

Abstract
The activation of sterol regulatory element binding proteins (SREBPs) is regulated by insulin-induced genes-1 and -2 (Insig-1 and Insig-2) and SCAP. We previously reported that feeding R-α-lipoic acid (LA) to Zucker Diabetic Fatty (ZDF) rats improves severe hypertriglyceridemia. In this study, we investigated the role of cAMP-responsive element-binding protein H (CREBH) in the lipid-lowering mechanism of LA and its involvement in the SREBP-1c and Insig pathway. Incubation of McA cells with LA (0.2 mM) or glucose (6 mM) stimulated activation of CREBH. LA treatment further induced mRNA expression of Insig-1 and Insig-2a, but not Insig-2b, in glucose-treated cells. In vivo, feeding LA to obesity-induced hyperlipidemic ZDF rats activated hepatic CREBH and stimulated transcription and translation of Insig-1 and Insig-2a. Activation of CREBH and Insigs induced by LA suppressed processing of SREBP-1c precursor into nuclear SREBP-1c, which subsequently inhibited expression of genes involved in fatty acid synthesis, including FASN, ACC and SCD-1, and reduced triglyceride contents in both glucose-treated cells and ZDF rat livers. Additionally, LA treatment also decreased abundances of very-low-density lipoprotein (VLDL)-associated apolipoproteins, apoB100 and apoE, in glucose-treated cells and livers of ZDF rats, leading to decreased secretion of VLDL and improvement of hypertriglyceridemia. This study unveils a novel molecular mechanism whereby LA lowers triglyceride via activation of hepatic CREBH and increased expression of Insig-1 and Insig-2a to inhibit de novo lipogenesis and VLDL secretion. These findings provide novel insight into the therapeutic potential of LA as an anti–hypertriglyceridemia dietary molecule.
Keywords: apolipoproteins, cell signaling, dyslipidemias, sterol regulatory element-binding proteins, triglyceride metabolism, very low density lipoprotein
1. Introduction
Hyperlipidemia is closely related to the pathogenesis of a cluster of chronic metabolic diseases, including fatty liver disease, insulin resistance, type-2 diabetes and atherosclerosis. Cyclic AMP-responsive element-binding protein H (CREBH) is a transcription factor localized to the ER membrane and selectively expressed in the liver and small intestine [1, 2]. Nutritionally, CREBH is induced by FAs (fatty acids) [3–5] and fasting, and suppressed by refeeding [3, 4]. Accumulating evidence has demonstrated that CREBH is fundamentally involved in glucose and lipid metabolism, including gluconeogenesis, hepatic lipid synthesis, FA oxidation, and lipoprotein metabolism [6–8]. Human subjects with nonsynonymous and insertional mutations within the CREBH gene suffer from severe hypertriglyceridemia [9]. Depletion of CREBH induces hypertriglyceridemia in mice under fasting conditions [3], with plasma TG specifically increased in the VLDL fraction. Reduced lipoprotein lipase activity has been proposed to be a contributing factor to the hypertriglyceridemia observed in CREBH-null mice [9]. However, the role of CREBH in lipid metabolism is not fully understood.
The sterol responsive element-binding proteins (SREBPs) are master transcription factors of lipid metabolism. In liver, the SREBP-1c and SREBP-2 isoforms mainly regulate hepatic FA and cholesterol synthesis, respectively. Upon exposure to low levels of cellular sterol, activation of SREBPs is regulated via a group of ER-resident proteins consisting of insulin-induced gene-1 and -2 (Insig-1 and -2) and SCAP [10]. Insig-2 exists as two isoforms, Insig-2a and -2b, with Insig-2a predominantly expressed in liver and Insig-2b expressed ubiquitously. Expression of both isoforms is regulated by distinct mRNA splicing within the 5′-UTR, which eventually produces a common mRNA that encodes identical proteins [11, 12].
R-α-lipoic acid (LA) is enzymatically synthesized from octanoic acid in the mitochondria of most prokaryotic and eukaryotic microorganisms. It plays a vital role in mitochondrial metabolism by acting as a critical co-factor for α-ketoacid dehydrogenases. Although LA is naturally synthesized in sufficient amounts, many studies have shown that LA oral supplements have therapeutic effects for a variety of pathophysiological conditions, including diabetic complications and hypertension [13, 14]. Recently, LA has been reported to reduce plasma TG in animal models [15–18] and human subjects. Diets containing LA dose-dependently decreased hepatic TG and cholesterol concentrations in rats [19]. In Zucker Diabetic Fatty (ZDF) rats, a rodent model in which SREBP-1c expression and lipogenesis are known to be abnormally high [20] and develops hypertriglyceridemia after the age of 7 weeks, feeding a regular chow diet supplemented with LA at a dose of 2.4 g/kg diet from the age of 5 weeks prevented the development of hyperlipidemia and maintained plasma TG levels at a level comparable to lean counterparts [16]. In addition to preventing hypertriglyceridemia, LA corrected blood lipid levels once TG had become elevated [15, 17]. Downregulation of genes involved in hepatic long-chain FA and TG synthesis has been proposed to play a role in the anti-hypertriglyceridemic action of LA [15, 17].
