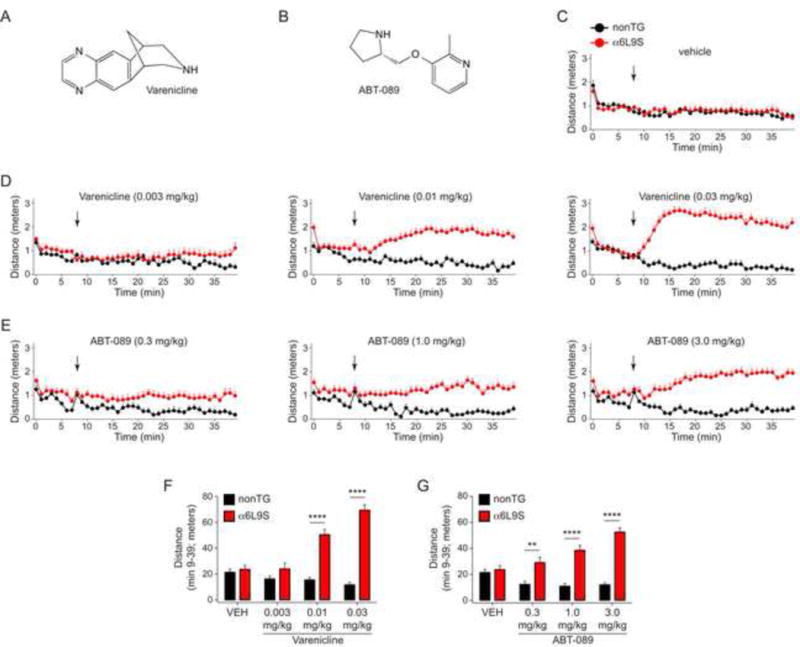
A) Structure of varenicline.
B) Structure of ABT-089.
C) Control vehicle injections in α6L9S (n=16) and nonTG (n=8) mice. α6L9S or nonTG littermate mice were placed in a fresh home cage and locomotor activity was recorded. After 8 min to establish baseline locomotor activity, mice were injected with vehicle (saline) and immediately returned to the cage for an additional 30 min of monitoring.
D) Locomotor activation in α6L9S mice by varenicline using procedures described in (C). Locomotor activation in α6L9S or nonTG littermate mice was measured following injection with the indicated dose of varenicline (0.03, 0.01, or 0.003 mg/kg; i.p.).
E) Locomotor activation in α6L9S mice by ABT-089 using procedures described in (C). Locomotor activation in α6L9S or nonTG littermate mice was measured following injection with the indicated dose of ABT-089 (0.3, 1.0, or 3.0 mg/kg; i.p.).
F) Quantification of varenicline-elicited locomotor activity in α6L9S (n=16) and nonTG (n=8) mice. Total distance traveled between and including minutes 9 to 39 from the experiment described in (D) was summed for each mouse and averaged across all mice in each group. Mean distance traveled from minute 9 to 39 is plotted for each varenicline dose and each strain (α6L9S and nonTG). Mann-Whitney U-test: ****p<0.0001
G) Quantification of ABT-089-elicited locomotor activity in α6L9S (n=16) and nonTG (n=8) mice. Total distance traveled between and including minutes 9 to 39 from the experiment described in (E) was summed for each mouse and averaged across all mice in each group. Mean distance traveled from minute 9 to 39 is plotted for each ABT-089 dose and each strain (α6L9S and nonTG). Mann-Whitney U-test: **p<0.01 (actual: p=0.0014), ****p<0.0001
Next, we tested ABT-089 for its ability to stimulate locomotor activity in α6L9S mice. ABT-089 (Fig. 6B; also know as pozanicline) is an investigational drug with partial agonist properties at α4β2 and α6*. In particular, ABT-089 has been reported to possess differential pharmacology at distinct αCtxMII-sensitive nAChRs; it is a partial agonist at a high-affinity α6* subtype and a full agonist at a low-affinity α6* subtype (Marks et al., 2009). We speculated that if ABT-089 supported locomotor activity at low doses, this could help confirm its activity at a high-affinity α6* subtype. ABT-089 was administered i.p. to α6L9S and nonTG mice and distance traveled was measured. Unlike varenicline, which stimulated locomotor activity at doses such as 0.01 mg/kg, higher doses of ABT-089 were required to stimulate similar locomotor responses in α6L9S mice. Doses of 1.0 mg/kg and 3.0 mg/kg ABT-089 were required to elicit appreciable locomotor activation in α6L9S mice (Fig. 6E and G). These results, combined with experiments employing varenicline, confirm that partial agonists at α6* nAChRs are capable of strongly stimulating locomotor activity in the α6L9S system.
3.2. Synaptic Plasticity in VTA DA Neurons
Nicotine exposure is known to alter synaptic plasticity at VTA DA neurons (Saal et al., 2003, Gao et al., 2010, Jin et al., 2011, Mao et al., 2011). Previously, we demonstrated that incubating brain slices from naïve animals with low concentrations of nicotine causes α6* nAChR activation and enhanced AMPAR function in VTA DA neurons (Engle et al., 2013, Engle et al., 2015). In this study, we sought to determine whether in vivo activation of α6* nAChRs is sufficient to support changes in VTA DA neuron excitability. We tested the ability of in vivo nicotine administration to act selectively through α6* nAChRs to enhance AMPAR function on the surface of these cells. α6L9S and nonTG control littermate mice were injected (i.p.) with nicotine or vehicle. After 60 min, mice were sacrificed and brain slices were prepared (Fig. 7A). AMPAR function was measured using local application of AMPA to the recorded cell using a puff-pipette as previously described and validated (Engle et al., 2012, Engle et al., 2013, Engle et al., 2015). Based on the dose-range found to be sufficient to stimulate locomotor activity in α6L9S mice (0.02 to 0.05 mg/kg; Fig. 2), α6L9S and nonTG mice were injected with 0.03 mg/kg nicotine or vehicle. This dose was sufficient to enhance AMPA-evoked currents on the surface of VTA DA neurons in α6L9S mice, but was below threshold in nonTG mice (Fig. 7B and C). A relatively higher dose of nicotine (0.17 mg/kg) was needed to increase the amplitude of AMPA-evoked currents on the surface of VTA DA neurons from nonTG mice (Fig. 7B and C). Thus, a single systemic exposure to nicotine can act through α6* nAChRs to enhance glutamatergic transmission in VTA DA neurons.
