

Abstract
MT1-MMP (MMP14) is a collagenolytic enzyme located at the cell surface and implicated in extracellular matrix (ECM) remodeling. Mmp14−/− mice present dwarfism, bone abnormalities, and premature death. We demonstrate herein that the loss of MT1-MMP also causes cardiac defects and severe metabolic changes, and alters the cytoskeleton and the nuclear lamina structure. Moreover, the absence of MT1-MMP induces a senescent phenotype characterized by up-regulation of p16INK4a and p21CIP1/WAF1, increased activity of senescence-associated β-galactosidase, generation of a senescence-associated secretory phenotype, and somatotroph axis alterations. Consistent with the role of retinoic acid signaling in nuclear lamina stabilization, treatment of Mmp14−/− mice with all-trans retinoic acid reversed the nuclear lamina alterations, partially rescued the cell senescence phenotypes, ameliorated the pathological defects in bone, skin, and heart, and extended their life span. These results demonstrate that nuclear architecture and cell senescence can be modulated by a membrane protease, in a process involving the ECM as a key regulator of nuclear stiffness under cell stress conditions.
Keywords: aging, extracellular matrix, metalloprotease, nuclear lamina, retinoids
Introduction
The extracellular matrix (ECM) provides an attachment substrate for most cells, but its composition and stiffness are also able to modify many cellular characteristics (Eyckmans et al, 2011). Furthermore, certain ECM proteins can limit the access of cells to specific growth factors, by sequestering them within the ECM (Bergers et al, 2000). Therefore, the ECM constitutes an essential player during development and differentiation. Due to the dynamic nature of the ECM, a cellular response to different stimuli can result in a massive remodeling of this matrix. This response can be slow, by modifying its composition through secretion of alternative ECM proteins, or fast, by activating specific proteases which cleave and modify the ECM surrounding the cell.
Matrix metalloproteinases (MMPs) constitute a group of extra- or pericellular proteases with the ability to process virtually all components of the ECM (Fanjul-Fernandez et al, 2010; Kessenbrock et al, 2010). Among them, collagenases represent a small subgroup of MMPs which precisely cleave fibrillar collagens and initiate collagen degradation and ECM remodeling. Collagenase-1, collagenase-2, and collagenase-3 (MMP-1, MMP-8, and MMP-13) were initially discovered by their ability to cleave collagen in different cell types (Overall & Lopez-Otin, 2002). However, the generation of mice deficient in specific MMPs has revealed that membrane-bound MT1-MMP (MMP14) is a powerful collagenase, necessary for cell invasion through collagen-rich ECMs (Sabeh et al, 2004). The importance of MT1-MMP in cell function has also been reinforced by the analysis of animals lacking different MMPs. Thus, mutant mice deficient in any of the three secreted collagenases (Mmp1a, Mmp8, and Mmp13) are viable and fertile (Balbin et al, 2003; Fanjul-Fernandez et al, 2013; Inada et al, 2004). In contrast, loss of mouse MT1-MMP (Mmp14) results in a complex phenotype that leads to premature death as soon as 3 weeks after birth (Holmbeck et al, 1999; Zhou et al, 2000). Mmp14-deficient mice show dwarfism, osteopenia, and severe connective tissue abnormalities, as well as defects in adipose tissue formation and alveolar development (Atkinson et al, 2005; Chun et al, 2006). Nevertheless, the systemic effects caused by the loss of MT1-MMP and the effect of a deficient ECM remodeling on cell structure and function are largely unknown.
The ECM and the cytoskeleton constitute dynamic structures which are tightly interconnected at the plasma membrane by integrins (Hynes, 2002). These heterodimeric proteins are able to bind specific ECM components at the outer surface and, at the same time, recruit adaptor proteins to the cytoplasmic side, thus linking integrins with components of the cytoskeleton and intracellular signaling networks. The cytoskeleton is connected to the nucleus by a series of proteins that constitute the LINC complex (linker of the nucleoskeleton and the cytoskeleton) (Crisp et al, 2006; Mellad et al, 2011). This intricate network of proteins and filaments provides mechanosensor properties to cells, allowing them to adapt to different forms of mechanical stress (Martins et al, 2012). Genetic inactivation of the murine genes encoding lamin A or other components of the nuclear envelope causes progeroid syndromes characterized by short life span, lipodystrophy, and cardiac and skeletal abnormalities, as well as inability to produce a functional ECM (Hernandez et al, 2010; Osorio et al, 2011; Pendas et al, 2002). It is remarkable that some of these phenotypes have also been reported in Mmp14-deficient mice (Chun et al, 2006; Holmbeck et al, 1999; Zhou et al, 2000). Interestingly, depletion of the Sun-1 protein from the LINC complex in lamin A-deficient progeroid animals extends their longevity and ameliorates most pathological phenotypes (Chen et al, 2012), underscoring the importance of the nucleus–cytoskeleton connection in premature aging and cellular senescence. It is also remarkable the recent finding that ECM stiffness increases the relative abundance of lamin A through a process modulated by the retinoid receptor signaling pathway (Swift et al, 2013), reflecting the critical role of lamin A in mechanosensing (Buxboim et al, 2014).
Herein, we show that the abnormal proteolytic processing of ECM components in Mmp14-deficient mice triggers signaling events that alter the nuclear lamina and cytoskeleton structure. Moreover, loss of Mmp14 elicits a cellular senescence response characterized by major metabolic changes. We also show that the Mmp14-mediated proteolytic activity prevents the senescent phenotype of Mmp14-deficient cells, providing evidence of a cellular senescence response caused by abnormal ECM remodeling. Finally, we report that treatment of Mmp14-null mice with retinoic acid rescues some of their senescent features and increases their life span.
Results
Generation and characterization of a new strain of Mmp14-deficient mice
We generated mice deficient in Mmp14 by deleting exons 4 and 5, encoding the MT1-MMP catalytic domain (Supplementary Fig S1A). MT1-MMP protein could not be detected in Mmp14−/− tissues by Western blot analysis, and a reduction in the activation of pro-MMP2 (a substrate for MT1-MMP) was observed by gelatin zymography (Supplementary Fig S1B). In agreement with previous studies (Holmbeck et al, 1999; Zhou et al, 2000), Mmp14−/− mice were smaller than their wild-type littermates and showed a reduced growth which was evident as soon as 4 days after birth (Supplementary Fig S1C and D). These mutant mice failed to grow and their mean survival was 14 days (Supplementary Fig S1E) (Zhou et al, 2000). Mmp14 deficiency also resulted in a severe skeletal phenotype which has been thoroughly investigated (Holmbeck et al, 1999; Zhou et al, 2000). We also found prominent cranial sutures in Mmp14-null mice (Supplementary Fig S1F) and a marked impairment of the collagenolytic activity in Mmp14-null fibroblasts (Supplementary Fig S1G). A more detailed analysis of these mutant mice allowed us to identify other alterations, including an enlargement of the distal airways and alveoli, the accumulation of extravasated blood cells in the lung matrix of Mmp14−/− mice, and the presence of inflammatory cells around biliar ducts, which were not detected in wild-type mice (Supplementary Fig S1H). We also observed that the skeletal muscle structure was altered in Mmp14-deficient mice as assessed by the detection of changes in the activity of several mitochondrial enzymes such as succinate dehydrogenase and by the accumulation of collagen fibers in muscle from Mmp14−/− mice (Supplementary Fig S1I). Furthermore, muscle fiber length was 30% shorter in Mmp14−/− animals than that in control mice (29 versus 20 μm, P < 0.05) (Supplementary Fig S1J). Together, and despite the clear skeletal abnormalities observed in Mmp14−/− mice, these data show additional alterations in non-skeletal tissues which might contribute to the early death of these animals.