In the present study we identify the molecular mechanism by which LA inhibits hepatic TG synthesis and VLDL secretion. Specifically, we demonstrate that LA induces hepatic CREBH expression and activation and increases transcription and translation of Insig-2a and Insig-1 both in vitro and in vivo. In turn, the increased abundance of Insig-1 and Insig-2a sequesters hepatic SREBP-1c in the ER and hinders its activation, preventing SREBP-1c-dependent TG synthesis. Inhibition of TG synthesis therefore reduces lipid substrate availability for VLDL biogenesis, leading to reduced secretion of VLDL-apoB and improvement of systemic hypertriglyceridemia.
2. Materials and Methods
2.1 Animal protocols
Obese 7-week old male Zucker rats (GmiCrl-fa/fa) were purchased from Charles River Laboratories (Wilmington, MA). Rats were acclimated for two weeks after arrival, housed in individual cages at an ambient temperature of 22 ± 2°C with a 12:12-hr light–dark cycle and free access to water and food (Purina 5008: 56.4% calories from carbohydrates, 26.8% calories from protein, 16.7% from fat; Dyets, Bethlehem, PA). The 9-week old rats were then randomly assigned to one of two feeding groups for two weeks: Purina 5008 + LA (3 g R-α-lipoic acid/kg diet; MAK Wood, Grafton, WI) or pair-fed Purina 5008. Food was provided between 1 and 3 pm in two-day feeding rations along with MilliQ water, and food intake, water intake, and body weight were recorded every second day. LA consumption was thus estimated to approximate 200 mg/kg of body weight per day. At the end of the feeding trial, rats were fasted for three hours and blood was collected in EDTA-coated tubes, then centrifuged at 12,000×g for 1 min to collect plasma, which was then stored at −80°C. Rats were then anesthetized using isoflurane and livers were excised and weighed, then frozen in liquid nitrogen and stored at −80°C. Animals were handled in accordance with the Institutional Animal Care and Use Committee (IACUC), University of Nebraska-Lincoln, and all experiments were conducted in conformity with the Public Health Service (PHS) Policy on Humane Care and Use of Laboratory Animals.
2.2 Cell culture
The hepatic tumor cell line McA-RH7777, originally derived from the Buffalo rat, was obtained from America Type Culture Collection (Manassas, VI) and maintained in DMEM containing 10% FBS, 1.0 g/L glucose and 0.5% penicillin/streptomycin. Cells were grown in T75 flasks at 37°C, 5% CO2, with the media changed every other day and cells passaged every 3–4 days. Twenty-four hours prior to treatment, cells were seeded onto 6-well plates (0.25 × 106) and maintained overnight in DMEM containing 4.5 g/L glucose. The next day, 6 mM glucose was added to the glucose treated groups (glucose group, Glu) while the control group was kept in DMEM with 4.5 g/L glucose. Cells were then treated with 0.2 mM LA for 48 hours, a concentration that can be achieved in human blood plasma after ingestion of a supplement [21]. LA was dissolved in ethanol to prepare a 100 mM stock solution, and glucose was dissolved in water to prepare a 1 M solution. Palmitic acid (PA) was prepared as previously described [22]. For cell transfection, 1.5 g of plasmid DNAs were transfected into McA cells as previously described [23].
2.3 PCR
Total RNA was isolated from tissues and cells using TRIzol (Life Technologies, Grand Island, NY). RNA integrity was confirmed using a NanoDrop 2000 (Wilmington, DE). First strand cDNA was synthesized with oligo(dT) and random primers using a High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Life Technologies). PCR was performed on a Bio-Rad T100 Thermo Cycler (Hercules, CA). Fold changes in mRNA induction were determined after normalization of the mRNA levels to internal control 18S ribosomal RAN levels. Primer sequences will be provided upon request.
2.4 Immunoblot analysis
Liver tissue and cells were washed with ice cold PBS and lysed with cold lysis buffer. Plasma protein was also prepared using lysis buffer. Total protein amounts in liver tissue and cell lysates were determined by Bradford dye-binding method (Bio-Rad) and immunoblot analysis was performed. The following antibodies were used: anti-apoB, anti-apoE, and anti-albumin (Midland Bioproducts, Boone, IA); anti-β-actin (Sigma-Aldrich, St. Louis, MO); anti-CREBH, anti-SREBP-1, anti-SREBP-2, anti-Insig-1, and anti-Insig-2 (Santa Cruz, Dallas, TX); and anti-histone H3 (Thermo Scientific, Waltham, MA). Signals were detected using enhanced chemiluminescence (Pierce, Rockford, IL).
2.5 Determination of lipid contents in cells
Total TG and cholesterol were extracted from cells using a hexane:isopropanol (2:3) solvent extraction mixture. Briefly, 1 mL of the solvent was added to each well for 30 min then collected; this was then repeated with another 0.5 mL of the solvent. The solvent was then allowed to evaporate and the dried lipids were dissolved in 100% ethanol. TG and cholesterol concentrations were detected using an Enzymatic/GPO endpoint method (Pointe Scientific, Canton, MI). Cells were lysed in 50 mM NaOH for protein assay by Bradford dye-binding method (Bio-Rad). Total TG and cholesterol contents in the cells are shown as ratios relative to total cellular protein (mg/g).