Figure 7. Systemic nicotine acts through α6-containing nAChRs to enhance AMPA receptor function in VTA DA neurons.
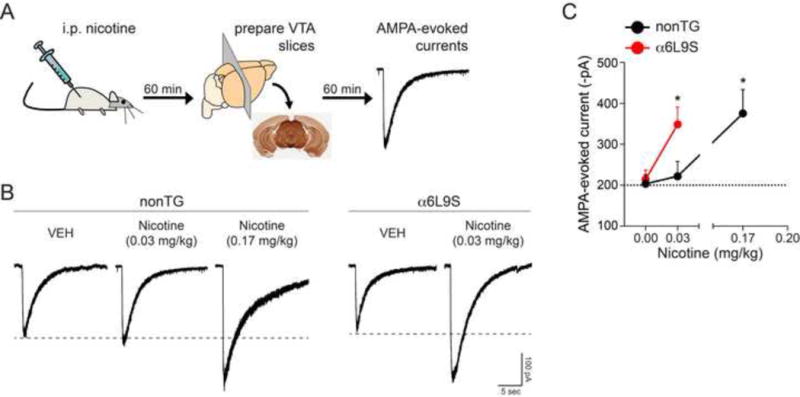
A) Experimental design. α6L9S mice were injected (i.p.) with nicotine at the indicated dose. Sixty minutes after nicotine injection, mice were used to prepare brain slices for patch clamp recording in VTA DA neurons. AMPAR currents were elicited by locally puffing AMPA onto the cell body of the recorded neuron and recording inward currents in voltage clamp mode.
B) A dose of nicotine (0.03 mg/kg) similar to the dose used to elicit locomotor activation in α6L9S mice is sufficient to enhance AMPAR currents in VTA DA neurons. Representative AMPA-evoked currents from α6L9S and nonTG mice injected with the indicated dose of nicotine are shown.
C) Quantification of AMPA-evoked current responses in VTA DA neurons from α6L9S and nonTG mice injected with the indicated dose of nicotine. Mean peak AMPA-evoked currents for each group/treatment are plotted. Mann-Whitney U-test: *p<0.05 (actual: α6L9S saline vs. nicotine 0.03 mg/kg, p=0.0293; nonTG saline vs. nicotine 0.17 mg/kg, p=0.019). (nonTG: VEH n=4; nicotine (0.03 mg/kg) n=7; nicotine (0.17 mg/kg) n=6; α6L9S: VEH n=6; nicotine (0.03 mg/kg) n=8)
Last, we tested the hypothesis that systemic, low-dose nicotine administration in α6L9S mice acts through ventral midbrain α6* nAChRs to enhance AMPA-evoked currents. Prior to nicotine (0.03 mg/kg; i.p.) challenge, the VTA of α6L9S mice was infused with αCtxMII (10 pmol) or vehicle (Fig. 8A). Brain slices were prepared and AMPA-evoked currents were measured as in Fig. 7. The nicotine-elicited increase in AMPAR function was abolished when αCtxMII was infused into the VTA (Fig. 8B and C). αCtxMII infusion paired with a vehicle injection did not show any change over baseline. Injection sites in the VTA were verified post-hoc (Fig. 8D). These data, along with Fig. 7, demonstrate that in vivo activation of VTA α6* nAChRs is sufficient to drive synaptic plasticity changes in VTA DA neurons that are known to be important for locomotor sensitization and reward behavior.
Figure 8. Inhibition of α6-containing nAChRs in VTA blocks AMPAR enhancement by systemic nicotine.
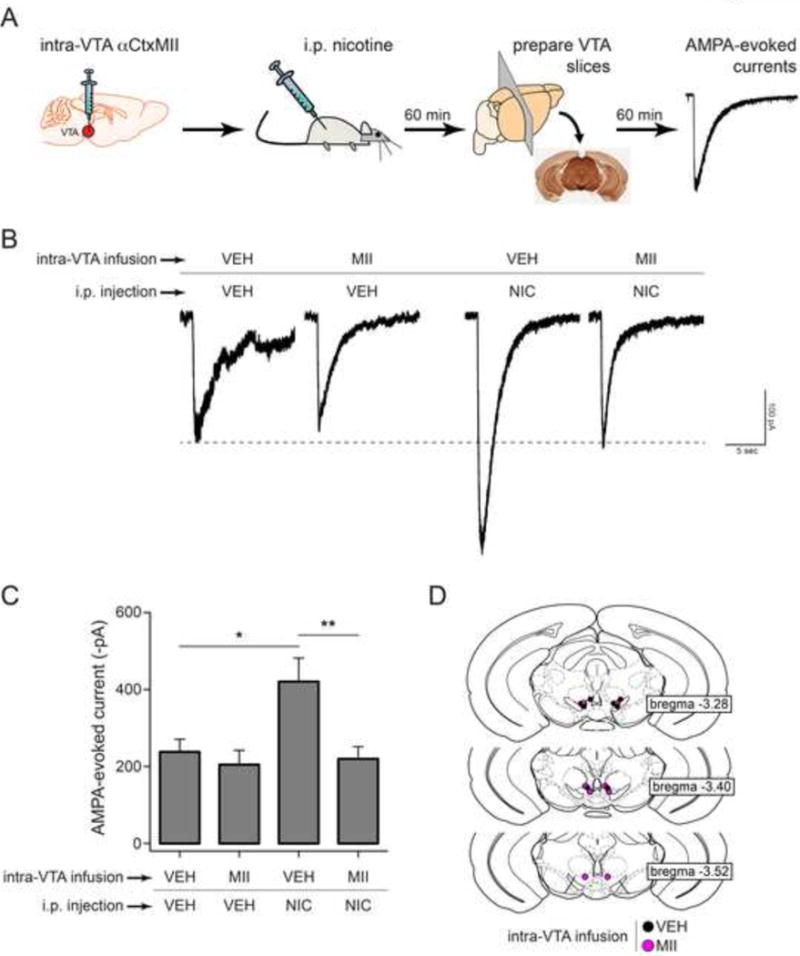
A) Experimental design. α6L9S mice were cannulated and vehicle or αCtxMII (10 pmol) was infused into the VTA. Following VTA infusion, mice were injected i.p. with saline or nicotine (0.03 mg/kg). Sixty min later, brain slices were prepared for recording. AMPA-evoked currents were elicited by locally puffing AMPA onto the cell body of the recorded neuron and recording inward cation currents in voltage clamp mode.