Loss of MT1-MMP induces cellular senescence
Notably, we also observed that Mmp14-null mice exhibit cardiovascular defects which have not been previously reported in other strains of Mmp14-null mice. We found a thickened muscular wall of the right ventricle and ventricular septum hypertrophy in 16-day-old Mmp14−/− hearts (Fig1A; Supplementary Fig S2A), suggesting abnormal cardiac function in these animals. These cardiac abnormalities were accompanied by a strong accumulation of type I collagen in hearts from Mmp14−/− mice as determined by picrosirius red staining to visualize collagen. Quantitative PCR analysis of different factors involved in cardiomyocyte function revealed that hearts from Mmp14−/− mice accumulate β-myosin heavy chain (β-MHC/Myh7), gap junction connexin 40 (Cx40) and NKx2.5 (Supplementary Fig S1K–M). These findings reflect either an improper maturation of cardiomyocytes during heart development or the presence of cardiac stress in these animals (Lowes et al, 1997). Interestingly, Mmp14−/− mice also exhibited an accumulation of sudan black staining in heart, which detects the complex lysosomal aggregate known as lipofucsin, that usually accumulates in aged tissues, suggesting the occurrence of a senescence process in their cardiac tissue (Georgakopoulou et al, 2013) (Fig1B; Supplementary Fig S2B). The finding of these cardiac defects and cell senescence features in mice lacking MT1-MMP led us to evaluate the putative presence of senescence in other tissues from these mutant animals. Analysis of the senescence-associated β-galactosidase (SA-β-Gal) activity, a biomarker for senescence and aging cells, demonstrated that it was clearly augmented in kidney from Mmp14−/− mice (Fig1C; Supplementary Fig S2C), reflecting an increased number of senescent cells in vivo. Similarly, higher SA-β-Gal activity was observed in adipose tissue and fibroblasts derived from Mmp14-deficient mice (Fig1C; Supplementary Fig S2C). To further confirm the senescence phenotype, we performed a BrdU incorporation assay to corroborate the lack of proliferation in these fibroblasts (Fig1D), and an immunofluorescence assay of HP1γ, a senescence-associated heterochromatin protein that is accumulated in the foci of senescence cells (Fig1E). We next analyzed different molecular mediators implicated in the senescence process. The tumor suppressor protein p16INK4a, a marker of cellular senescence (Collado & Serrano, 2010; Krishnamurthy et al, 2004), showed a strong accumulation in fibroblasts derived from Mmp14−/− mice (Fig1F). Gene expression analysis also revealed that the cyclin-dependent kinase inhibitor 1A (p21CIP1/WAF1), a direct target of p53 involved in senescence processes (Baker et al, 2013), was overexpressed in Mmp14−/− muscles (Fig1G). Thus, quantitative RT–PCR analysis showed that p21CIP1/WAF1 expression was 11-fold higher in muscles from Mmp14−/− mice when compared with Mmp14+/+ animals (P < 0.01) (Fig1G). Likewise, Mmp14 deficiency also caused a 6-fold (P < 0.01) and a 2-fold (P < 0.05) increase in p21CIP/WAF1 expression in kidney and liver, respectively (Supplementary Fig S3A and B), suggesting that cellular senescence is induced in different tissues from these mutant mice.
Figure 1.

Lack of MT1-MMP activates a cellular senescence signaling process and causes alterations in the somatotroph axis
- A Cardiac defects and accumulation of collagen fibers in Mmp14-deficient mice. H&E and picrosirius red staining of hearts from 15-day-old mice.
- B Sudan black staining counterstained with nuclear fast red to visualize senescence activity in hearts from Mmp14-deficient mice (40× magnification, and right panel shows a detailed view of the area indicated on the left panel).
- C SA-β-Gal activity was assayed in kidney, adipose tissue (dashed square and H&E staining to confirm the adipose tissue), and fibroblasts from control and Mmp14−/− mice (n = 3) for each condition.
- D Percentage of cells with positive staining for BrdU.
- E Senescence heterochromatin foci were visualized by HP-1γ immunostaining. The number of foci per cell is represented. A representative image is shown.
- F Western blot (left panel) and RT–qPCR analysis (right panel) of p16INK4a in control and Mmp14−/− fibroblasts.
- G Gene expression analysis of p21CIP1/WAF1 by RT–qPCR in muscle from 15-day-old control and Mmp14−/− mice.
- H Absence of MT1-MMP reduces cell proliferation. Control and Mmp14−/− cells (1 million cells), passage 1, were seeded onto plates and every 3 days cell populations were counted. The graphic shows the log2 of the number of million cells for each population. The experiment was done in triplicate using fibroblasts from three wild-type and three mutant mice.
- I Plasma samples from 15-day-old Mmp14−/− and Mmp14+/+ mice were collected and pooled, and levels of IL-6 were measured by ELISA (n = 12). Each sample was measured in triplicate.
- J Blood glucose concentrations in Mmp14+/+ (n = 12) and Mmp14−/− (n = 12) of 15-day-old mice.
- K, L Plasma concentration of IGF-1 and GH was measured in 15-day-old Mmp14−/− mice (n = 4) and their wild-type littermates (n = 4). Concentrations were normalized to the mean control.
Data information: Mean values are represented and error bars indicate SD (*P < 0.05, **P < 0.01; two-tailed Student’s t-test).
Further analysis of putative senescent features revealed that Mmp14−/− fibroblasts exhibited a significant proliferative decrease, when compared with wild-type fibroblasts (Fig1H). Additionally, and consistent with the fact that MT1-MMP triggers anti-inflammatory responses (Shimizu-Hirota et al, 2012), we observed signs of a chronic inflammatory response in Mmp14−/− mice, characterized by a significant increase in plasma levels of interleukin-6 (Fig1I) and the chemokine CXCL-1 (Supplementary Fig S3C). These results support the occurrence of a senescence-associated secretory phenotype in Mmp14−/− animals, as previously described in other mouse models of cellular senescence (Osorio et al, 2012; Rodier et al, 2009; Tchkonia et al, 2013). Furthermore, and in agreement with recent data showing that the TGF-β1 pathway mediates paracrine senescence and influences senescence in vivo (Acosta et al, 2013), we observed that Mmp14-deficient mice show a significant increase in TGF-β1 levels (Supplementary Fig S3D).
Finally, we examined telomere length in cells from Mmp14-null mice, as telomere shortening is believed to promote cell senescence and it has been proposed to be a hallmark of biological aging (Lopez-Otin et al, 2013). By using a quantitative PCR method, we observed a significant reduction in telomeric TRF (terminal restriction fragment) in muscles from Mmp14-null mice (Supplementary Fig S3E). Collectively, these findings indicate that the loss of MT1-MMP induces a cell senescence process, which may contribute to explain the phenotypic alterations observed in Mmp14-null mice.
Loss of MT1-MMP causes profound metabolic changes
We next explored whether non-cell-autonomous alterations detected in mouse models with cellular senescence phenotypes could also be present in Mmp14-null mice. We found that Mmp14−/− mice are hypoglycemic, with blood glucose concentration three times lower in these mutant mice when compared to Mmp14+/+ littermates (71 versus 227 mg/dl, P < 0.01) (Fig1J). Due to the short age of these mice, glucose levels were determined before weaning. Nevertheless, these alterations in blood glucose could not be attributed to differences in access to food, as animals from both genotypes had visible milk in their stomachs.
Next, we observed that lack of MT1-MMP causes a profound alteration of the somatotroph axis, a major regulator of longevity from nematodes to man (Lopez-Otin et al, 2013; Niedernhofer et al, 2006; Russell & Kahn, 2007). Circulating levels of insulin-like growth factor 1 (IGF-1) were drastically reduced in plasma from Mmp14−/− animals in comparison with wild-type mice (73 versus 608 pg/ml, P < 0.01) (Fig1K). As IGF-1 synthesis is mainly regulated by circulating growth hormone (GH), we also measured plasma GH concentration in these animals with the finding of very high levels of circulating GH in Mmp14−/− mice when compared to wild-type littermates (13 versus 2.3 pg/ml, P < 0.05) (Fig1L). Furthermore, expression of miR-1, which targets Igf1 and is altered during premature aging (Mariño et al, 2010), was dramatically increased up to 159-fold in the liver of Mmp14−/− animals when compared with wild-type mice (P < 0.01) (Supplementary Fig S3F). Taken together, these results demonstrate that, in addition to cell intrinsic abnormalities, the loss of MT1-MMP causes profound systemic alterations which closely resemble those previously described in different models of cell senescence and premature aging.