3. Results
3.1 LA induces expression of CREBH, Insig-1, and Insig-2a and activates CREBH in vitro
Previous reports have shown that treating mouse primary hepatocytes [6, 24], human Huh7 cells, and rat H4IIE liver cells [25] with free FAs, such as oleate or palmitic acid (PA), induces CREBH expression. To determine whether PA has a similar impact in our in vitro cell model, we treated McA cells with 0.25 mM PA for 48 hours and found that PA significantly induced CREBH mRNA expression. Treating the cells with PA plus 0.2 mM LA for 48 hours also induced significant expression of CREBH (Figure 1A). To further dissect the inhibitory impact of LA on FA synthesis and the role of CREBH in this process, we turned to a glucose-induced lipogenic cell culture model. Instead of directly supplying cells with free FAs, we cultured McA cells in glucose (6 mM) to promote de novo lipogenesis from citrate [26]. This would thus allow us to study the effect of LA on de novo TG synthesis from acetyl-CoA, an essential substrate in lipid synthesis. Similar to what we observed with FA treatment, glucose treatment (6 mM) for 48 hours induced significant CREBH mRNA expression and the presence of LA had an additive effect (Figure 1B). Treating McA cells with LA significantly activated CREBH, indicated by the increased abundance of N-terminal CREBH (CREBH-N), which is the active nuclear form of CREBH, while the protein mass of full-length CREBH (CREBH-F) was maintained at a comparable level between treated and untreated cells (Figure 1C). To further investigate the potential association between CREBH and SREBPs, we examined the Insig proteins, as they are critical regulators of SREBP signaling. Treatment with LA significantly increased mRNA expression of both Insig-1 and Inisig-2a in glucose-treated cells (Figure 1D). In contrast, the presence of LA had no detectable effect on Insig-2b mRNA expression (Figure 1D). The relationship between CREBH and Insig-1/2a was further investigated by overexpressing CREBH cDNAs in McA cells and measuring Insig-1 and Insig-2a mRNAs. As shown in Figure 1E, compared to the mock transfection, forced expression of CREBH enhanced Insig-2a mRNA transcripts but not Insig-1, indicating that the increases in Insig-1 and Insig-2a mRNAs upon LA treatment may be regulated by different mechanisms. These results suggest that LA treatment induces mRNA transcription and activation of CREBH, as well as increases mRNA levels of Insig-1 and Insig-2a. Activation of CREBH may exert an effect on Insig-2a mRNA expression under conditions of glucose-derived de novo lipogenesis.
Figure 1.

LA induces expression of CREBH, Insig-1, and Insig-2a in McA cells. (A) McA cells were treated with PA (0.25 mM) and LA (0.2 mM) for 48 hours, as indicated, and total RNA was extracted to detect mRNA expression of CREBH. Transcript levels were normalized to internal control 18S ribosomal RAN. (B) McA cells were cultured in normal medium (Control, CTL), glucose (6 mM) or glucose plus LA (0.2 mM) for 48 hours. Total RNA was extracted to detect mRNA expression of CREBH. Transcript levels were normalized to internal control 18S ribosomal RAN. (C) McA cells were treated the same as in (B). Protein masses of full-length (F) and nuclear (N) CREBH were detected by immunoblot analysis of cell lysates. Immunoblots were quantified by densitometry and CREBH-F was normalized to β-actin, while CREBH-N was normalized to Histone H3. (D) Total RNA prepared as in (B) was used to determine the mRNA expression of Insig-1, Insig-2a, and Insig-control vector (Mock) or a plasmid expressing WT CREBH (CREBH WT). Forty-eight hours after transfection, cells were collected, total RNA was extracted, and mRNA levels of Insig-1 and Insig-2a were determined by q-PCR. Results are shown as means ± SD for two experiments performed in triplicate. *P<0.05, **P<0.01, ***P<0.001
3.2 LA induces expression and activation of CREBH and Insigs in the liver of ZDF rats
It has been reported that overexpression of exogenous Insig-1 and Insig-2 in ZDF rats suppresses hepatic lipogenesis [20]. Previously, we also showed that ZDF rats develop severe hypertriglyceridemia at the age of 7 weeks, which is markedly improved by LA feeding [15, 17]. To determine whether the CREBH-Insigs pathway activated by LA is involved in the TG-lowering mechanism of LA, we measured CREBH and Insigs in LA-fed ZDF rats. Since the chow diet used was rich in carbohydrates, substrates for lipid synthesis were primarily derived from carbohydrates, thus ensuring comparability between the in vitro and in vivo models. Similar to LA-treated McA cells, LA feeding significantly increased hepatic CREBH mRNA levels (Figure 2A). Protein level of the active nuclear form of CREBH was also significantly increased in the livers of LA-fed ZDF rats, while the protein abundance of full-length CREBH remained unchanged (Figure 2B). Furthermore, hepatic Insig-1 and Insig-2a mRNAs were concomitantly increased by LA feeding, whereas Insig-2b was unaffected (Figure 2C). The increase in Insig-2a mRNA translated into a significant increase in Insig-2 protein (Figure 2D). Surprisingly, this was not the case with Insig-1, as we were unable to detect an increase in Insig-1 protein mass despite increased Insig-1 mRNA (Figure 2D). This may be due to the relative short lifetime of Insig-1 protein. These data suggest that LA feeding induces mRNA transcription and activation of CREBH in the livers of ZDF rats, which is associated with increased expression of hepatic Insig-1 and Insig-2a.
