

Abstract
Objective:
To evaluate the expression of activating and inhibitory Fc-gamma receptors (FcγRs) before and during clinically effective therapy with IV immunoglobulin (IVIg) in patients with chronic inflammatory demyelinating polyneuropathy (CIDP).
Methods:
Peripheral blood leukocyte subsets, including classical CD14highCD16− and nonclassical inflammatory CD14lowCD16+ monocytes as well as naive CD19+CD27− and memory CD19+CD27+ B cells, were obtained at baseline and monitored at 2 and 4–8 weeks after initiation of IVIg therapy.
Results:
Compared with healthy donors matched by age and sex, patients with CIDP showed increased expression levels of the activating high-affinity FcγR1 on CD14highCD16− (p < 0.001) and CD14lowCD16+ monocytes (p < 0.001). Expression of the activating low-affinity FcγRIIA was increased on CD14lowCD16+ monocytes (p = 0.023). Conversely, expression of the inhibitory FcγRIIB was reduced on naive (p = 0.009) and memory (p = 0.002) B cells as well as on CD14highCD16− monocytes (p = 0.046). Clinically effective IVIg therapy partially restored deregulated FcγR expression on B cell subsets and monocytes.
Conclusions:
The FcγR regulatory system is disturbed in patients with CIDP. Balancing activating vs inhibitory FcγR expression might provide a clinical benefit for patients with CIDP.
Humoral immune responses, which link innate and adaptive immunity, are believed to play a central role in mediating peripheral nerve injury and represent important therapeutic targets in chronic inflammatory demyelinating polyneuropathy (CIDP). Both sera and immunoglobulin G (IgG) molecules from patients with CIDP induce peripheral demyelination in susceptible animals1 and inhibit nerve conduction in several models of peripheral neuropathies.2 Plasma exchange therapy, i.e., removal of humoral immune factors, as well as IV immunoglobulin (IVIg) are first-line therapies in patients with CIDP.3,4