B) Representative AMPA-evoked currents from α6L9S mice injected/infused with the indicated drugs are shown.
C) Mean peak AMPA-evoked currents for each group shown in (B) are plotted. Mann-Whitney U-test: *p<0.05 (actual: p=0.0205), **p<0.01 (actual: p=0.003). (VEH/VEH: n=7, MII/VEH: n=7, VEH/NIC: n=8, MII/NIC: n=8)
D) Cannula location for each mouse in groups indicated in (B) is shown.
4. Discussion
4.1. α6* nAChRs in VTA mediate locomotor activation
In this study, we began by re-assessing previous studies showing that low doses of nicotine stimulate locomotor activity in α6L9S mice. Whereas these previous studies utilized a low-resolution locomotor activity system, the current study employed video tracking software to monitor distance traveled and velocity. The current data are very consistent with results published in 2008 (Drenan et al., 2008a). The magnitude and duration of nicotine-elicited locomotor responses are comparable between the two studies (Drenan et al., 2008a). Transgenes in transgenic animals are known to undergo silencing, resulting in reduced transgene expression (Mehtali et al., 1990). α6L9S mice harbor 10 or more copies of the α6L9S transgene (Drenan et al., 2008a), but the present results suggest that there has been no appreciable functional loss of α6L9S transgene expression compared to mice in the original study.
Mouse behavior, including behaviors resulting from transgene expression, are also known to be sensitive to genetic background. The α6L9S mice were originally generated using FVB/N embryos, and each generation has been back-crossed to C57BL/6. Whereas mice used to generate behavioral data in the original study were back-crossed less than 10 times to C57BL/6, mice in the present study have been back-crossed to C57BL/6 20 times or more and therefore contain more C57BL/6 allelic DNA. Our data showing that locomotor activation in α6L9S mice is comparable to the original study imply that the locomotor phenotype in α6L9S mice appears quite stable on the C57BL/6 background.
Although α6 nAChR subunit expression is sparse in brain compared to α4 subunits, there are still several brain areas displaying measurable α6 subunit expression. α6 subunits are expressed in noradrenergic neurons of the locus coeruleus (Drenan et al., 2008a), which send projections to brain areas such as the cerebral cortex and hippocampus (Azam and McIntosh, 2006). α6 subunits are also expressed by a subset of retinal ganglion cells, a sparse population of neurons in the dorsal lateral geniculate nucleus of the thalamus, and are widely expressed in several layers of superior colliculus (Mackey et al., 2012). α6 subunits are also expressed in medial habenula to interpeduncular nucleus pathway (Henderson et al., 2014, Shih et al., 2014), and have recently been shown to play an important role in nociception via their expression in spinal cord (Wieskopf et al., 2015). Although the DA system is the most logical candidate to mediate nicotine-elicited locomotor activation, several of these areas also have a plausible link to the locomotor system. Additionally, many studies probing the role of α6* nAChRs rely heavily on αCtxMII, which has modest affinity for α3* nAChRs (Cartier et al., 1996). Expression of α3* nAChRs in midbrain DA neurons has not been completely ruled out (Azam et al., 2002), and could theoretically play a role in some nicotine-mediated responses. Thus, we employed intracranial manipulations in α6L9S mice to probe the role of the VTA and α6* nAChRs in mediating the locomotor phenotype we observed.
Nicotine delivered into the VTA of α6L9S mice is sufficient to recapitulate the effect of systemic nicotine in the locomotor activity assay, and co-infusion of nicotine with αCtxMII blocks the action of nicotine alone (Fig. 5). Thus, no other brain areas or pathways that express α6 subunits besides the VTA are required to produce locomotor activity in α6L9S mice. Our results build upon previous results in rat brain showing that intracerebroventricular infusion of α6 nAChR antisense oligos blocks nicotine-stimulated locomotor activation (le Novere et al., 1999). Our data are also complementary to data presented in another study; Gotti and colleagues demonstrated that αCtxMII-sensitive nAChRs in VTA are required for nicotine-elicited locomotor activation, DA release in nucleus accumbens, and nicotine self-administration in rat (Gotti et al., 2010). Our data are also consistent with previous results showing that re-expression of α6 subunits in the VTA of α6 knockout mice is sufficient to restore acute nicotine self-administration (Pons et al., 2008). It should be noted, however, that the generalizability of these results to nicotine-induced modulation of locomotor activity in wild type mice is limited. C57BL/6 wild type mice exhibit locomotor suppression at moderate (~0.5 mg/kg) nicotine doses (Drenan et al., 2008a). The mechanism behind this suppression is not clear, but could involve a dominant role for GABA release in the mesolimbic DA system via α4β2 nAChRs. By “bypassing” α4β2 activation on GABA neurons by the use of hypersensitive α6* nAChRs expressed only on DA neurons, locomotor results in α6L9S mice may support this conclusion regarding GABA release and nicotine-mediated locomotor suppression.