Figure 2.

LA increases hepatic CREBH and Insig-2 expression in ZDF rats. Livers were isolated from 11-wk old ZDF rats fed an LA-supplemented diet for two weeks and from pair-fed controls. Total RNA was extracted from liver tissues to detect mRNA expression of (A) CREBH, as well as (C) Insig-1, Insig-2a, Insig-2b. Transcript levels were normalized to internal control 18S ribosomal RAN. Protein mass of (B) full-length (F) and nuclear (N) CREBH, as well as (D) Insig-1 and Insig-2, were detected by immunoblot analysis of homogenized liver tissues. Immunoblots were quantified by densitometry and normalized to β-actin, except for CREBH-N, which was normalized to Histone H3. Results are shown as means ± SD (n=4/group). *P<0.05, **P<0.01, ***P<0.001.
3.3 LA inhibits activation of the SREBP-1c pathway in glucose-treated McA cells and ZDF rats
Activation of Insig-1 and Insig-2a signaling upon exposure to LA treatment prompted us to further investigate de novo lipid synthesis pathways in glucose-treated McA cells. We first determined whether LA inhibits SREBP-1c activation since this is the SREBP isoform that regulates free FA synthesis in the liver. Expressions of full-length SREBP-1c precursor (SREBP-1c-p) protein, as well as the active nuclear form of SREBP-1c (SREBP-1c-n), were both significantly decreased by LA treatment (Figure 3A). To further investigate another SREBP family member, SREBP-2, which regulates cholesterol metabolism, we found that the protein abundances of full-length (SREBP-2-p) and active nuclear SREBP-2 (SREBP-2-n) were not affected by LA (Figure 3A). Consistent with these in vitro observations, in the livers of the LA-treated ZDF rats, both the full-length and nuclear forms of SREBP-1 were significantly decreased upon LA treatment, compared to pair-fed untreated controls (Figure 3B). Neither the full-length nor the nuclear form of SREBP-2 was downregulated by LA feeding (Figure 3B). The regulatory effect of LA on SREBP-1c appeared to be at the post-transcriptional level, as no alteration was detected in SREBP-1c mRNA between the LA-treated and non-treated groups (Suppl. Figure 1). These data therefore indicate that the increased expression of Insig-1 and Insig-2a induced by LA was capable of sequestering and retaining SREBP-1c in the ER, thereby hindering its activation by proteolytic cleavage in the Golgi complex.
Figure 3.

LA reduces hepatic full-length and nuclear SREBP-1 protein expression in McA cells and ZDF rats. (A) McA cells were cultured in either glucose (6 mM) alone or glucose (6 mM) plus LA (0.2 mM) for 48 hours. Protein masses of precursor (p) and nuclear (n) SREBP-1c and SREBP-2 were detected by immunoblot analysis of cell lysates. (B) Livers were isolated from 11-wk old ZDF rats fed an LA-supplemented diet for two weeks and from pair-fed controls. Protein mass of precursor and nuclear SREBP-1c as well as nuclear SREBP-2 were detected by immunoblot analysis of liver tissues. Immunoblots were quantified by densitometry and precursor proteins were normalized to β-actin, while nuclear proteins were normalized to Histone H3. Results are shown as means ± SD for two experiments performed in triplicate and n of 4 per group for the animal study. *P<0.05.
3.4 LA suppresses TG synthesis in glucose-induced lipogenic McA cells
To further investigate the underpinnings of lipogenesis in McA cells with or without LA treatment, we measured the mRNAs of a number of SREBP-1c target genes that are involved in hepatic de novo FA synthesis, including fatty acid synthase (FASN), acetyl-CoA carboxylase (ACC), and stearoyl-CoA desaturase-1 (SCD-1). In accordance with the suppression of SREBP-1c upon LA treatment, transcription of FASN, ACC, and SCD-1 mRNAs was decreased in LA-treated McA cells relative to vehicle-treated cells (Figures 4A). Further assessment of cellular lipid contents revealed that LA treatment significantly decreased TG levels in McA cells compared to control cells (Figure 4B). In the presence of LA, cholesterol contents were slightly reduced, but not to a level that was statistically significant (Figure 4B). These results suggest that under carbohydrate-mediated pro-lipogenic conditions, LA specifically inhibits de novo TG synthesis through modulation of hepatic SREBP-1c and Insig-1/Insig-2 signaling without significant abatement of cholesterol synthesis through SREBP-2.