IgG-mediated effector functions are mediated through interaction of the antibodies' Fc fragment with cellular Fc-gamma receptors (FcγRs) expressed by innate immune cells and B lymphocytes.5,6 The family of FcγRs consists of several activating members (FcγRIA, IIA, IIC, and IIIA in humans) and one inhibitory member (FcγRIIB). FcγR-mediated effector functions are determined by signals derived from both activating and inhibitory FcγRs since both classes of receptors are expressed by most hematopoietic cells.5–8 We previously reported that compared with demographically matched healthy controls, patients with CIDP showed lower FcγRIIB expression levels on B cells and upregulated FcγRIIB expression on CD19+ B cells and, in some patients, on CD14+ monocytes and following IVIg treatment.9 Here, we investigated the expression profile of activating and inhibitory FcγRs in patients with CIDP before and during IVIg therapy.
METHODS
Patients.
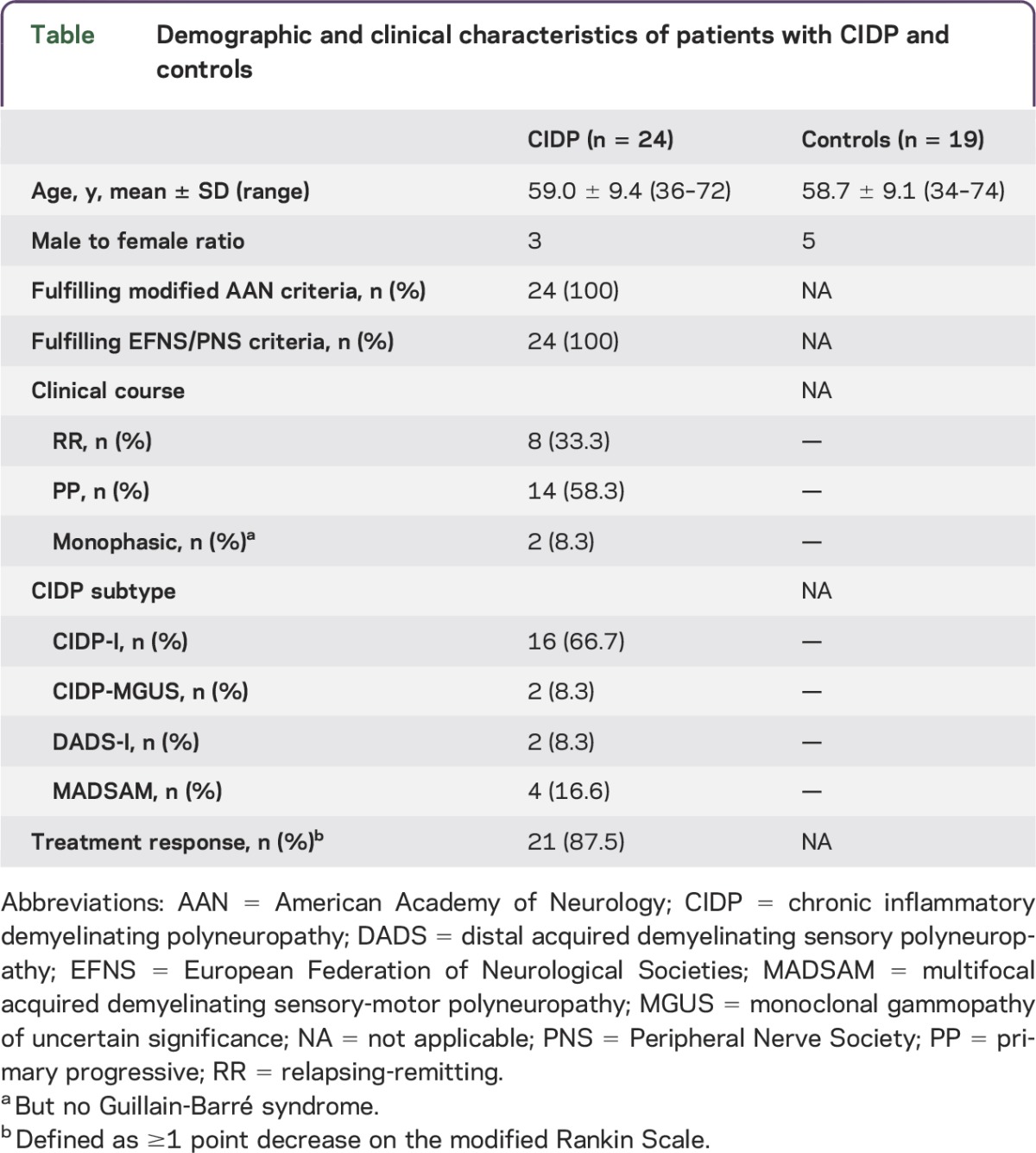
Patients with CIDP (n = 24) and healthy controls matched by age and sex (n = 19) were recruited between 2010 and 2013 at the Department of Neurology, University of Marburg, Germany (table). All patients fulfilled the European Federation of Neurological Societies/Peripheral Nerve Society diagnostic criteria for CIDP. They were prospectively followed during IVIg therapy (2 g/kg over 5 days) using the modified Rankin Scale to monitor clinical treatment response.9,10 Improvement on the modified Rankin Scale within 4 weeks after IVIg treatment was defined as a positive treatment response. Patients did not receive IVIg therapy or immunosuppressive treatment including corticosteroids before study entry. Peripheral blood mononuclear cell samples were collected at each visit and obtained before, 2 weeks after, and 8 weeks after treatment with IVIg.
Table.
Demographic and clinical characteristics of patients with CIDP and controls
Standard protocol approvals, registrations, and patient consents.
The university's institutional review board approved the study according to the Declaration of Helsinki, good clinical practice, and German law (file reference 46/00).
Flow cytometry.