Mmp14 deficiency alters nuclear envelope structure and cytoskeleton organization
The fact that mesenchymal tissues appeared to be more affected than other tissues by the lack of MT1-MMP likely reflects the sensitivity of these tissues to mechanical tensions created by their interactions with the ECM (Buxboim et al, 2010). Microscopic analysis of Mmp14−/− fibroblasts revealed profound aberrations in the nuclear envelope. Confocal microscopy with antibodies against the nuclear lamina component lamin A/C showed an abnormal morphology of the nucleus, including the presence of blebs and herniations of the nuclear lamina in Mmp14−/− fibroblasts, which were not observed in Mmp14+/+ cells (Fig2A).
Figure 2.

Structural and morphological alterations of the nuclear envelope and cytoskeleton in Mmp14-deficient mice
- A Immunofluorescence analysis of the nuclear envelope architecture in Mmp14−/− mice stained with an anti-lamin A/C antibody and counterstained with DAPI. The plot represents the percentage of nuclei with alterations, blebbing, or irregular shape in Mmp14+/+ and Mmp14−/− fibroblasts.
- B Western blot analysis of proteins of the nuclear envelope from Mmp14+/+ and Mmp14−/− fibroblasts using specific antibodies against lamin A/C, nesprin-3, Sun-1, and Sun-2. β-actin was used as a loading control.
- C–E Transcriptional analyses by RT–qPCR of nesprin-3, Sun1, and Sun2 were performed in skeletal muscle from control and Mmp14−/− mice. A GAPDH probe was used to normalize the expression level.
- F Immunofluorescence staining of γH2AX foci in fibroblasts from Mmp14−/− and control mice (left panel). The percentage of nuclei that were positive for γH2AX is represented in the graphic (right panel). Three independent experiments were carried out for each genotype.
- G Alteration of cytoskeleton structure around the nucleus in Mmp14−/− fibroblasts compared to wild-type cells. Confocal microscopy of cytoskeleton filaments, tubulin, F-actin (phalloidin), and vimentin stained in red; the nuclear lamin was stained with an anti-lamin A/C antibody (green) and counterstained with DAPI (blue). All the images in the z-stack were used to generate a maximum intensity projection (63× magnification).
Data information: Mean values are represented and error bars indicate SD (*P < 0.05, **P < 0.01; two-tailed Student’s t-test).
Nuclear envelope abnormalities are a common feature of several progeroid syndromes caused by mutations which lead to an abnormal maturation of prelamin A (Agarwal et al, 2003; De Sandre-Giovannoli et al, 2003; Dechat et al, 2008; Eriksson et al, 2003; Gordon et al, 2014; Varela et al, 2008). Mmp14−/− fibroblasts showed marked differences in lamin A levels when compared with cells from wild-type mice, as revealed by Western blot analysis (Fig2B). The connection between nucleus and cytoskeleton involves a series of LINC complex proteins, including nesprins and Sun proteins located in the nuclear membrane (Crisp et al, 2006). Nesprin-3 is an important member of this complex that links the nuclear envelope to intermediate filaments (Ketema & Sonnenberg, 2011). Analysis of nesprin-3 levels revealed a significant increase in this LINC component in Mmp14−/− cells when compared to wild-type cells, both at the protein and at the transcriptional level (Fig2B and C). In addition, the expression of two other components of the LINC complex, Sun-1 and Sun-2, was also slightly altered in muscles from Mmp14-null animals (Fig2B, D and E). These results suggest a deficient connectivity between nuclear envelope and cytoskeleton in mice lacking MT1-MMP, which likely contributes to the nuclear abnormalities observed in these animals.
Previous studies have shown that the nuclear stress caused by alterations in the nuclear envelope is frequently accompanied by DNA damage, which triggers a DNA damage response (Saha et al, 2013). Accordingly, immunofluorescence staining for the histone γH2AX, an early marker of cell response to DNA damage, revealed that Mmp14−/− fibroblasts displayed an increased number of DNA damage foci when compared to control fibroblasts (Fig2F). These nuclear structure abnormalities found in Mmp14−/− cells were also accompanied by marked alterations in the cytoskeletal organization around the nucleus (Fig2G). Immunofluorescence analysis of cytoskeleton filaments showed that Mmp14+/+ cells presented a well-organized structure of the cytoskeleton around the nucleus. However, we observed a marked reduction in the number of actin fibers in the perinuclear and nuclear region of Mmp14−/− fibroblasts (Fig2G). Furthermore, vimentin was also altered in Mmp14−/− cells, displaying an irregular distribution characterized by its marked accumulation at one side of the nucleus and its complete absence in other areas (Fig2G). We also observed a diminished number of tubulin filaments in the nuclear region (Fig2G). These alterations were similar to those reported in cells deficient in nesprin-3 (Postel et al, 2011).
Then, and because the connection between ECM and cytoskeleton is mainly mediated by cell adhesion molecules, we evaluated the putative occurrence of changes in cell adhesion proteins in tissues from Mmp14-null mice. By using RT–qPCR, we found that the expression of integrin β6 was drastically reduced in Mmp14−/− muscles when compared to wild-type animals (0.059 versus 1, P < 0.01) (Supplementary Fig S3G). By contrast, other molecules implicated in cell adhesion, such as osteopontin, showed a marked increase in Mmp14−/− muscle (8.6 versus 1, P < 0.01) (Supplementary Fig S3H). These alterations were accompanied by changes in the activation of downstream effectors, such as the focal adhesion kinase (FAK). Western blot analysis of muscle tissue from Mmp14−/− mice and wild-type littermates showed that phosphorylation of Tyr397 in FAK was increased in Mmp14−/− muscles compared to Mmp14+/+ controls, while total levels of FAK were similar in both genotypes (Supplementary Fig S3I). Taken together, these results indicate that the loss of MT1-MMP causes marked alterations in both nuclear envelope structure and cytoskeletal organization.
MT1-MMP proteolytic activity is required for maintaining cell integrity
To further address the role of MT1-MMP in regulating the cytoskeletal and nuclear structure, we transduced Mmp14-deficient fibroblasts, seeded onto collagen plates, with expression vectors for either wild-type MT1-MMP or a catalytically inactive mutant (E217Q) of this metalloprotease. We observed that overexpression of wild-type MT1-MMP partially rescued the cytoskeletal abnormalities in a significant fraction (60 versus 25%, P < 0.05) of Mmp14-deficient cells, but infection with a catalytically inactive mutant failed to significatively rescue this phenotype (Fig3A), demonstrating that the proteolytic activity of MT1-MMP is required for this process. To determine whether MT1-MMP-mediated remodeling of the ECM is able to induce the phenotypic alterations observed in Mmp14-null mice, we placed Mmp14−/− fibroblasts on a collagen matrix that was previously cultured for 48 h with either wild-type or Mmp14-deficient fibroblasts, which were detached afterward. Mmp14−/− fibroblasts were grown for 48 h on these two collagen matrices and then analyzed (Fig3B). We found that when collagen was precultured on the presence of Mmp14+/+ cells, this collagen matrix was able to reduce in more than 50% the number of Mmp14−/− cells with cytoskeletal abnormalities, while collagen precultured with Mmp14−/− cells failed to rescue this phenotype (Fig3B). These results support the idea that deficient ECM remodeling is responsible for both the nuclear abnormalities and cellular senescence phenotype observed in Mmp14-null mice.
Figure 3.

The proteolytic activity of MT1-MMP is required for maintaining cell integrity
- 293T cells were transfected with retroviral vectors (pMX-GFP) containing the wild-type MT1-MMP or a catalytically inactive mutant (E217Q). Mmp14−/− fibroblasts were infected with retroviral particles containing wild-type MT1-MMP or a catalytically inactive mutant (E217Q). Then, cells were seeded onto collagen plates, cultured for 2 days, and immunostained with vimentin (red) to visualize the fibers (100× magnifications). Right panel represents the percentage of cells with fibers around the nuclear region (n = 50 cells).
- Alterations in the cytoskeleton structure are rescued after ECM exchange. Fibroblasts obtained from either Mmp14+/+ or Mmp14−/− animals were seeded onto collagen plates and cultured for 2 days. Then, cells were detached and Mmp14−/− cells were seeded onto plates with an ECM derived from either Mmp14+/+ fibroblasts (ECM+/+) or Mmp14−/− fibroblasts (ECM−/−). Immunofluorescence analyses were performed using a vimentin antibody (right panel, 100× magnification). Middle panel represents the percentage of cells containing fibers around the nucleus after co-culturing (n = 50 cells).