Figure 4.

LA suppresses FA synthesis genes and reduces cellular TG and cholesterol levels in McA cells. McA cells were cultured in high glucose (4.5 g/L) DMEM with the addition of either glucose (6 mM) alone or glucose (6 mM) plus LA (0.2 mM) for 48 hours. (A) Total RNA was extracted to detect mRNA expression of FASN, ACC, and SCD-1 reductase. Transcript levels were normalized to internal control 18S ribosomal RAN. (B) Media was removed and total lipid was extracted from cells to measure TG and cholesterol concentrations. Results are shown as means ± SD for one representative experiment performed in triplicate. Two experiments were performed in triplicate and showed similar results. *P<0.05.
3.5 LA reduces intracellular and secreted VLDL-associated apolipoproteins and improves systemic hyperlipidemia
The inhibitory effect of LA on TG synthesis prompted us to further investigate VLDL metabolism in our experimental models. Since apoB is a critical structural protein in the biogenesis of VLDL and its cellular abundance is associated with proper lipidation occurring at the co- or post-translational steps [27], we reasoned that reduced availability of lipids by LA treatment would create a lipid-poor environment that would prompt the degradation of apoB, thereby diminishing VLDL assembly and secretion. Indeed, protein levels of cellular apoB100 as well as secreted VLDL-apoB100 in the culture medium were significantly decreased in the LA-treated cells compared to the untreated cells (Figure 5A). Moreover, the abundance of apoE, another VLDL-associated apolipoprotein that is involved in VLDL secretion, was also decreased by LA [28] (Figure 5A). In vivo, LA feeding significantly reduced protein levels of apoB100 and apoE in the livers of ZDF rats (Figure 5B). This inhibitory effect occurred specifically for the VLDL-associated apolipoproteins, as protein expression of albumin, another liver secreted protein, was not affected by LA treatment (Figures 5A and B). In addition, LA had no detectable effect on the mRNA levels of microsomal triglyceride transfer protein (MTP), an ER chaperone that mediates protein lipidation of apoB during VLDL assembly (Suppl. Figure 2). Decreased hepatic apoB and apoE upon LA feeding resulted in a reduction in VLDL secretion, indicated by the significantly reduced levels of plasma apoB and apoE (Figure 5C). We further observed that mRNA transcripts of LDL-receptor, a receptor responsible for the uptake of LDL remnants into hepatocytes, were also decreased in the livers of LA-treated ZDF rats (Figure 5D). Taken together, these results suggest that LA curbs TG-rich VLDL assembly and secretion by lowering apoB and apoE availability.
Figure 5.
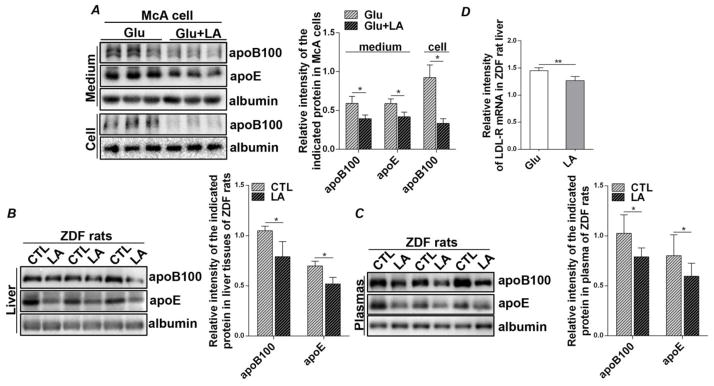
LA reduces intracellular levels and secretion of VLDL-associated apolipoproteins in McA cells and ZDF rats. (A) McA cells were cultured in either glucose (6 mM) alone or glucose (6 mM) plus LA (0.2 mM) for 48 hours. Protein mass of apoB100 and apoE were detected by immunoblot analysis of cell lysates and culture medium. Results are shown as means ± SD for two experiments performed in triplicate. (B, C) Three-hour fasted blood was collected and livers were isolated from 11-wk old ZDF rats fed an LA-supplemented diet for two weeks and from pair-fed controls. Protein mass of apoB100 and apoE were detected by immunoblot analysis of (B) liver tissues and (C) plasma. Results are shown as means ± SD (n=4/group). Immunoblots were quantified by densitometry and normalized to albumin. (D) Total RNA was extracted from liver tissues to detect mRNA expression of LDL-receptor. *P<0.05, **P<0.01.
4. Discussion
This study demonstrated that LA activates CREBH and upregulates Insig-2a both in vitro and in vivo. CREBH activation by LA may contribute to the ability of this bioactive food compound to inhibit the proteolytic cleavage and nuclear translocation of SREBP-1c under glucose conditions, thereby repressing the transcription of lipogenic genes and lipidation of VLDL. LA-dependent activation of the transcription factor CREBH may represent the missing piece in the molecular mechanism by which LA ameliorates hepatic steatosis and hypertriglyceridemia.