FcγR expression on naive and memory B cells as well as on classical and nonclassical inflammatory monocyte subsets was quantified by flow cytometry. Fluorochrome-labeled antibodies were purchased from BioLegend (San Diego, CA; CD3-PE-Cy5 [clone HIT3a], HLA-DR-Pacific Blue [L243], CD19-AlexaFluor700 [HIB19], CD64-APC-Cy7 [10.1]), BD Biosciences Pharmingen (San Diego, CA; CD11c-PE-Cy7 [B-Ly6], CD27-PE [M-T271]), STEMCELL Technologies (Vancouver, British Columbia, Canada; CD32A-FITC [IV.3]), eBioscience (San Diego, CA; CD16-e-fluor605 [eBioCB16], CD123-PerCPCy5.5 [6H6]), Invitrogen (Waltham, MA; CD14-QDot655 [TüK4], CD56-PE-Texas-Red [MEM-188]), and Miltenyi Biotec (Bergisch Gladbach, Germany; BDCA-1-PE [AD5-14H12], BDCA-3-PE [AD5-8E7]). The CD32B (FcγRIIB)-specific monoclonal antibody clone 2B6 was in-house purified and coupled to Alexa Fluor 647. Dead cells were excluded using Fixable Aqua Dead Cell Stain Kit (Invitrogen). Cells were suspended in phosphate-buffered saline +0.01% sodium azide (fluorescence-activated cell sorting [FACS] buffer) containing the fluorochrome-labeled antibodies and incubated for 30 minutes on ice. Cells were washed, suspended in FACS buffer, and acquired using BD LSR Fortessa. All analysis was performed with FlowJo 9 (Tree Star Inc., Ashland, OR).
Statistics.
Statistical data analysis for cross-sectional studies was performed using the Mann-Whitney U test. The Wilcoxon signed rank test was used for longitudinal analyses. p Values less than 0.05 were considered statistically significant.
RESULTS
Deregulated FcγR expression in patients with CIDP.
Expression of the activating FcγRI and FcγRIIA and the inhibitory FcγRIIB was determined on circulating CD19+CD27− naive and CD19+CD27+ memory B cells as well as on classical CD14highCD16− and inflammatory CD14lowCD16+ monocytes in untreated patients with CIDP and demographically matched healthy blood donors (table). No statistically significant differences were detectable in the frequencies of the aforementioned immune cell subsets. Consistent with earlier reports,9 patients with CIDP showed reduced expression levels of FcγRIIB on B cells (figure 1A). The reduction in FcγRIIB expression was stronger in the CD19+CD27+ memory (p = 0.002) compared with the CD19+CD27− naive (p = 0.009) B cell compartment due to a failure of patients with CIDP to upregulate or maintain upregulation of FcγRIIB as B cells become memory cells. FcγRIIB expression was also reduced in classical CD14+CD16− monocytes (p = 0.046) and tended to be lower in CD16+ monocytes (p = 0.216). Expression of the high-affinity activating FcγRI (CD64), not expressed on B cells, was increased in both monocyte subsets in patients with CIDP (figure 1B). Expression of the low-affinity FcγRIIA was increased on CD14lowCD16+ monocytes. Thus, expression levels of inhibitory vs activating FcγRs are deregulated in untreated patients with CIDP.
Figure 1. Deregulated inhibitory and activating FcγR expression in CIDP.

Peripheral blood mononuclear cells of patients with chronic inflammatory demyelinating polyneuropathy (CIDP) and age-matched healthy donors (HD) were stained with fluorochrome-coupled antibodies. Fc-gamma receptor (FcγR) expression levels were measured on B cell and monocyte subsets by flow cytometry. (A) Median fluorescence intensity (MFI) of FcγRIIB on naïve (CD27−) and memory (CD27+) B cells, and classical (CD14highCD16−) and nonclassical inflammatory (CD14lowCD16+) monocytes. (B) FcγRI and (C) FcγRIIa MFI on classical (CD14highCD16−) and inflammatory (CD14lowCD16+) monocytes. Statistics were performed using the Mann-Whitney U test.
Effect of IVIg therapy on deregulated FcγR expression.