- The extracellular activity of MT1-MMP is required for maintaining a correct cytoskeleton structure around the nucleus. Mmp14−/− fibroblasts were transduced with pMX-Tomato-expressing vector (Red) and co-cultured in collagen plates for 2 days in the presence of either Mmp14+/+ or Mmp14−/− fibroblasts, and immunofluorescence analysis was performed using a vimentin antibody conjugated to Alexa488 (40× magnification).
- The percentage of cells with fibers around the nuclear region is shown. Only cells with no fibers around the nuclei were considered for the containing.
- SA-β-Gal activity was assayed in co-cultured cells, and the number of positive cells was evaluated. Data represent the mean of 10 random fields for each condition.
Data information: Mean values are represented and error bars indicate SD (*P < 0.05, **P < 0.01; two-tailed Student’s t-test).
Finally, we compared the cytoskeleton organization in pMX-Tomato-expressing Mmp14−/− fibroblasts co-cultured with either Mmp14−/− or Mmp14+/+ fibroblasts (Fig3C). Co-culture of Mmp14−/− fibroblasts with Mmp14−/− cells did not alter the phenotype of these cells, which showed an abnormal distribution of intermediate filaments around the nucleus, while co-culture with wild-type fibroblasts was sufficient to partially rescue the cytoskeletal abnormalities observed in Mmp14-deficient cells (Fig3D). Moreover, the senescence phenotype observed in Mmp14−/− fibroblasts was partially rescued when these cells were co-cultured with Mmp14+/+ cells (Fig3E). Altogether, these data rule out the hypothesis that the Mmp14-dependent organization of the cytoskeleton and the cellular senescence caused by loss of MT1-MMP was due to potential intracellular activities of this protease (Golubkov et al, 2005), and support an extracellular activity responsible for this effect.
Retinoic acid ameliorates nuclear alterations and increases life span of Mmp14-null mice
Recent reports have linked ECM stiffness with the accumulation of lamin A in the nucleus, which drives nuclear translocation of the retinoic acid receptor beta to stimulate LMNA transcription, whereas treatment with all-trans retinoic acid (ATRA) agonists decreases lamin A levels (Swift et al, 2013). Although retinoids were first described as inhibitors of MMP production (Dutta et al, 2010; Rankin et al, 2013; Schroen & Brinckerhoff, 1996), further studies demonstrated that these compounds induce the expression of several MMP family members, such as collagenase-3 and MT1-MMP, in chondrocytic cells (Jimenez et al, 2001; Zhang & Ross, 2013). We asked whether ATRA treatment could restore the cellular alterations observed in Mmp14-deficient mice. To explore this possibility, fibroblasts were treated with 1 μM ATRA for 2 days and, then, the cellular abnormalities of Mmp14-deficient mice were analyzed. We observed a remarkable reduction in the nuclear alterations characteristic of cells from these mutant mice (Fig4A), and this reduction was accompanied by a decrease in the number of senescent cells in vivo as demonstrated by SA-β-Gal assay (Fig4B). Furthermore, the cyclin-dependent kinase inhibitor 1A (p21CIP1/WAF1), whose expression is up-regulated in Mmp14−/− mice, was significantly reduced after ATRA treatment (Fig4C). Together, these results reflect an ATRA-mediated improvement in the senescence phenotype of Mmp14-null mice. We also observed that ATRA treatment partially restored the marked alterations in the cytoskeletal organization around the nucleus in these Mmp14−/− cells (Fig4D and E; Supplementary Fig S4A and B). Thus, this treatment increased the percentage of cells containing fibers around the nucleus in more than 50% (Fig4D). Immunofluorescence analysis of cytoskeleton filaments showed that ATRA-treated Mmp14−/− cells presented a much more organized structure of the cytoskeleton around the nucleus than that present in non-treated Mmp14−/− cells (Fig4E; Supplementary Fig S4A and B). We also detected an increase in the number of actin fibers in the perinuclear and nuclear region (Fig4E). Moreover, we observed the presence of a higher number of vimentin and tubulin filaments around the nuclear region in ATRA-treated Mmp14−/− cells (Supplementary Fig S4A and B). These results suggest that MT1-MMP plays a pivotal role in shaping the intracellular structure and organization of the cells through a signaling pathway influenced by retinoids.
Figure 4.

ATRA treatment partially restores the cellullar defects observed in Mmp14-deficient mice
- A Cells were seeded onto a collagen matrix, treated with ATRA, and stained with DAPI. The plot represents the percentage of cells with nuclear abnormalities in control or ATRA-treated fibroblasts for each genotype (n = 50 cells, 10 random fields). Each experiment was performed with cells derived from three independent primary cultures.
- B Cells for each genotype were treated with 1 μM ATRA for 2 days, and SA-β-Gal assays were performed. A representative image is shown on the right panel.
- C RT–qPCR expression of p21 was analyzed in muscles from treated or control mice for each genotype (n = 4 mice).
- D, E Immunofluorescence analysis of cytoskeleton filaments was performed in fibroblast cell lines. Cells were seeded onto collagen matrix and treated with ATRA for 2 days. Actin stress fibers were visualized using phalloidin–TRITC, and the nuclear structure was marked with anti-nesprin-3 conjugated with Alexa488, and counterstained with DAPI. All the images in the z-stack were used to generate a maximum intensity projection (63× magnification). A representative cell is shown in the bottom panel. Cells containing fibers around the nucleus were quantified by analyzing 10 random fields. Each experiment was done in triplicate (n = 3).
Data information: Mean values are represented and error bars indicate SD (*P < 0.05; **P < 0.01; two-tailed Student’s t-test).
We finally analyzed whether ATRA treatment could modify the expression of several components of the LINC complex that were altered in Mmp14-deficient mice. We observed that ATRA-treated Mmp14−/− mice restored the expression of lamin A to levels similar to those of wild-type littermates (Supplementary Fig S5A–D). ATRA treatment also restored expression of other components of the LINC complex, including nesprin-3 and Sun proteins (Supplementary Fig S5A–D). Furthermore, the up-regulation of inflammatory signaling pathway observed in Mmp14-null mice was reduced after ATRA treatment in these mice (Supplementary Fig S5E).
On this basis, together with the finding that nuclear envelope abnormalities are associated with changes in retinoic acid signaling (Swift et al, 2013), we decided to examine whether retinoic acid treatment could have some biological effect on Mmp14-deficient mice. Thus, we treated control and Mmp14-deficient mice with ATRA, and the animals were monitored during their lifetimes. Interestingly, we found that this treatment improved body weights and significantly extended life span of mutant mice when compared with untreated Mmp14-null mice (Fig5A and B). The mean survival of ATRA-treated Mmp14-null mice was extended from 14 days to 23 days (P < 0.01) (Fig5B). A representative photograph of 14-day-old ATRA-treated and control Mmp14−/− mice is shown in Fig5C. Notably, ATRA-treated Mmp14-null mice exhibit a marked increase in cartilage resorption and a reorganization of the growth plate, when compared with the untreated mice which show profound deficiencies in bone development (Fig5D). We also observed that, after ATRA treatment, Mmp14-null mice presented a significant increase in hypodermis thickness (Fig5E and F; Supplementary Fig S5F), thus ameliorating the characteristic skin abnormalities found in these mutant mice. These results demonstrate that ATRA treatment significantly increases life span and ameliorates some phenotypic defects present in Mmp14-null mice. We also observed that ATRA treatment of Mmp14-null mice reduced SA-β-Gal activity in adipose tissue (Fig5G), as well as the senescence process observed in cardiac tissues (Fig5H). Our results suggest that the absence of MT1-MMP activity may generate a mechanical tension that affects the LINC complex, and therefore, the nuclear structure and these alterations can be partially ameliorated by retinoic acid treatment.
Figure 5.