This is the first report showing that activation of CREBH by LA represses TG lipid synthesis through inhibition of SREBP-1c. Previously, CREBH knockout mice were shown to develop hypertriglyceridemia as a result of impaired clearance of plasma TG rather than exacerbated secretion of TG-rich lipoproteins from the liver [9]. This is also the first report showing that CREBH is upregulated transcriptionally by a dietary organosulfur compound. Importantly, LA-induced upregulation of CREBH mRNA was accompanied by a rise in CREBH protein, particularly the nuclear, transcriptionally competent form of CREBH. Although historically recognized as an antioxidant able to reduce various oxidized targets, including other antioxidants such as ascorbate or glutathione, LA does not appear to act as an antioxidant here. At the doses ingested from a supplemented diet and because it is very rapidly metabolized, LA does not accumulate to sufficient cellular levels (low μM) to have a lasting impact similar to that of traditional antioxidants (e.g., ascorbate or tocopherol) [13, 14]. Rather, the short window of activity afforded by exogenous LA suggests that it acts transiently on cell signaling pathways involved in lipid homeostasis.
SREBPs are major transcription factors that control the biosynthesis of cholesterol, FA, and TG in the liver [29]. Their activation is regulated by sister proteins in the ER, Insig-1 and liver-specific Insig-2, whose expressions are regulated by different signaling pathways. Nutritional factors, such as fasting and refeeding, regulate Insig gene expression, which has been proposed to be associated with the fluctuation in plasma insulin levels induced by nutritional factors, particularly carbohydrates. In addition, expression of Insig-1 is induced by insulin stimulation and active SREBPs. In contrast, protein level of Insig-2 is downregulated by insulin [30]. Another nutritional factor that has been proposed to regulate Insig protein is sterol. Low level of sterol induces ubiquitination of Insig-1 and reduces Insig-1 protein level [29]. Research continually strives to identify new inhibitors of SREBPs and targeting Insig proteins may offer therapeutic alternatives to managing hyperlipidemia and fatty liver disease.
In the present study, we provide new evidence that Insig-2a and CREBH expressions are concomitantly increased by LA both in vitro and in vivo. Since overexpression of CREBH in McA cells enhanced gene expression of Insig-2a, but not Insig-1 or Insig-2b, we propose that Insig-2a is a target gene of CREBH. The CREBH-Insig-2a pathway may be a novel pathway that is specifically associated with SREBP-1c to control hepatic FA and TG synthesis. Research is currently being undertaken in our laboratory to test this hypothesis.
Intraperitoneal administration of LA (100 mg/kg body weight) has been reported to activate hepatic AMPK signaling, which in turn inhibits SREBP-1c mRNA expression. However, this signaling was only sustained for a very short time frame, as AMPK phosphorylation returned to baseline within 3 days [31]. The transient effect of LA on AMPK indicates that there is/are (an) alternative AMPK-independent pathway(s) that mediate(s) LA’s effect on hepatic steatosis [31]. Indeed, Timmers et al. reported that LA supplementation could prevent lipid accumulation associated with feeding a high-fat diet to Wistar rats, independently of AMPK [32]. In our study, at the end of a 2-week diet trial, we were unable to observe any alteration on AMPK phosphorylation or mRNA transcription of SREBP-1c in the liver tissues of untreated and treated groups [17], suggesting that the anti-SREBP effect of LA, at this time point, was mediated by a mechanism other than AMPK activation. That is, LA negatively regulated SREBP-1c activation at the post-transcriptional level by inducing the expression of Insig proteins to sequester the precursor of SREBP-1c in the ER. Perhaps this is a major pathway that mediates the anti-lipogenic property of LA in a prolonged treatment.
In addition to its impact on hepatic TG synthesis, the anti-hypertriglyceridemia property of LA was further reflected through its impact on the metabolism of the lipoprotein VLDL. The assembly and secretion of VLDL takes place in the hepatic ER and is regulated by multiple factors. Co- and post-translational lipidation of apoB mediated by MTP is a critical step in VLDL assembly [33–35] and intracellular abundance of apoB protein is mainly regulated by the availability of lipid substrates, including TG, cholesterol, and phospholipids. High levels of intracellular lipids stabilize apoB and stimulate the assembly and secretion of VLDL, whereas when intracellular lipids are decreased, apoB is subject to degradation through proteasomal and nonproteasomal pathways, thereby reducing VLDL secretion [27]. Since LA treatment downregulates apoB protein level without altering the mRNA expression of either apoB (data not shown) or MTP in McA cells or livers of ZDF rats, the decrease in apoB protein upon LA treatment is very likely caused by increased protein degradation due to the low level of lipids in the LA-treated hepatocytes. Taken together, our findings provide novel insights into the TG-lowering mechanism of LA and further support of its potential as a therapeutic approach in the treatment of metabolic hyperlipidemia.
Supplementary Material
Acknowledgments
We gratefully acknowledge the contributions of Anjeza Pashaj, Xiaohua Yi, and Mengna Xia during the care and feeding of the animals as well as sample preparation. This work was supported by Tobacco Settlement Funds and a Layman Seed Grant to R. Moreau, and a P20 GM104320-01A1 grant from NIH, Hatch funds from USDA/NIFA and a Faculty Seed Grant to Q. Su.