4.2. α6*-mediated locomotor activity by nAChR partial agonists
To further explore α6* nAChR function, and to better describe the individual α6* nAChR subtypes that may mediate locomotor activation in α6L9S mice, we used two additional agonists in addition to nicotine. Varenicline, in addition to its known partial agonist activity at α4β2 nAChRs (Coe et al., 2005, Rollema et al., 2007), has recently been shown to posses high affinity for and partial agonist activity at α6* nAChRs in the DA system. Using agonist-evoked DA release from mouse striatal synaptosomes, Grady and colleagues measured varenicline-induced release at αCtxMII-resistant (α4β2 and α4α5β2 nAChRs) and αCtxMII-sensitive (α4α6β2β3, α4α6β2, α6β2β3, and α6β2 nAChRs) receptors (Grady et al., 2010). Varenicline is a high-affinity partial agonist at both subtypes (26% of nicotine and 50 nM EC50 at αCtxMII-resistant nAChRs; 39% of nicotine and 75 nM EC50 at αCtxMII-sensitive nAChRs) (Grady et al., 2010). Similar studies in rat and monkey synaptosomes indicate varenicline may posses slight selectivity for αCtxMII-sensitive vs. -resistant nAChRs (Bordia et al., 2012). Varenicline stimulated locomotor activity in α6L9S mice at doses that did not affect locomotion in nonTG mice (Fig. 6), but with kinetics that differed from those of nicotine. The latency to reach peak distance traveled was greater for varenicline compared to nicotine. For example, 0.05 mg/kg nicotine caused mice to reach peak distance traveled within ~2 minutes whereas mice injected with 0.03 mg/kg varenicline took ~8 minutes to reach peak distance traveled. Peak distance traveled, once achieved by α6L9S mice, was similar for nicotine and varenicline. However, the duration of agonist-stimulated locomotor activity differed for the two drugs. Nicotine-induced locomotor activity fell back to baseline levels within ~30 minutes for all doses of nicotine tested, whereas varenicline-induced locomotor activity remained elevated at ~30 minutes after injection. These kinetic differences could be due to full versus partial agonism for the two drugs, differences in metabolism and bioavailability, or some combination thereof. In mice, nicotine is metabolized within 7–10 minutes (Matta et al., 2007), whereas varenicline levels persist for much longer (Obach et al., 2006).
ABT-089, also known as pozanicline, was developed as a nAChR subtype-selective partial agonist for the treatment of neurological disorders (Lin et al., 1997). Indeed, ABT-089 has cognition-enhancing properties (Decker et al., 1997, Lin et al., 1997), improves attention (Prendergast et al., 1998), reduces nicotine intake (Lee et al., 2014), reduces L-Dopa-induced dyskinesias in Parkinson’s disease models (Zhang et al., 2014), and has shown efficacy in human clinical trials for attention deficit hyperactivity disorder (Wilens et al., 2006). Although originally characterized as an α4β2-specific ligand, later pharmacology studies demonstrated significant activity at α6* nAChRs. Using striatal DA release assays, Marks and colleagues demonstrated that ABT-089 is a high-potency (EC50=0.11 μM) partial agonist (36% of nicotine) at one αCtxMII-sensitive nAChR subtype, and is a low-potency (28 μM) full agonist (98% of nicotine) at another αCtxMII-sensitive subtype (Marks et al., 2009). Marks and colleagues speculated that because α4α6* nAChRs exhibit high sensitivity to nicotine and ACh (Salminen et al., 2007), ABT-089 may be acting at this subtype to mediate its actions at low (nanomolar) concentrations (Marks et al., 2009). To determine what nAChR subtype ABT-089 acts on in vivo, we were interested to determine whether ABT-089 would activate locomotor activity at similar doses as varenicline. Although capable of stimulating locomotor activity specifically in α6L9S mice and not in nonTG littermates, much higher doses of ABT-089 were required relative to varenicline doses. These results suggest that varenicline and ABT-089 may act on different α6* nAChR subtypes in vivo. ABT-089 did share similar properties with varenicline with regard to latency to reach peak distance traveled and the duration of locomotor responses (Fig. 6E). Although the differences in kinetics between nicotine-mediated locomotor activation and activity induced by either of varenicline or ABT-089 are speculated to be due to the partial agonist properties of these ligands, this has not been shown at α6L9S* nAChRs. Mutations in nAChR transmembrane α-helices are known to alter the pharmacology of ligands, in some cases converting partial agonists to full agonists, or antagonists to agonists (Bertrand et al., 1992). Kinetic differences in locomotor activation could be fully accounted for by differences in metabolism between nicotine and varenicline/ABT-089.
4.3. VTA α6* nAChRs and synaptic plasticity
To connect our in vivo locomotor activity data, which measures α6* nAChR activation in VTA, with cellular and molecular events that are relevant to nicotine addiction, we recorded from VTA DA neurons using brain slice patch clamp electrophysiology. In vivo exposure to nicotine, along with other drugs of abuse such as cocaine and alcohol, results in long-term potentiation (LTP) of excitatory glutamatergic inputs to VTA DA neurons (Ungless et al., 2001, Saal et al., 2003). Such changes in LTP are typically measured by recording AMPA/NMDA ratios, as enhanced AMPAR function is often the driver of LTP in VTA (Wolf et al., 2004). These changes in synaptic plasticity underlie important drug-induced behavioral changes, including behavioral sensitization, reward memories, and self-administration. With regard to nicotine exposure, it is critical to determine which nAChR subtype(s) mediate this effect. Doing so could lead to novel therapeutic approaches for treating nicotine addiction and/or addiction to other drugs of abuse.