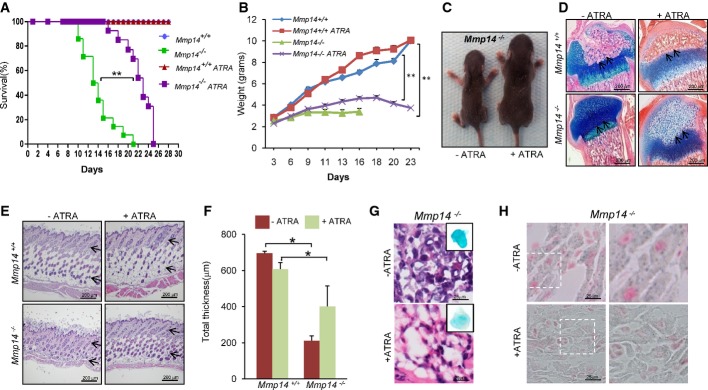
All-trans retinoic acid increases life span and ameliorates the structural defects observed in Mmp14-null mice
Mice were treated with ATRA (0.5 mg/kg) or vehicle every 2 days, and body weight and life span were monitored.
- Kaplan–Meier survival plots for control and Mmp14-deficient mice treated with ATRA (n = 13) (**P < 0.01 for all the comparisons, log-rank Mantel–Cox test).
- Cumulative plot of body weight versus age. Dots represents mean value (n = 13 for each genotype) (**P < 0.001).
- Representative photographs of 14-day-old control or ATRA-treated Mmp14−/− mice.
- Alcian blue staining of femurs from control and Mmp14-null mice treated with ATRA. Sections were obtained using an Olympus BX53 (4× magnification).
- H&E staining of skin from control or ATRA-treated mice. Black arrows indicate the dermis and the subcutaneous fat layer.
- The thickness of the skin was determined by counting five random fields that were measured, and the mean of each is represented (n = 4 mice per genotype) (*P < 0.05; two-tailed Student’s t-test).
- SA-β-Gal activity was assayed in adipose tissue from control and Mmp14−/− mice (n = 3) for each condition.
- Sudan black staining counterstained with nuclear fast red to visualize senescence activity in hearts from Mmp14-null mice after ATRA treatment (40× magnification). A detailed view is shown on the right panel.
Data information: Mean values are represented and error bars indicate SD (*P < 0.05; **P < 0.01 two-tailed Student’s t-test).
Discussion
In this work, we found that a MT1-MMP-dependent signaling between a defective ECM and the cell nucleus activates a senescence response that could explain some of the phenotypes caused by the loss of Mmp14 in mouse. This senescence process involves p16INK4a and p21CIP1/WAF1 and is also characterized by a series of archetypal senescent features, such as the presence of marked nuclear envelope abnormalities, the occurrence of a reduced proliferative potential, the induction of a chronic DNA damage response, and the triggering of a senescence-associated secretory phenotype which involves the production of several inflammatory factors. We also show that this senescence program can be partially reversed by interventions on retinoid receptor signaling pathways, as demonstrated by the fact that treatment with ATRA increases life span and restores some of the phenotypic alterations observed in Mmp14-deficient mice.
Interestingly, most molecular and cellular features observed in Mmp14−/− mice have also been described in different human progeroid syndromes, including HGPS and Nestor–Guillermo progeria syndrome (NGPS) (Cabanillas et al, 2011; Gordon et al, 2014; Puente et al, 2011), as well as in animal models of premature aging (Osorio et al, 2011; Pendas et al, 2002; Sullivan et al, 1999). In these cases, mutations in genes coding for nuclear envelope proteins, such as lamin A/C and BAF, or for enzymes involved in their maturation, such as Zmpste24 metalloproteinase, cause nuclear abnormalities. The unexpected similarities between Mmp14-deficient mice and all these progeroid models include cell-autonomous processes, such as nuclear envelope abnormalities, increased DNA damage, and the activation of a cell senescence program. In addition, mice lacking MT1-MMP share a series of systemic alterations with different models of premature aging, including metabolic and somatotroph axis changes, as well as the occurrence of elevated SA-β-Gal activity in different tissues.
It is well established that the absence of MT1-MMP proteolytic activity at the cellular surface impairs ECM remodeling, preventing the completion of different developmental programs, including skeletal growth and formation of connective and adipose tissues (Chun et al, 2006; Holmbeck et al, 1999). Beyond these previous studies, we demonstrate in this work that the pericellular proteolytic activity of MT1-MMP plays a pivotal role in shaping the intracellular structure and organization. In this regard, we have found that the loss of MT1-MMP alters the expression of several components of the LINC complex, including nesprin-3 and Sun proteins. It is likely that the mechanical tension generated by the absence of MT1-MMP activity is transmitted by the cytoskeleton and affects nesprin-3 interactions, including its binding to Sun proteins at the nuclear envelope or to plectin at intermediate filaments (Ketema & Sonnenberg, 2011). The relevance of the LINC complex in connecting ECM and nuclear envelope is further reinforced by the recent implication of Sun-1 in the development of progeroid syndromes. Thus, in a mouse model of HGPS, proliferative arrest of postnatal fibroblasts is caused by their inability to produce a functional ECM (Hernandez et al, 2010), while loss of Sun1 in these mice partially rescues their progeroid phenotypes and extends their longevity (Chen et al, 2012). Consistent with these findings, our results provide evidence that the loss of MT1-MMP likely generates a transduction force that modifies the nuclear envelope structure in a process in which several LINC complex components are altered.
The similarities observed between Mmp14-null mice and different progeroid models, both in phenotype changes and in altered pathways, highlight the potential role of MT1-MMP in the senescence process. Interestingly, the expression of genes encoding ECM proteins or ECM remodeling enzymes is substantially altered in normal and pathological aging (de la Rosa et al, 2013; Hernandez et al, 2010; Lopez-Otin et al, 2013), being of especial relevance the observed down-regulation of MT1-MMP in HGPS cells (Csoka et al, 2004), which provides additional support to our proposal that this metalloproteinase modulates senescence processes.
The results presented herein link, for the first time, the activity of an ECM remodeling protease to the nuclear architecture organization. Our studies indicate that the process by which MT1-MMP is able to impact cell function is not due to the proteolytic processing of soluble factors. Conversely, we have found that ECM exchange experiments are able to restore a normal cytoskeletal structure in Mmp14−/− cells, demonstrating that MT1-MMP-remodeled ECM is required for maintaining a normal cellular structure. In the absence of MT1-MMP activity, the mechanical forces shaping the cell are altered, generating an abnormal tension in the cytoskeleton. We have also shown that the expression of integrin β6 is altered in skeletal muscle from Mmp14−/− mice. Notably, the recent generation of another conditional knockout model for Mmp14 has demonstrated the role of this protease in skeletal stem cell commitment in mouse (Tang et al, 2013). In these mutant animals, an integrin β1-dependent signaling cascade mediates the commitment of skeletal stem cells, reinforcing our proposal that integrin receptors are involved in the outside-in signaling initiated by MT1-MMP remodeling of the ECM.
Recent studies have described an association between ECM stiffness and nuclear accumulation of lamin A, which leads to the activation of retinoid signaling pathways (Swift et al, 2013). Moreover, age-related alterations in vitamin A metabolism have been described in rats and humans (Feart et al, 2005). Interestingly, we have found that treatment of Mmp14-deficient mice with ATRA increased life span and rescued some of the cell senescence and progeroid phenotypes characteristic of these mutant mice. These results suggest that the changes in the nuclear envelope structure caused by the loss of MT1-MMP affect the expression of different genes implicated in the normal function of the cell.
In summary, our results reveal a novel function for the pericellular collagenase MT1-MMP in maintaining the functional structure of the nuclear envelope and preventing cellular senescence. The proteolytic activity of MT1-MMP is required to remodel the ECM, and the mechanical properties of the ECM have an important impact on the nuclear lamina structure by mechanotransduction, in a process involving integrins, the cytoskeleton, and components of the LINC complex. Accordingly, in the absence of MT1-MMP, there are profound alterations in the nuclear architecture and the cytoskeleton, which induce a cellular senescence response and contribute to the development of severe phenotypic abnormalities. Finally, the finding that ATRA ameliorates cell senescence features and extends life span in Mmp14-null mice opens the possibility of exploring the putative therapeutic relevance of retinoids in several pathological conditions associated with dysregulated senescence processes, including devastating progeroid syndromes such as HGPS and NGPS.