Studies in several mouse autoimmune disease models demonstrated that the anti-inflammatory activity of IVIg requires the presence and upregulation of the inhibitory FcγRIIB.11–13 Here, we analyzed expression levels of FcγRIIB compared with the activating FcγRI and FcγRIIA in patients with CIDP before as well as 2 and 4–8 weeks following IVIg therapy. The clinical response rate to IVIg therapy was 87.5%, as defined by an improvement in disability within 4 weeks after IVIg therapy.10,14 FcγIIB expression was induced on both naive and memory B cells 2 weeks after IVIg therapy (p = 0.007 and 0.004, respectively) (figure 2A). After 4–8 weeks, no further induction of FcγRIIB expression on B cells was observed for most patients (naive B cells: further induction in 7/20 [35.0%]; memory B cells: further induction in 5/17 [29.4%]), but levels remained significant compared with baseline values (p = 0.002 and 0.047 for naive and memory B cells, respectively). None of the patients with a clinical response to the first dose of IVIg (2 g/kg for 2–3 days) deteriorated before initiation of maintenance therapy (1 g/kg for 1–2 days), which was started 4–8 weeks after the first dose of IVIg, suggesting that sustained upregulation of FcγRIIB after 4–8 weeks is associated with a clinical response to IVIg therapy. Three patients did not show improvement on the modified Rankin Scale 4 weeks after the first course of IVIg therapy. These patients received a second course of IVIg therapy based on observations that some patients require at least 2 courses of IVIg to determine whether they are responding to treatment.13,15 Indeed, these patients improved after a second course of IVIg therapy and thereafter received long-term maintenance therapy. FcγIIB expression was also upregulated on naive and memory B cells in those patients with an initially poor response to IVIg (figure 2). On monocyte subsets, FcγRIIB expression was upregulated in some patients (CD14highCD16−: 16/23 [69.6%] after 2 weeks, 15/22 [68.2%] after 4–8 weeks; CD14lowCD16+: 10/19 [52.6%] after 2 weeks, 9/20 [45.0%] after 4–8 weeks), but the overall difference was not statistically significant (figure 2A). IVIg therapy was associated with a transient downregulation of the activating FcγRI on inflammatory CD14lowCD16+ monocytes after 2 weeks (p < 0.001) (figure 2B). After 4–8 weeks, FcγRI levels did not differ significantly from baseline values (p = 0.18). Monocyte expression levels of the low-affinity activating FcγRIIA were not significantly regulated by IVIg therapy (figure 2C).
Figure 2. IVIg therapy partially restores FcγR expression on B cells and monocytes.

Flow cytometry analysis of Fc-gamma receptor (FcγR) expression on peripheral blood mononuclear cells of treatment-naïve patients with chronic inflammatory demyelinating polyneuropathy (CIDP) as well as 2 and 4–8 weeks after a single dose of IV immunoglobulin (IVIg) (2 g/kg) treatment. Data points for individual patients are connected by lines. Three patients did not show improvement on the modified Rankin Scale 4 weeks after the first course of IVIg therapy (red lines) but improved after a second course of IVIg and thereafter received long-term maintenance therapy. For some patients not all data points were available. Changes of FcγRIIB (A) as well as FcγRI (B) and FcγRIIA (C) median fluorescence intensity following IVIg treatment are shown relative to baseline (untreated). Statistics were performed using the Wilcoxon signed rank test.
DISCUSSION
Balanced signaling through activating and inhibitory FcγRs regulates innate and adaptive immune responses, and impairment of the FcγR regulatory system is associated with an increased susceptibility to autoimmune disease.5 Mice deficient in activating FcγR signaling are resistant to the induction of various autoimmune disease models. Mice lacking the inhibitory FcγRIIB spontaneously develop autoimmune disorders,16 and restoration of decreased FcγRIIB expression on activated B cells in autoimmune-susceptible mice restores immunologic tolerance.6 We found that patients with CIDP show increased expression levels of the activating FcγRI and FcγRIIA on monocytes but decreased expression levels of the inhibitory FcγRIIB on monocytes and B cells. IVIg therapy partially restored deregulated FcγR expression levels.