Our results in α6L9S mice indicate that systemic administration of α6-selective doses of nicotine (0.03 mg/kg; Fig. 7B and C) is sufficient to enhance AMPAR function on VTA DA neurons. This result was directly ascribed to α6* nAChRs, as αCtxMII infusion into VTA blocked the effect of systemic nicotine (Fig. 8B and C). These data are consistent with our previous results. When naïve brain slices containing VTA were treated with nicotine, AMPAR function (Engle et al., 2013) and AMPA/NMDA ratios (Engle et al., 2015) were enhanced via activation of α6* nAChRs. These prior studies also demonstrated, in naïve slices, that α4 subunits and NMDAR activity are also required for α6*-mediated increases in AMPAR activity (Engle et al., 2013). This enhanced AMPAR activity was due to an increase in the potency of agonists at the AMPAR, not because such agonists had increased efficacy (Engle et al., 2013). Ca2+/calmodulin kinase II activity was also required for this effect (unpublished observations), but α7 nAChR activity was not (Engle et al., 2013). Future studies on in vivo α6* nAChR activation signaling in VTA DA neurons could help to determine whether the same signaling mechanisms operating in naïve slice experiments also drive the effects we report here. Taken together, our results isolating α6* nAChR activity in VTA using local infusion of αCtxMII reinforce the notion that the VTA is a critical site for the cellular changes induced by nicotine exposure.
4.4. Conclusions
In summary, using behavioral and cellular measures of α6* nAChR function, we have demonstrated that the VTA is a key site of action for nicotine at α6* nAChRs. Our results also show that varenicline is capable of strongly modulating α6* nAChR activity, whereas ABT-089 has lower-potency at these receptors. Given the ability of varenicline to foster smoking cessation, further research should be done to discriminate its actions at α4β2 versus α6* nAChRs. Finally, these results highlight the potential utility of the α6L9S mouse model, particularly its ligand-activated locomotor phenotype, in future drug discovery efforts targeting the α6* nAChR.
Highlights.
A transgenic mouse strain (α6L9S) expressing hypersensitive α6* nAChRs was studied
VTA α6* nAChRs are required for nicotine-induced locomotion in α6L9S mice
Intra-VTA nicotine recapitulates effects of systemic nicotine on locomotion
nAChR partial agonists also activate locomotion in α6L9S mice
Nicotine-mediated synaptic plasticity changes require VTA α6* nAChRs
Acknowledgments
This work was supported by grants from the National Institutes of Health (DA030396 and DA035942 to Ryan M. Drenan, and GM103801 and GM48677 to J. Michael McIntosh). Staci Engle was supported by fellowships/awards from Purdue University (Frederick N. Andrews Fellowship, John Davisson Endowment Research Award). We thank members of the Drenan laboratory for helpful technical assistance and discussion. ABT-089 was a generous gift from Abbvie.
Abbreviations
- αCtxMII
α-conotoxin MII
- ACh
acetylcholine
- AMPA
α-amino-3-hydroxy-5-methyl-isoxazolepropionic acid
- AMPAR
α-amino-3-hydroxy-5-methyl-isoxazolepropionic acid receptor
- DA
dopamine
- NAc
nucleus accumbens
- NAcs
nucleus accumbens shell
- nAChR
nicotinic acetylcholine receptor
- SNc
substantia nigra pars compacta
- VTA
ventral tegmental area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Azam L, McIntosh JM. Characterization of nicotinic acetylcholine receptors that modulate nicotine-evoked [3H]norepinephrine release from mouse hippocampal synaptosomes. Mol Pharmacol. 2006;70:967–976. doi: 10.1124/mol.106.024513. [DOI] [PubMed] [Google Scholar]
- Azam L, Winzer-Serhan UH, Chen Y, Leslie FM. Expression of neuronal nicotinic acetylcholine receptor subunit mRNAs within midbrain dopamine neurons. J Comp Neurol. 2002;444:260–274. doi: 10.1002/cne.10138. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Devillers-Thiery A, Revah F, Galzi JL, Hussy N, Mulle C, Bertrand S, Ballivet M, Changeux JP. Unconventional pharmacology of a neuronal nicotinic receptor mutated in the channel domain. Proc Natl Acad Sci U S A. 1992;89:1261–1265. doi: 10.1073/pnas.89.4.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolam JP, Wainer BH, Smith AD. Characterization of cholinergic neurons in the rat neostriatum. A combination of choline acetyltransferase immunocytochemistry, Golgi-impregnation and electron microscopy. Neuroscience. 1984;12:711–718. doi: 10.1016/0306-4522(84)90165-9. [DOI] [PubMed] [Google Scholar]
- Bordia T, Hrachova M, Chin M, McIntosh JM, Quik M. Varenicline is a potent partial agonist at α6β2* nicotinic acetylcholine receptors in rat and monkey striatum. J Pharmacol Exp Ther. 2012;342:327–334. doi: 10.1124/jpet.112.194852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunzell DH, Boschen KE, Hendrick ES, Beardsley PM, McIntosh JM. α-Conotoxin MII-sensitive nicotinic acetylcholine receptors in the nucleus accumbens shell regulate progressive ratio responding maintained by nicotine. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2009;35:665–673. doi: 10.1038/npp.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachope R, Mateo Y, Mathur BN, Irving J, Wang HL, Morales M, Lovinger DM, Cheer JF. Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep. 2012;2:33–41. doi: 10.1016/j.celrep.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, McIntosh JM. A new α-conotoxin which targets α3β2 nicotinic acetylcholine receptors. J Biol Chem. 1996;271:7522–7528. doi: 10.1074/jbc.271.13.7522. [DOI] [PubMed] [Google Scholar]