Materials and Methods
Generation of conditional knockout mice
We generated a conditional Mmp14 construct by cloning a 14.5-kb fragment obtained from BAC bMQ-414L9, into the pL253 plasmid corresponding to murine Mmp14 (from 2 kb upstream of exon 1 to 2.5 kb downstream of exon 10). We also introduced two loxP sites flanking exons 4 and 5, using the recombineering technology as previously described (Chan et al, 2007). The retrieval vector was constructed by cloning two short homologous arms generated by PCR amplification of bMQ-414L9 BAC DNA with the following primer sets: 5′-ATCAAGTCATTTTCCCCACCAC-3′ and 5′-CTGAGCTGTGCATGGTCTCTT-3′ for the 5′-homologous arm, and 5′-ATCCAGAGGGAATTCTGCAAAG-3′ and 5′-GACCTTATGACCACGAAGGCA-3′ for the 3′-homologous arm. These arms were first cloned into pL253 and sequenced, then cut with SalI/EcoRI (5′RV) or BamHI/NotI (3′RV), and subcloned into SalI/EcoRI/NotI-digested pL452 plasmid. The linearized vector was used to retrieve a 14.5-kb fragment from BAC bMQ-414L9 by gap repair in EL350 cells. We next inserted a loxP site between exons 3 and 4, and a FRT-PGK-Neo-FRT-LoxP cassette between exons 5 and 6 to generate the Mmp14-flox/flox construct. The linearized vector was electroporated into EL350 bacterial cells using a Bio-Rad electroporator set to 1.75 kV, 25 μF, 200 Ω. After selection with different antibiotics, the final targeting vector was obtained and used for homologous recombination in ES cells. ES cells were injected into C57BL/6 blastocysts to generate chimeras. Once germ line transmission of the targeted allele was confirmed by PCR, we crossed the descendent with FLP transgenic mice to delete the NeoR cassette. The final targeted allele contains a single loxP site located at introns 3–4 of Mmp14 and a FRT/loxP site at introns 5–6. We next crossed these loxP/loxP mice with Cre transgenic mice obtained from Jackson Laboratory No 006054, to generate the Mmp14+/− mice from which, by subsequent crossing, we generated the Mmp14−/− mice used for these studies. Mmp14 conditional mice were backcrossed with C57BL/6J mice for five generations and wild-type littermates served as controls.
Animal experiments
All the animal experiments were performed in accordance with the guidelines of the Committee for Animal Experimentation of the Universidad de Oviedo. Mice were treated intraperitoneally with all-trans retinoic acid (Sigma) at a concentration of 0.5 mg/kg and starting at day 3 after birth. Control littermates were treated with vehicle. For histological analysis, samples were extracted and fixed in 4% paraformaldehyde in PBS.
Retroviral infection
HEK-293T cells were transfected with pMX-GFP plasmids (Cell Biolabs, Inc) containing either wild-type full length MT1-MMP or a site-specific mutant (E217Q) affecting the active site, together with a pCL-Eco package system, kindly provided by Dr. J. M. Silva (Columbia University, New York, USA). In brief, a mixture of 1 μg of the desired plasmid and 1 μg of each retroviral helper was transfected using Lipofectamine Plus (Invitrogen), following manufacturer’s instructions. Medium was removed 24 h after transfection, and fresh medium was added to the plate. Cell supernatants were collected at 24 and 48 h, cleared by centrifugation for 10 min and filtered through a 0.45-μm sterile filter. Fresh isolated fibroblasts from Mmp14−/− mice (passage 0) were incubated with the supernatant with 8 mg/ml polybrene for 2 days at 37°C. Cell transduction was evaluated after 3 days in culture using a fluorescence microscope.
RNA extraction and RT–qPCR
RNA was extracted from different tissues and cells using TRIzol (Invitrogen), following manufacturer’s instructions. Samples were quantified and evaluated for purity (260/280 nm ratio) with a NanoDrop ND-1000 spectrophotometer. cDNA was synthesized with 1–4 μg of total RNA with the ThermoScript RT–PCR system (Invitrogen). RT–qPCR was performed in triplicate for each experimental condition using either TaqMan PCR Master Mix or SYBR Green PCR Master Mix (Applied Biosystems), according to the manufacturer’s instructions. To normalize mRNA levels, GAPDH or ACTB probes were used. The primers used are listed in Supplementary Table S1.
microRNA analysis
miR-1 expression analysis was performed as previously described (Ugalde et al, 2011). For RT–qPCR analysis, total RNA was prepared using miRVANA™ miRNA isolation Kit (Ambion, Austin, TX) and RNA samples were quantified and evaluated as mentioned above. miRNA detection was performed using TaqMan miRNA expression assays (Applied Biosystems). Briefly, 10 ng of total RNA was reverse-transcribed using TaqMan miRNA reverse transcription kit (Applied Biosystems) and PCR-amplified using an Applied Biosystems 7300 Real-Time PCR system. As an internal control, miRNA expression was normalized to snoRNA202 for mouse samples, using TaqMan Gene Expression Assays (Applied Biosystems). All protocols were carried out according to the manufacturer’s instructions.
Histological analysis
Tissues were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) and stored in 70% ethanol. Fixed tissues were embedded in paraffin by standard procedures. Blocks were sectioned (5 μm) and stained with hematoxylin and eosin (H&E). In other experiments, samples were treated with 15% sucrose in PBS for 4 h, and then with 30% sucrose solution overnight, embedded in Tissue-Tek OCT compound (Sakura Fine Technical Co. Ltd.), and stored at −70°C. Finally, samples were sectioned at 5 μm thickness with a cryostat (CM3050 S; Leica). For muscle studies, samples were obtained from quadriceps and dissected under the stereoscopic microscope. Using a thin gauge, fibers were oriented transversally and snap-frozen on liquid nitrogen-freezing isopentane. Then, 5-μm sections were obtained with a cryostat and stored at −70°C until used. Histochemical techniques including H&E, Masson trichrome, SDH, NADH, PAS, picrosirius red, and sudan black were performed following manufacturer’s instructions, while studies on muscle were carried out as previously described (Valdes-Mas et al, 2014). For Alcian blue staining of forepaws, tissues were decalcified, deparaffinized, stained with Alcian blue solution pH 2.5, and counterstained with eosin. Representative pictures were taken using an Olympus BX53 microscope with a DP73 digital camera using the analysis software CellSens. For Alcian blue–Alizarin red skulls staining, skin and organs were removed, fixed in 95% EtOH for 2 days. Then, skulls were incubated with Alcian blue solution for 3 days. The staining was washed with 95% EtOH, cleared with 2% KOH for 24 h, and finally stained with 0.03% Alizarin red for 24 h and cleared with a 1% KOH-20% glycerol solution.
Zymography
MMP-2 activity was analyzed by gelatin zymography as previously described (Gutierrez-Fernandez et al, 2007). Liver extracts from Mmp14+/+ and Mmp14−/− mice were homogenized in a modified RIPA lysis buffer (100 mM Tris/HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.1% SDS) containing complete protease inhibitor cocktail without EDTA (Roche, Mannheim, Germany), and protein content was quantified using the BCA method (Pierce, Rockford, IL, USA). Proteins (5 μg) were separated in an 8% SDS–PAGE gel containing 0.2% gelatin. Gels were washed twice for 30 min with 2.5% Triton X-100 and incubated at 37°C for 4 h in the presence of 10 mM Tris (pH 8.0) and 5 mM CaCl2, and stained with Coomassie Brilliant Blue to reveal gelatinolytic bands.
Blood and plasma parameters
Blood glucose was measured with an Accu-Check glucometer (Roche Diagnostics) using blood extracted after decapitation. To obtain serum, blood was centrifuged immediately after collection at 3,000 g at 4°C and the supernatant was collected and stored at −20°C until analysis. Serum IGF-1 and GH were determined by using the Quantikine ELISA kit (R&D systems) and the Linco ELISA kit, respectively. For quantification of IL-6 and CXCL1 cytokines, a Quantikine ELISA kit (R&D Systems) was used. All protocols were performed according to the manufacturer’s instructions.