Nonfunctional FcγRIIB variants or decreased FcγRIIB expression have been shown to be associated with the development and severity of systemic lupus erythematosus (SLE).6,17 In line with our previous study in an independent cohort of patients with CIDP, we found that untreated patients with CIDP show lower FcγRIIB expression levels on naive and memory B cells and fail to upregulate or maintain upregulation of FcγRIIB as B cells progressed from the naive to the memory compartment.9 In addition, monocyte expression of FcγRIIB is impaired in patients with CIDP. A potential explanation for the lower expression levels of FcγRIIB is functionally relevant single nucleotide polymorphisms in the FcγRIIB promoter, which are associated with autoimmune phenotypes9,18–20 and increased in frequency in patients with CIDP.9
Increased monocyte expression of the activating FcγRI was recently identified as a potential biomarker for disease activity in patients with SLE and rheumatoid arthritis (RA), in whom monocyte expression levels of FcγRI are significantly higher in active disease compared with inactive disease.21 Our data indicate that deregulated monocyte expression of FcγRI is not confined to patients with SLE and RA. FcγRI expression is induced by interferon-α (IFN-α), among other cytokines, and increased FcγRI levels on monocytes derived from patients with SLE were attributed to higher IFN-α production in these patients.21 A role for IFN-α in the pathogenesis of CIDP is less well established, although some studies report increased levels of type I IFN signaling in patients with CIDP22 or development of CIDP associated with IFN-α therapy.23–29 Activation of FcγRI on monocytes triggers differentiation into immature dendritic cells that induce autoreactive T cell responses and has therefore been implicated in mediating tissue injury in antibody-mediated autoimmune diseases.30 FcγRI-mediated activation and differentiation of myeloid cells might also contribute to peripheral nerve damage in CIDP, where myeloid cells are believed to be the main local effector cells.2
The analysis of concomitant expression of activating and inhibitory FcγRs indicates that the FcγR regulatory system, rather than expression levels of a single receptor, is disturbed in patients with CIDP. Altogether, these data suggest that deregulated expression of activating vs inhibitory FcγR expression might lower the activation threshold for myeloid cells and B cells,7,8 thereby contributing to susceptibility to CIDP.5
In animal models of immune thrombocytopenic purpura, RA, and nephrotoxic nephritis, IVIg administration results in an upregulation of FcγRIIB surface expression on effector macrophages or enhanced recruitment of FcγRIIB-expressing myeloid cells at the site of inflammation in vivo.11,31,32 Moreover, the clinical efficacy of IVIg therapy was shown to be dependent on the presence of the inhibitory FcγRIIB.31 Our data suggest that restoration of deregulated activating vs inhibitory FcγR expression might contribute to the clinical efficacy of IVIg in patients with CIDP. Notably, upregulation of the inhibitory FcγRIIB on B cells and downmodulation of the activating FcγRI on CD14lowCD16+ inflammatory monocytes were stronger after 2 weeks compared with 4–8 weeks in most patients, suggesting that a single treatment course of IVIg does not lead to sustained restoration of FcγR expression levels in patients with CIDP.
There are limitations to our study. First, due to the relatively small number of patients and the high response rate to IVIg therapy, patients were not stratified for disease severity and treatment response. FcγR expression profiling could potentially assist in the development of biomarkers indicative of disease activity and treatment response,33 but this requires validation in larger independent cohorts. Second, we did not monitor FcγR levels beyond 8 weeks following therapy. Repeated infusions of IVIg are generally necessary to maintain a clinical benefit,4,15 and it is tempting to speculate that FcγR expression levels might return to pretreatment levels beyond an observation period of 8 weeks. Nevertheless, our findings should provide incentive to conduct larger prospective investigations to examine FcγR expression in leukocyte subsets in patients with CIDP.
Our study provides evidence for a deregulated FcγR expression profile in treatment-naive patients with CIDP that might contribute to disease susceptibility. The validity of FcγR expression profiling as a biomarker for disease progression and treatment response remains to be evaluated. Therapeutic strategies that aim at balancing deregulated FcγR expression levels might provide a clinical benefit for patients with CIDP.
ACKNOWLEDGMENT
The authors thank the patients for their continuous cooperation. I.Q. was supported by a scholarship provided by the Austrian Academy of Sciences. F.N. is supported by the Deutsche Forschungsgemeinschaft (DFG-TRR130).
GLOSSARY
- CIDP
chronic inflammatory demyelinating polyneuropathy
- FcγR
Fc-gamma receptor
- FACS
fluorescence-activated cell sorting
- IFN
interferon
- IgG
immunoglobulin G
- IVIg
IV immunoglobulin
- RA
rheumatoid arthritis
- SLE
systemic lupus erythematosus
AUTHOR CONTRIBUTIONS
Isaak Quast: study concept and design, acquisition of data, analysis and interpretation, critical revision of the manuscript for important intellectual content. Flavio Cueni: acquisition of data, analysis and interpretation. Dr. Falk Nimmerjahn: analysis and interpretation, critical revision of the manuscript for important intellectual content. Dr. Björn Tackenberg: study concept and design, analysis and interpretation, critical revision of the manuscript for important intellectual content. Dr. Jan Lünemann: study concept and design, analysis and interpretation, critical revision of the manuscript for important intellectual content, study supervision.