- Champtiaux N, Han ZY, Bessis A, Rossi FM, Zoli M, Marubio L, McIntosh JM, Changeux JP. Distribution and pharmacology of α6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J Neurosci. 2002;22:1208–1217. doi: 10.1523/JNEUROSCI.22-04-01208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J, Sands SB, Davis TI, Lebel LA, Fox CB, Shrikhande A, Heym JH, Schaeffer E, Rollema H, Lu Y, Mansbach RS, Chambers LK, Rovetti CC, Schulz DW, Tingley FD, 3rd, O’Neill BT. Varenicline: an α4β2 nicotinic receptor partial agonist for smoking cessation. Journal of medicinal chemistry. 2005;48:3474–3477. doi: 10.1021/jm050069n. [DOI] [PubMed] [Google Scholar]
- Cohen BN, Mackey ED, Grady SR, McKinney S, Patzlaff NE, Wageman CR, McIntosh JM, Marks MJ, Lester HA, Drenan RM. Nicotinic cholinergic mechanisms causing elevated dopamine release and abnormal locomotor behavior. Neuroscience. 2012;200:31–41. doi: 10.1016/j.neuroscience.2011.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker MW, Bannon AW, Curzon P, Gunther KL, Brioni JD, Holladay MW, Lin NH, Li Y, Daanen JF, Buccafusco JJ, Prendergast MA, Jackson WJ, Arneric SP. ABT-089 [2-methyl-3-(2-(S)-pyrrolidinylmethoxy)pyridine dihydrochloride]: II. A novel cholinergic channel modulator with effects on cognitive performance in rats and monkeys. J Pharmacol Exp Ther. 1997;283:247–258. [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Steele AD, McKinney S, Patzlaff NE, McIntosh JM, Marks MJ, Miwa JM, Lester HA. Cholinergic modulation of locomotion and striatal dopamine release is mediated by α6α4* nicotinic acetylcholine receptors. J Neurosci. 2010;30:9877–9889. doi: 10.1523/JNEUROSCI.2056-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Whiteaker P, McClure-Begley T, McKinney S, Miwa JM, Bupp S, Heintz N, McIntosh JM, Bencherif M, Marks MJ, Lester HA. In vivo activation of midbrain dopamine neurons via sensitized, high-affinity α6* nicotinic acetylcholine receptors. Neuron. 2008a;60:123–136. doi: 10.1016/j.neuron.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Lester HA. Insights into the neurobiology of the nicotinic cholinergic system and nicotine addiction from mice expressing nicotinic receptors harboring gain-of-function mutations. Pharmacol Rev. 2012;64:869–879. doi: 10.1124/pr.111.004671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Nashmi R, Imoukhuede P, Just H, McKinney S, Lester HA. Subcellular trafficking, pentameric assembly, and subunit stoichiometry of neuronal nicotinic acetylcholine receptors containing fluorescently labeled α6 and β3 subunits. Mol Pharmacol. 2008b;73:27–41. doi: 10.1124/mol.107.039180. [DOI] [PubMed] [Google Scholar]
- Engle SE, Broderick HJ, Drenan RM. Local application of drugs to study nicotinic acetylcholine receptor function in mouse brain slices. J Vis Exp. 2012:e50034. doi: 10.3791/50034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle SE, McIntosh JM, Drenan RM. Nicotine and ethanol cooperate to enhance ventral tegmental area AMPA receptor function via α6-containing nicotinic receptors. Neuropharmacology. 2015;91:13–22. doi: 10.1016/j.neuropharm.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle SE, Shih PY, McIntosh JM, Drenan RM. α4α6β2* nicotinic acetylcholine receptor activation on ventral tegmental area dopamine neurons is sufficient to stimulate a depolarizing conductance and enhance surface AMPA receptor function. Mol Pharmacol. 2013;84:393–406. doi: 10.1124/mol.113.087346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley R, Cragg SJ. Presynaptic nicotinic receptors: a dynamic and diverse cholinergic filter of striatal dopamine neurotransmission. Br J Pharmacol. 2008;153(Suppl 1):S283–297. doi: 10.1038/sj.bjp.0707510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley R, Maubourguet N, David V, Eddine R, Evrard A, Pons S, Marti F, Threlfell S, Cazala P, McIntosh JM, Changeux JP, Maskos U, Cragg SJ, Faure P. Distinct contributions of nicotinic acetylcholine receptor subunit α4 and subunit α6 to the reinforcing effects of nicotine. Proc Natl Acad Sci U S A. 2011;108:7577–7582. doi: 10.1073/pnas.1103000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Jin Y, Yang K, Zhang D, Lukas RJ, Wu J. Mechanisms involved in systemic nicotine-induced glutamatergic synaptic plasticity on dopamine neurons in the ventral tegmental area. J Neurosci. 2010;30:13814–13825. doi: 10.1523/JNEUROSCI.1943-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Guiducci S, Tedesco V, Corbioli S, Zanetti L, Moretti M, Zanardi A, Rimondini R, Mugnaini M, Clementi F, Chiamulera C, Zoli M. Nicotinic acetylcholine receptors in the mesolimbic pathway: primary role of ventral tegmental area α6β2* receptors in mediating systemic nicotine effects on dopamine release, locomotion, and reinforcement. J Neurosci. 2010;30:5311–5325. doi: 10.1523/JNEUROSCI.5095-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Drenan RM, Breining SR, Yohannes D, Wageman CR, Fedorov NB, McKinney S, Whiteaker P, Bencherif M, Lester HA, Marks MJ. Structural differences determine the relative selectivity of nicotinic compounds for native α4β2*-, α6β2*-, α3β4*- and α7-nicotine acetylcholine receptors. Neuropharmacology. 