Isolation and culture of fibroblasts
Fibroblasts were isolated from 2-week-old wild-type (Mmp14+/+) or Mmp14-deficient (Mmp14−/−) mice as previously described (Varela et al, 2005). In brief, tissues were minced, digested with collagenase–dispase (4 mg/ml) for 45 min at 37°C and then inactivated with complete media. The following day, cells were separated using a 100 μM cell strainer and plated in a 6-well plate. Cells were maintained in DMEM supplemented with 15% FBS, 1M HEPES, sodium pyruvate, non-essential amino acids, L-glutamine, and β-mercaptoethanol. For co-culture experiments, 12-well plates were coated with 150 μg of rat tail collagen type I (Roche) and 1 × 104 Mmp14−/− cells transfected with pMX-Tomato vector (Mmp14−/−-pMX-Tomato) or a mixture of Mmp14−/−-pMX-Tomato (5 × 103) and Mmp14+/+ cells (5 × 103) were seeded onto the plates. After 2 days in culture, cells were fixed and immunofluorescence assays were performed. For studies involving the exchange of the ECM, plates were coated with collagen as described above, and 3 × 105 Mmp14+/+ and Mmp14−/− cells were seeded onto the plates for 2 days. Then, cells were detached using 1 mM EDTA and counted and the same amount of Mmp14−/− cells (1 × 104) were seeded onto plates containing either wild-type Mmp14 ECM+/+ or Mmp14 KO ECM−/−. After 2 days in culture, cells were fixed and immunofluorescence assays were performed.
Collagen degradation assay
Twenty-four-well plates were coated with 100 μg/well of collagen type I (Roche) and 5 × 104 fresh isolated fibroblasts from either Mmp14+/+ or Mmp14−/− mice were seeded in 35 μl of medium in the center of each well and allowed to adhere as previously described. Then, medium was replaced, and plates were washed with serum-free medium and incubated with 10 ng/mL of PDGF-BB, GM6001 (25 mM), or BB-94 (5 mM) for 5 days. Cells were removed using trypsin–EDTA 0.025%, and collagen was stained with Coomassie Brilliant Blue. The amount of newly synthesized collagen (acid- and pepsin-soluble collagen) levels was quantified using the Sircol™ Collagen Assay (Biocolor, Life Science) according to manufacturer’s instructions.
Cell senescence, BrdU incorporation, and cell proliferation analysis
Cell senescence was assessed by detecting the senescence-associated β-galactosidase activity at pH 6.0 (SA-β-Gal) using a commercial kit (Cell Signaling). Staining was performed in 6-well plates and in vivo tissues, and the SA-βGal-positive cells were detected by phase contrast microscopy using a Zeiss Axiovert 200M fluorescence microscope (Zeiss). Quantification was performed by counting the positive cells present in 20 independent fields of view at 10× magnification, and images were captured and processed using Adobe Photoshop CS3. For BrdU incorporation assay, cells were incubated with medium containing 10 μM BrdU for 18 h. Upon BrdU removal, cells were fixed and analyzed by immunofluorescence. To visualize BrdU, cells were treated with 2 M HCl before blocking buffer to open up DNA double strand and to expose the BrdU epitope. To measure cell proliferation, cells were seeded onto plates at 1 × 106 cell density, and after 3 days, cells were detach and counted using a Neubauer chamber.
Western blot analysis
For protein extraction, cultured cells were washed twice with 1× PBS and resuspended in RIPA buffer (100 mM Tris pH 7.4, 150 mM NaCl, 10 mM EDTA, 1% Triton X-100, 1% deoxycholic acid, and 0.1% SDS) supplemented with complete protease inhibitor cocktail without EDTA (Roche), sodium orthovanadate (1 mM), and NaF (1 mM). Tissues were snap-frozen in liquid nitrogen. Frozen tissues (∼50 mg in each sample) were homogenized in 300 μL of 100 mM Tris–HCl (pH 7.4), 2% SDS, and 50 mM EDTA with a Polytron homogenizer. Protein concentration was evaluated with the bicinchoninic acid technique (Pierce BCA protein assay kit). Proteins were run in SDS–PAGE gels, transferred to nitrocellulose membranes, blocked with 5% non-fat dry milk in TBS-T buffer (20 mM Tris pH 7.4, 150 mM NaCl, 0.05% Tween 20), and incubated overnight at 4°C with the following primary antibodies: MT1-MMP (ab 38971), nesprin-3 (ab123031), Sun-1 (ab74758), and Sun-2 (ab87036) (Abcam); FAK and FAKY397 (Cell Signaling); p16 (sc-373695) and lamin A/C (sc-20681) (Santa Cruz Biotechnology); and β-actin and tubulin (AC-40, Sigma). Primary antibodies were detected with horseradish peroxidase-conjugated species-specific secondary antibodies (Jackson Immunoresearch or Thermo Scientific) with Luminata Forte Western HRP Substrate (Millipore) using a LAS-300 (Fujifilm).
Immunofluorescence analysis
For immunofluorescence analysis, cells were fixed in 4% paraformaldehyde solution, rinsed in PBS, and permeabilized with 0.5% Triton X-100. For γH2AX staining, the permeabilization step was performed with 0.1% sodium citrate and 0.1% Triton X-100 as previously described (Osorio et al, 2011). Cells were incubated with primary antibodies diluted in 10% goat serum in 1× PBS for 1–3 h at room temperature. After washing with PBS-T, slides were incubated with 1:500 Alexa 488-, 546-, or 633-conjugated secondary antibody (Life Technologies) for 1 h at 25°C. After washing, nuclei were counterstained with DAPI (Roche), and slides were mounted in VectaShield mounting medium (Vector Laboratories). Micrographs were obtained with an Axioplan-2 Zeiss fluorescent microscope (Zeiss), and images were captured with a charge-coupled device camera (Photometrics SenSys). Nuclei from Mmp14−/− and Mmp14+/+ fibroblasts were counted (n = 200 for each experiment). To determine the actin stress fiber distribution in the cell and the intermediate filaments, all the images at the precise planes in the z-stack were used to generate a maximum intensity projection. Cells were classified based on the presence of short actin fragments around the nucleus, disturbed or collapsed perinuclear vimentin network organization, or the absence of fibers over the nuclei. The following antibodies were used: vimentin-XP® (#5741, Cell Signaling), alpha-tubulin (CBL270, Millipore), lamin A/C (sc-20681 and sc-6215 Santa Cruz Biotechnology), γH2AX (JBW301 Millipore), nesprin-3 (ab-57397, Abcam), BrdU (ab63626, Abcam), and HP1γ (MAB3450, Millipore).
Statistical analysis
All experimental data are reported as means. We performed statistical analysis of the differences between mouse cohorts, or between wild-type and KO cells with the nonparametric Student’s t-test. Kaplan–Meier survival analysis was performed using the MedCalc statistical package. We used MS Excel or GraphPad Prism software for calculations and expressed the results as the means ± SDs.
Acknowledgments
We are very grateful to Drs. J.M.P. Freije, A. R. Folgueras, A. Ramsay, and M. Mittelbrunn for helpful comments; Drs. J. Cadiñanos and P. Liu for reagents; Drs. M.A. Guervós and M.T. Fernández for assistance with the Confocal Microscopy and Histological Analysis; and D. A. Puente, S. Alvarez, F. Rodríguez, R. Feijoo, and A. Moyano for excellent technical assistance. This work was supported by grants from MINECO and RTICC (Spain). The IUOPA was supported by Obra Social Cajastur-Asturias. C.L-O. is an Investigator of the Botin Foundation supported by Banco Santander through its Santander Universities Global Division.