STUDY FUNDING
The study was supported by a Baxter BioScience Grant (BT09-000143).
DISCLOSURE
I. Quest received research support from Austrian Academy of Sciences. F. Cueni reports no disclosures. F. Nimmerjahn received research support from German Research Foundation. B. Tackenberg is on the scientific advisory boards for CSL Behring, Grifols, Biogen, Novartis, Bayer Healthcare, Genzyme, and UCB; received speaker honoraria from CSL Behring, Grifols, Octapharma, Biogen, Novartis, Bayer Healthcare, and Genzyme; has consulted for CSL Behring, Grifols, Biogen, Novartis, Bayer Healthcare, Genzyme, and UCB; and received research support from Biogen and Novartis. J. Lünemann received research support from Baxter Research Grant Program, Swiss National Research Foundation, and Swiss MS Foundation. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Yan WX, Taylor J, Andrias-Kauba S, Pollard JD. Passive transfer of demyelination by serum or IgG from chronic inflammatory demyelinating polyneuropathy patients. Ann Neurol 2000;47:765–775. [PubMed] [Google Scholar]
- 2.Meyer zu Horste G, Hartung HP, Kieseier BC. From bench to bedside—experimental rationale for immune-specific therapies in the inflamed peripheral nerve. Nat Clin Pract Neurol 2007;3:198–211. [DOI] [PubMed] [Google Scholar]
- 3.Dalakas MC. Mechanisms of action of IVIg and therapeutic considerations in the treatment of acute and chronic demyelinating neuropathies. Neurology 2002;59:S13–S21. [DOI] [PubMed] [Google Scholar]
- 4.Hughes RA, Donofrio P, Bril V, et al. ; ICE Study Group. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol 2008;7:136–144. [DOI] [PubMed] [Google Scholar]
- 5.Takai T. Roles of Fc receptors in autoimmunity. Nat Rev Immunol 2002;2:580–592. [DOI] [PubMed] [Google Scholar]
- 6.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 2008;8:34–47. [DOI] [PubMed] [Google Scholar]
- 7.Boruchov AM, Heller G, Veri MC, Bonvini E, Ravetch JV, Young JW. Activating and inhibitory IgG Fc receptors on human DCs mediate opposing functions. J Clin Invest 2005;115:2914–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brownlie RJ, Lawlor KE, Niederer HA, et al. Distinct cell-specific control of autoimmunity and infection by FcgammaRIIb. J Exp Med 2008;205:883–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tackenberg B, Jelcic I, Baerenwaldt A, et al. Impaired inhibitory Fcgamma receptor IIB expression on B cells in chronic inflammatory demyelinating polyneuropathy. Proc Natl Acad Sci USA 2009;106:4788–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Swieten JC, Koudstaal PJ, Visser MC, Schouten HJ, van Gijn J. Interobserver agreement for the assessment of handicap in stroke patients. Stroke 1988;19:604–607. [DOI] [PubMed] [Google Scholar]
- 11.Bruhns P, Samuelsson A, Pollard JW, Ravetch JV. Colony-stimulating factor-1-dependent macrophages are responsible for IVIG protection in antibody-induced autoimmune disease. Immunity 2003;18:573–581. [DOI] [PubMed] [Google Scholar]
- 12.Vermeulen M, van Doorn PA, Brand A, Strengers PF, Jennekens FG, Busch HF. Intravenous immunoglobulin treatment in patients with chronic inflammatory demyelinating polyneuropathy: a double blind, placebo controlled study. J Neurol Neurosurg Psychiatry 1993;56:36–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mendell JR, Barohn RJ, Freimer ML, et al. Randomized controlled trial of IVIg in untreated chronic inflammatory demyelinating polyradiculoneuropathy. Neurology 2001;56:445–449. [DOI] [PubMed] [Google Scholar]
- 14.Tackenberg B, Lunemann JD, Steinbrecher A, et al. Classifications and treatment responses in chronic immune-mediated demyelinating polyneuropathy. Neurology 2007;68:1622–1629. [DOI] [PubMed] [Google Scholar]
- 15.Latov N, Deng C, Dalakas MC, et al. Timing and course of clinical response to intravenous immunoglobulin in chronic inflammatory demyelinating polyradiculoneuropathy. Arch Neurol 2010;67:802–807. [DOI] [PubMed] [Google Scholar]