2010;58:1054–1066. doi: 10.1016/j.neuropharm.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson BJ, Srinivasan R, Nichols WA, Dilworth CN, Gutierrez DF, Mackey ED, McKinney S, Drenan RM, Richards CI, Lester HA. Nicotine exploits a COPI-mediated process for chaperone-mediated up-regulation of its receptors. The Journal of general physiology. 2014;143:51–66. doi: 10.1085/jgp.201311102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Yang K, Wang H, Wu J. Exposure of nicotine to ventral tegmental area slices induces glutamatergic synaptic plasticity on dopamine neurons. Synapse. 2011;65:332–338. doi: 10.1002/syn.20850. [DOI] [PubMed] [Google Scholar]
- le Novere N, Zoli M, Lena C, Ferrari R, Picciotto MR, Merlo-Pich E, Changeux JP. Involvement of α6 nicotinic receptor subunit in nicotine-elicited locomotion, demonstrated by in vivo antisense oligonucleotide infusion. Neuroreport. 1999;10:2497–2501. doi: 10.1097/00001756-199908200-00012. [DOI] [PubMed] [Google Scholar]
- Lee AM, Arreola AC, Kimmey BA, Schmidt HD. Administration of the nicotinic acetylcholine receptor agonists ABT-089 and ABT-107 attenuates the reinstatement of nicotine-seeking behavior in rats. Behavioural brain research. 2014;274:168–175. doi: 10.1016/j.bbr.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin NH, Gunn DE, Ryther KB, Garvey DS, Donnelly-Roberts DL, Decker MW, Brioni JD, Buckley MJ, Rodrigues AD, Marsh KG, Anderson DJ, Buccafusco JJ, Prendergast MA, Sullivan JP, Williams M, Arneric SP, Holladay MW. Structure-activity studies on 2-methyl-3-(2(S)-pyrrolidinylmethoxy) pyridine (ABT-089): an orally bioavailable 3-pyridyl ether nicotinic acetylcholine receptor ligand with cognition-enhancing properties. Journal of medicinal chemistry. 1997;40:385–390. doi: 10.1021/jm960233u. [DOI] [PubMed] [Google Scholar]
- Mackey ED, Engle SE, Kim MR, O’Neill HC, Wageman CR, Patzlaff NE, Wang Y, Grady SR, McIntosh JM, Marks MJ, Lester HA, Drenan RM. α6* Nicotinic Acetylcholine Receptor Expression and Function in a Visual Salience Circuit. J Neurosci. 2012;32:10226–10237. doi: 10.1523/JNEUROSCI.0007-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli-Engvall M, Evrard A, Pons S, Maskos U, Svensson TH, Changeux JP, Faure P. Hierarchical control of dopamine neuron-firing patterns by nicotinic receptors. Neuron. 2006;50:911–921. doi: 10.1016/j.neuron.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Mao D, Gallagher K, McGehee DS. Nicotine potentiation of excitatory inputs to ventral tegmental area dopamine neurons. J Neurosci. 2011;31:6710–6720. doi: 10.1523/JNEUROSCI.5671-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Wageman CR, Grady SR, Gopalakrishnan M, Briggs CA. Selectivity of ABT-089 for α4β2* and α6β2* nicotinic acetylcholine receptors in brain. Biochemical pharmacology. 2009;78:795–802. doi: 10.1016/j.bcp.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux JP, Evrard A, Cazala P, Cormier A, Mameli-Engvall M, Dufour N, Cloez-Tayarani I, Bemelmans AP, Mallet J, Gardier AM, David V, Faure P, Granon S, Changeux JP. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436:103–107. doi: 10.1038/nature03694. [DOI] [PubMed] [Google Scholar]
- Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, Gardiner PS, Grady SR, Heberlein U, Leonard SS, Levin ED, Lukas RJ, Markou A, Marks MJ, McCallum SE, Parameswaran N, Perkins KA, Picciotto MR, Quik M, Rose JE, Rothenfluh A, Schafer WR, Stolerman IP, Tyndale RF, Wehner JM, Zirger JM. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology. 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- McIntosh JM, Azam L, Staheli S, Dowell C, Lindstrom JM, Kuryatov A, Garrett JE, Marks MJ, Whiteaker P. Analogs of α-Conotoxin MII are selective for α6-containing nicotinic acetylcholine receptors. Mol Pharmacol. 2004;65:944–952. doi: 10.1124/mol.65.4.944. [DOI] [PubMed] [Google Scholar]
- Mehtali M, LeMeur M, Lathe R. The methylation-free status of a housekeeping transgene is lost at high copy number. Gene. 1990;91:179–184. doi: 10.1016/0378-1119(90)90086-7. [DOI] [PubMed] [Google Scholar]
- Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, Huang Q, McClure-Begley T, Lindstrom JM, Labarca C, Collins AC, Marks MJ, Lester HA. Chronic nicotine cell specifically upregulates functional α4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci. 2007;27:8202–8218. doi: 10.1523/JNEUROSCI.2199-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obach RS, Reed-Hagen AE, Krueger SS, Obach BJ, O’Connell TN, Zandi KS, Miller S, Coe JW. Metabolism and disposition of varenicline, a selective α4β2 acetylcholine receptor partial agonist, in vivo and in vitro. Drug metabolism and disposition: the biological fate of chemicals. 2006;34:121–130. doi: 10.1124/dmd.105.006767. [DOI] [PubMed] [Google Scholar]