Footnotes
Conflicts of interest
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ginsberg HN, Zhang YL, Hernandez-Ono A. Metabolic syndrome: Focus on dyslipidemia. Obesity. 2006;14:41s–9s. doi: 10.1038/oby.2006.281. [DOI] [PubMed] [Google Scholar]
- 2.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 3.Asada R, Kanemoto S, Kondo S, Saito A, Imaizumi K. The signalling from endoplasmic reticulum-resident bZIP transcription factors involved in diverse cellular physiology. J Biochem. 2011;149:507–18. doi: 10.1093/jb/mvr041. [DOI] [PubMed] [Google Scholar]
- 4.Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13:365–76. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 5.Bailey D, O’Hare P. Transmembrane bZIP transcription factors in ER stress signaling and the unfolded protein response. Antioxid Redox Signal. 2007;9:2305–21. doi: 10.1089/ars.2007.1796. [DOI] [PubMed] [Google Scholar]
- 6.Zhang C, Wang G, Zheng Z, Maddipati KR, Zhang X, Dyson G, et al. Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology. 2012;55:1070–82. doi: 10.1002/hep.24783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kondo S, Murakami T, Tatsumi K, Ogata M, Kanemoto S, Otori K, et al. OASIS, a CREB/ATF-family member, modulates UPR signalling in astrocytes. Nat Cell Biol. 2005;7:186–94. doi: 10.1038/ncb1213. [DOI] [PubMed] [Google Scholar]
- 8.Liang G, Audas TE, Li Y, Cockram GP, Dean JD, Martyn AC, et al. Luman/CREB3 induces transcription of the endoplasmic reticulum (ER) stress response protein Herp through an ER stress response element. Mol Cell Biol. 2006;26:7999–8010. doi: 10.1128/MCB.01046-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JH, Giannikopoulos P, Duncan SA, Wang J, Johansen CT, Brown JD, et al. The transcription factor cyclic AMP-responsive element-binding protein H regulates triglyceride metabolism. Nat Med. 2011;17:812–5. doi: 10.1038/nm.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Llarena M, Bailey D, Curtis H, O’Hare P. Different mechanisms of recognition and ER retention by transmembrane transcription factors CREB-H and ATF6. Traffic. 2010;11:48–69. doi: 10.1111/j.1600-0854.2009.00997.x. [DOI] [PubMed] [Google Scholar]
- 11.Yabe D, Brown MS, Goldstein JL. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci USA. 2002;99:12753–8. doi: 10.1073/pnas.162488899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 13.Shay KP, Moreau RF, Smith EJ, Smith AR, Hagen TM. Alpha-lipoic acid as a dietary supplement: molecular mechanisms and therapeutic potential. Biochim Biophys Acta. 2009;1790:1149–60. doi: 10.1016/j.bbagen.2009.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Packer L, Kraemer K, Rimbach G. Molecular aspects of lipoic acid in the prevention of diabetes complications. Nutrition. 2001;17:888–95. doi: 10.1016/s0899-9007(01)00658-x. [DOI] [PubMed] [Google Scholar]
- 15.Pashaj A, Yi XH, Xia MN, Canny S, Riethoven JJM, Moreau R. Characterization of genome-wide transcriptional changes in liver and adipose tissues of ZDF (fa/fa) rats fed R-alpha-lipoic acid by next-generation sequencing. Physiological Genomics. 2013;45:1136–43. doi: 10.1152/physiolgenomics.00138.2013. [DOI] [PubMed] [Google Scholar]
- 16.Butler JA, Hagen TM, Moreau R. Lipoic acid improves hypertriglyceridemia by stimulating triacylglycerol clearance and downregulating liver triacylglycerol secretion. Arch Biochem Biophys. 2009;485:63–71. doi: 10.1016/j.abb.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yi X, Pashaj A, Xia M, Moreau R. Reversal of obesity-induced hypertriglyceridemia by (R)-alpha-lipoic acid in ZDF (fa/fa) rats. Biochem Biophys Res Commun. 2013;439:390–5. doi: 10.1016/j.bbrc.2013.08.063. [DOI] [PubMed] [Google Scholar]
- 18.Finlay LA, Michels AJ, Butler JA, Smith EJ, Monette JS, Moreau RF, et al. R-alpha-lipoic acid does not reverse hepatic inflammation of aging, but lowers lipid anabolism, while accentuating circadian rhythm transcript profiles. Am J Physiol Regul Integr Comp Physiol. 2012;302:R587–R97. doi: 10.1152/ajpregu.00393.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huong DT, Ide T. Dietary lipoic acid-dependent changes in the activity and mRNA levels of hepatic lipogenic enzymes in rats. Br J Nutr. 2008;100:79–87. doi: 10.1017/S0007114507876227. [DOI] [PubMed] [Google Scholar]