- 16.Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity 2000;13:277–285. [DOI] [PubMed] [Google Scholar]
- 17.Mackay M, Stanevsky A, Wang T, et al. Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLE. J Exp Med 2006;203:2157–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su K, Li X, Edberg JC, Wu J, Ferguson P, Kimberly RP. A promoter haplotype of the immunoreceptor tyrosine-based inhibitory motif-bearing FcgammaRIIb alters receptor expression and associates with autoimmunity. II. Differential binding of GATA4 and Yin-Yang1 transcription factors and correlated receptor expression and function. J Immunol 2004;172:7192–7199. [DOI] [PubMed] [Google Scholar]
- 19.Olferiev M, Masuda E, Tanaka S, Blank MC, Pricop L. The role of activating protein 1 in the transcriptional regulation of the human FCGR2B promoter mediated by the -343 G -> C polymorphism associated with systemic lupus erythematosus. J Biol Chem 2007;282:1738–1746. [DOI] [PubMed] [Google Scholar]
- 20.Blank MC, Stefanescu RN, Masuda E, et al. Decreased transcription of the human FCGR2B gene mediated by the -343 G/C promoter polymorphism and association with systemic lupus erythematosus. Hum Genet 2005;117:220–227. [DOI] [PubMed] [Google Scholar]
- 21.Kikuchi-Taura A, Yura A, Tsuji S, et al. Monocyte CD64 expression as a novel biomarker for the disease activity of systemic lupus erythematosus. Lupus Epub 2015 Mar 24. [DOI] [PubMed]
- 22.Madia F, Frisullo G, Nociti V, et al. pSTAT1, pSTAT3, and T-bet as markers of disease activity in chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst 2009;14:107–117. [DOI] [PubMed] [Google Scholar]
- 23.Marzo ME, Tintoré M, Fabregues O, Montalbán X, Codina A. Chronic inflammatory demyelinating polyneuropathy during treatment with interferon-alpha. J Neurol Neurosurg Psychiatry 1998;65:604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meriggioli MN, Rowin J. Chronic inflammatory demyelinating polyneuropathy after treatment with interferon-alpha. Muscle Nerve 2000;23:433–435. [DOI] [PubMed] [Google Scholar]
- 25.Anthoney DA, Bone I, Evans TR. Inflammatory demyelinating polyneuropathy: a complication of immunotherapy in malignant melanoma. Ann Oncol 2000;11:1197–1200. [DOI] [PubMed] [Google Scholar]
- 26.Hirotani M, Nakano H, Ura S, et al. Chronic inflammatory demyelinating polyneuropathy after treatment with interferon-alpha. Intern Med 2009;48:373–375. [DOI] [PubMed] [Google Scholar]
- 27.Khiani V, Kelly T, Shibli A, Jensen D, Mohanty SR. Acute inflammatory demyelinating polyneuropathy associated with pegylated interferon alpha 2a therapy for chronic hepatitis C virus infection. World J Gastroenterol 2008;14:318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato-Motozaki Y, Komai K, Takahashi K, et al. Polyethylene glycol interferon alpha-2b-induced immune-mediated polyradiculoneuropathy. Intern Med 2009;48:569–572. [DOI] [PubMed] [Google Scholar]
- 29.Shiga K, Tanaka E, Isayama R, Mizuno T, Itoh K, Nakagawa M. Chronic inflammatory demyelinating polyneuropathy due to the administration of pegylated interferon alpha-2b: a neuropathology case report. Intern Med 2012;51:217–221. [DOI] [PubMed] [Google Scholar]
- 30.Tanaka M, Krutzik SR, Sieling PA, Lee DJ, Rea TH, Modlin RL. Activation of Fc gamma RI on monocytes triggers differentiation into immature dendritic cells that induce autoreactive T cell responses. J Immunol 2009;183:2349–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science 2001;291:484–486. [DOI] [PubMed] [Google Scholar]
- 32.Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 2006;313:670–673. [DOI] [PubMed] [Google Scholar]
- 33.Dalakas MC; Medscape. Advances in the diagnosis, pathogenesis and treatment of CIDP. Nat Rev Neurol 2011;7:507–517. [DOI] [PubMed] [Google Scholar]