- Pisani A, Bernardi G, Ding J, Surmeier DJ. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 2007;30:545–553. doi: 10.1016/j.tins.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Pons S, Fattore L, Cossu G, Tolu S, Porcu E, McIntosh JM, Changeux JP, Maskos U, Fratta W. Crucial role of α4 and α6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J Neurosci. 2008;28:12318–12327. doi: 10.1523/JNEUROSCI.3918-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers MS, Broderick HJ, Drenan RM, Chester JA. Nicotinic acetylcholine receptors containing α6 subunits contribute to alcohol reward-related behaviours. Genes Brain Behav. 2013;12:543–553. doi: 10.1111/gbb.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast MA, Jackson WJ, Terry AV, Jr, Decker MW, Arneric SP, Buccafusco JJ. Central nicotinic receptor agonists ABT-418, ABT-089, and (−)-nicotine reduce distractibility in adult monkeys. Psychopharmacology. 1998;136:50–58. doi: 10.1007/s002130050538. [DOI] [PubMed] [Google Scholar]
- Quik M, McIntosh JM. Striatal α6* nicotinic acetylcholine receptors: potential targets for Parkinson’s disease therapy. J Pharmacol Exp Ther. 2006;316:481–489. doi: 10.1124/jpet.105.094375. [DOI] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ. Nicotine amplifies reward-related dopamine signals in striatum. Nature neuroscience. 2004;7:583–584. doi: 10.1038/nn1244. [DOI] [PubMed] [Google Scholar]
- Rollema H, Chambers LK, Coe JW, Glowa J, Hurst RS, Lebel LA, Lu Y, Mansbach RS, Mather RJ, Rovetti CC, Sands SB, Schaeffer E, Schulz DW, Tingley FD, 3rd, Williams KE. Pharmacological profile of the α4β2 nicotinic acetylcholine receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacology. 2007;52:985–994. doi: 10.1016/j.neuropharm.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–582. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- Salminen O, Drapeau JA, McIntosh JM, Collins AC, Marks MJ, Grady SR. Pharmacology of α-Conotoxin MII-Sensitive Subtypes of Nicotinic Acetylcholine Receptors Isolated by Breeding of Null Mutant Mice. Mol Pharmacol. 2007;71:1563–1571. doi: 10.1124/mol.106.031492. [DOI] [PubMed] [Google Scholar]
- Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, Grady SR. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol. 2004;65:1526–1535. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- Shih PY, Engle SE, Oh G, Deshpande P, Puskar NL, Lester HA, Drenan RM. Differential expression and function of nicotinic acetylcholine receptors in subdivisions of medial habenula. J Neurosci. 2014;34:9789–9802. doi: 10.1523/JNEUROSCI.0476-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Marks MJ, Lester HA. Nicotine responses in hypersensitive and knockout α4 mice account for tolerance to both hypothermia and locomotor suppression in wild-type mice. Physiol Genomics. 2007;31:422–428. doi: 10.1152/physiolgenomics.00063.2007. [DOI] [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA. Nicotine activation of α4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Wang Y, Lee JW, Oh G, Grady SR, McIntosh JM, Brunzell DH, Cannon JR, Drenan RM. Enhanced synthesis and release of dopamine in transgenic mice with gain-of-function α 6* nAChRs. J Neurochem. 2013;129:315–327. doi: 10.1111/jnc.12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieskopf JS, Mathur J, Limapichat W, Post MR, Al-Qazzaz M, Sorge RE, Martin LJ, Zaykin DV, Smith SB, Freitas K, Austin JS, Dai F, Zhang J, Marcovitz J, Tuttle AH, Slepian PM, Clarke S, Drenan RM, Janes J, Al Sharari S, Segall SK, Aasvang EK, Lai W, Bittner R, Richards CI, Slade GD, Kehlet H, Walker J, Maskos U, Changeux JP, Devor M, Maixner W, Diatchenko L, Belfer I, Dougherty DA, Su AI, Lummis SC, Imad Damaj M, Lester HA, Patapoutian A, Mogil JS. The nicotinic alpha6 subunit gene determines variability in chronic pain sensitivity via cross-inhibition of P2X2/3 receptors. Science translational medicine. 2015;7:287ra272. doi: 10.1126/scitranslmed.3009986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilens TE, Verlinden MH, Adler LA, Wozniak PJ, West SA. ABT-089, a neuronal nicotinic receptor partial agonist, for the treatment of attention-deficit/hyperactivity disorder in adults: results of a pilot study. Biological psychiatry. 2006;59:1065–1070. doi: 10.1016/j.biopsych.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Wolf ME, Sun X, Mangiavacchi S, Chao SZ. Psychomotor stimulants and neuronal plasticity. Neuropharmacology. 2004;47(Suppl 1):61–79. doi: 10.1016/j.neuropharm.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Woolf NJ, Butcher LL. Cholinergic neurons in the caudate-putamen complex proper are intrinsically organized: a combined Evans blue and acetylcholinesterase analysis. Brain research bulletin. 1981;7:487–507. doi: 10.1016/0361-9230(81)90004-6. [DOI] [PubMed] [Google Scholar]
- Woolf NJ, Butcher LL. Cholinergic systems in the rat brain: III. Projections from the pontomesencephalic tegmentum to the thalamus, tectum, basal ganglia, and basal forebrain. Brain research bulletin. 1986;16:603–637. doi: 10.1016/0361-9230(86)90134-6. [DOI] [PubMed] [Google Scholar]
- Zhang D, Bordia T, McGregor M, McIntosh JM, Decker MW, Quik M. ABT-089 and ABT-894 reduce levodopa-induced dyskinesias in a monkey model of Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society. 2014;29:508–517. doi: 10.1002/mds.25817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sulzer D. Frequency-dependent modulation of dopamine release by nicotine. Nature neuroscience. 2004;7:581–582. doi: 10.1038/nn1243. [DOI] [PubMed] [Google Scholar]