- 20.Takaishi K, Duplomb L, Wang MY, Li J, Unger RH. Hepatic insig-1 or -2 overexpression reduces lipogenesis in obese Zucker diabetic fatty rats and in fasted/refed normal rats. Proc Natl Acad Sci USA. 2004;101:7106–11. doi: 10.1073/pnas.0401715101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carlson DA, Smith AR, Fischer SJ, Young KL, Packer L. The plasma pharmacokinetics of R-(+)-lipoic acid administered as sodium R-(+)-lipoate to healthy human subjects. Altern Med Rev. 2007;12:343–51. [PubMed] [Google Scholar]
- 22.Nakamura T, Furuhashi M, Li P, Cao H, Tuncman G, Sonenberg N, et al. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell. 2010;140:338–48. doi: 10.1016/j.cell.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su QZ, Tsai J, Xu E, Qiu W, Bereczki E, Santha M, et al. Apolipoprotein B100 Acts as a Molecular Link Between Lipid-Induced Endoplasmic Reticulum Stress and Hepatic Insulin Resistance. Hepatology. 2009;50:77–84. doi: 10.1002/hep.22960. [DOI] [PubMed] [Google Scholar]
- 24.Danno H, Ishii KA, Nakagawa Y, Mikami M, Yamamoto T, Yabe S, et al. The liver-enriched transcription factor CREBH is nutritionally regulated and activated by fatty acids and PPARalpha. Biochem Biophys Res Commun. 2010;391:1222–7. doi: 10.1016/j.bbrc.2009.12.046. [DOI] [PubMed] [Google Scholar]
- 25.Gentile CL, Wang D, Pfaffenbach KT, Cox R, Wei Y, Pagliassotti MJ. Fatty acids regulate CREBh via transcriptional mechanisms that are dependent on proteasome activity and insulin. Mol Cell Biochem. 2010;344:99–107. doi: 10.1007/s11010-010-0533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aas V, Kase ET, Solberg R, Jensen J, Rustan AC. Chronic hyperglycaemia promotes lipogenesis and triacylglycerol accumulation in human skeletal muscle cells. Diabetologia. 2004;47:1452–61. doi: 10.1007/s00125-004-1465-9. [DOI] [PubMed] [Google Scholar]
- 27.Rutledge AC, Su Q, Adeli K. Apolipoprotein B100 biogenesis: a complex array of intracellular mechanisms regulating folding, stability, and lipoprotein assembly. Biochem Cell Biol. 2010;88:251–67. doi: 10.1139/o09-168. [DOI] [PubMed] [Google Scholar]
- 28.Mensenkamp AR, Jong MC, van GH, van Luyn MJ, Bloks V, Havinga R, et al. Apolipoprotein E participates in the regulation of very low density lipoprotein-triglyceride secretion by the liver. J Biol Chem. 1999;274:35711–8. doi: 10.1074/jbc.274.50.35711. [DOI] [PubMed] [Google Scholar]
- 29.Gong Y, Lee JN, Lee PC, Goldstein JL, Brown MS, Ye J. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. 2006;3:15–24. doi: 10.1016/j.cmet.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 30.Yabe D, Komuro R, Liang G, Goldstein JL, Brown MS. Liver-specific mRNA for Insig-2 down-regulated by insulin: implications for fatty acid synthesis. Proc Natl Acad Sci USA. 2003;100:3155–60. doi: 10.1073/pnas.0130116100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park KG, Min AK, Koh EH, Kim HS, Kim MO, Park HS, et al. Alpha-Lipoic Acid Decreases Hepatic Lipogenesis Through Adenosine Mono phosphate-Activated Protein Kinase (AMPK)-Dependent and AMPK-Independent Pathways. Hepatology. 2008;48:1477–86. doi: 10.1002/hep.22496. [DOI] [PubMed] [Google Scholar]
- 32.Timmers S, Bosch dV-vd, Towler MC, Schaart G, Moonen-Kornips E, Mensink RP, et al. Prevention of high-fat diet-induced muscular lipid accumulation in rats by alpha lipoic acid is not mediated by AMPK activation. J Lipid Res. 2010;51:352–9. doi: 10.1194/jlr.M000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dashti N, Gandhi M, Liu X, Lin X, Segrest JP. The N-terminal 1000 residues of apolipoprotein B associate with microsomal triglyceride transfer protein to create a lipid transfer pocket required for lipoprotein assembly. Biochemistry. 2002;41:6978–87. doi: 10.1021/bi011757l. [DOI] [PubMed] [Google Scholar]
- 34.Kulinski A, Rustaeus S, Vance JE. Microsomal triacylglycerol transfer protein is required for lumenal accretion of triacylglycerol not associated with ApoB, as well as for ApoB lipidation. J Biol Chem. 2002;277:31516–25. doi: 10.1074/jbc.M202015200. [DOI] [PubMed] [Google Scholar]
- 35.Hussain MM, Rava P, Walsh M, Rana M, Iqbal J. Multiple functions of microsomal triglyceride transfer protein. Nutr Metab (Lond) 2012;9:14. doi: 10.1186/1743-7075-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.