Author contributions
AG-F and CS-V performed experimental work, data interpretation, and preparation of the manuscript. FGO performed experimental work and data interpretation. JG-A, CG, and AA performed experimental work. MSF-G performed histological analysis and AF performed animal procedures. XSP and CL-O supervised research and project planning, data interpretation, and preparation of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Supplementary Figure Legends
Review Process File
References
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978–990. doi: 10.1038/ncb2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- Atkinson JJ, Holmbeck K, Yamada S, Birkedal-Hansen H, Parks WC, Senior RM. Membrane-type 1 matrix metalloproteinase is required for normal alveolar development. Dev Dyn. 2005;232:1079–1090. doi: 10.1002/dvdy.20267. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Weaver RL, van Deursen JM. p21 both attenuates and drives senescence and aging in BubR1 progeroid mice. Cell Rep. 2013;3:1164–1174. doi: 10.1016/j.celrep.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbin M, Fueyo A, Tester AM, Pendas AM, Pitiot AS, Astudillo A, Overall CM, Shapiro SD, Lopez-Otin C. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet. 2003;35:252–257. doi: 10.1038/ng1249. [DOI] [PubMed] [Google Scholar]
- Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxboim A, Ivanovska IL, Discher DE. Matrix elasticity, cytoskeletal forces and physics of the nucleus: how deeply do cells ‘feel’ outside and in? J Cell Sci. 2010;123:297–308. doi: 10.1242/jcs.041186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxboim A, Swift J, Irianto J, Spinler KR, Dingal PC, Athirasala A, Kao YR, Cho S, Harada T, Shin JW, Discher DE. Matrix elasticity regulates lamin-A, C phosphorylation and turnover with feedback to actomyosin. Curr Biol. 2014;24:1909–1917. doi: 10.1016/j.cub.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabanillas R, Cadinanos J, Villameytide JA, Perez M, Longo J, Richard JM, Alvarez R, Duran NS, Illan R, Gonzalez DJ, Lopez-Otin C. Nestor-Guillermo progeria syndrome: a novel premature aging condition with early onset and chronic development caused by BANF1 mutations. Am J Med Genet A. 2011;155A:2617–2625. doi: 10.1002/ajmg.a.34249. [DOI] [PubMed] [Google Scholar]
- Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–57. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, Stahl PD, Hodzic D. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol. 2006;172:41–53. doi: 10.1083/jcb.200509124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csoka AB, English SB, Simkevich CP, Ginzinger DG, Butte AJ, Schatten GP, Rothman FG, Sedivy JM. Genome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosis. Aging Cell. 2004;3:235–243. doi: 10.1111/j.1474-9728.2004.00105.x. [DOI] [PubMed] [Google Scholar]
- Chan W, Costantino N, Li R, Lee SC, Su Q, Melvin D, Court DL, Liu P. A recombineering based approach for high-throughput conditional knockout targeting vector construction. Nucleic Acids Res. 2007;35:e64. doi: 10.1093/nar/gkm163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Chi YH, Mutalif RA, Starost MF, Myers TG, Anderson SA, Stewart CL, Jeang KT. Accumulation of the inner nuclear envelope protein Sun1 is pathogenic in progeric and dystrophic laminopathies. Cell. 2012;149:565–577. doi: 10.1016/j.cell.2012.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TH, Hotary KB, Sabeh F, Saltiel AR, Allen ED, Weiss SJ. A pericellular collagenase directs the 3-dimensional development of white adipose tissue. Cell. 2006;125:577–591. doi: 10.1016/j.cell.2006.02.050. [DOI] [PubMed] [Google Scholar]
- de la Rosa J, Freije JM, Cabanillas R, Osorio FG, Fraga MF, Fernandez-Garcia MS, Rad R, Fanjul V, Ugalde AP, Liang Q, Prosser HM, Bradley A, Cadinanos J, Lopez-Otin C. Prelamin A causes progeria through cell-extrinsic mechanisms and prevents cancer invasion. Nat Commun. 2013;4:2268. doi: 10.1038/ncomms3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Levy N. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22:832–853. doi: 10.1101/gad.1652708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A, Sen T, Chatterjee A. All-trans retinoic acid (ATRA) downregulates MMP-9 by modulating its regulatory molecules. Cell Adh Migr. 2010;4:409–418. doi: 10.4161/cam.4.3.11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyckmans J, Boudou T, Yu X, Chen CS. A hitchhiker’s guide to mechanobiology. Dev Cell. 2011;21:35–47. doi: 10.1016/j.devcel.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanjul-Fernandez M, Folgueras AR, Cabrera S, Lopez-Otin C. Matrix metalloproteinases: evolution, gene regulation and functional analysis in mouse models. Biochim Biophys Acta. 2010;1803:3–19. doi: 10.1016/j.bbamcr.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Fanjul-Fernandez M, Folgueras AR, Fueyo A, Balbin M, Suarez MF, Fernandez-Garcia MS, Shapiro SD, Freije JM, Lopez-Otin C. Matrix metalloproteinase Mmp-1a is dispensable for normal growth and fertility in mice and promotes lung cancer progression by modulating inflammatory responses. J Biol Chem. 2013;288:14647–14656. doi: 10.1074/jbc.M112.439893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feart C, Pallet V, Boucheron C, Higueret D, Alfos S, Letenneur L, Dartigues JF, Higueret P. Aging affects the retinoic acid and the triiodothyronine nuclear receptor mRNA expression in human peripheral blood mononuclear cells. Eur J Endocrinol. 2005;152:449–458. doi: 10.1530/eje.1.01858. [DOI] [PubMed] [Google Scholar]
- Georgakopoulou EA, Tsimaratou K, Evangelou K, Fernandez Marcos PJ, Zoumpourlis V, Trougakos IP, Kletsas D, Bartek J, Serrano M, Gorgoulis VG. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging (Albany NY) 2013;5:37–50. doi: 10.18632/aging.100527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubkov VS, Boyd S, Savinov AY, Chekanov AV, Osterman AL, Remacle A, Rozanov DV, Doxsey SJ, Strongin AY. Membrane type-1 matrix metalloproteinase (MT1-MMP) exhibits an important intracellular cleavage function and causes chromosome instability. J Biol Chem. 2005;280:25079–25086. doi: 10.1074/jbc.M502779200. [DOI] [PubMed] [Google Scholar]
- Gordon LB, Rothman FG, Lopez-Otin C, Misteli T. Progeria: a paradigm for translational medicine. Cell. 2014;156:400–407. doi: 10.1016/j.cell.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Fernandez A, Inada M, Balbin M, Fueyo A, Pitiot AS, Astudillo A, Hirose K, Hirata M, Shapiro SD, Noel A, Werb Z, Krane SM, Lopez-Otin C, Puente XS. Increased inflammation delays wound healing in mice deficient in collagenase-2 (MMP-8) FASEB J. 2007;21:2580–2591. doi: 10.1096/fj.06-7860com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez L, Roux KJ, Wong ES, Mounkes LC, Mutalif R, Navasankari R, Rai B, Cool S, Jeong JW, Wang H, Lee HS, Kozlov S, Grunert M, Keeble T, Jones CM, Meta MD, Young SG, Daar IO, Burke B, Perantoni AO, et al. Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev Cell. 2010;19:413–425. doi: 10.1016/j.devcel.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA, Mankani M, Robey PG, Poole AR, Pidoux I, Ward JM, Birkedal-Hansen H. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99:81–92. doi: 10.1016/s0092-8674(00)80064-1. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Inada M, Wang Y, Byrne MH, Rahman MU, Miyaura C, Lopez-Otin C, Krane SM. Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc Natl Acad Sci USA. 2004;101:17192–17197. doi: 10.1073/pnas.0407788101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez MJ, Balbin M, Alvarez J, Komori T, Bianco P, Holmbeck K, Birkedal-Hansen H, Lopez JM, Lopez-Otin C. A regulatory cascade involving retinoic acid, Cbfa1, and matrix metalloproteinases is coupled to the development of a process of perichondrial invasion and osteogenic differentiation during bone formation. J Cell Biol. 2001;155:1333–1344. doi: 10.1083/jcb.200106147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketema M, Sonnenberg A. Nesprin-3: a versatile connector between the nucleus and the cytoskeleton. Biochem Soc Trans. 2011;39:1719–1724. doi: 10.1042/BST20110669. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowes BD, Minobe W, Abraham WT, Rizeq MN, Bohlmeyer TJ, Quaife RA, Roden RL, Dutcher DL, Robertson AD, Voelkel NF, Badesch DB, Groves BM, Gilbert EM, Bristow MR. Changes in gene expression in the intact human heart. Downregulation of alpha-myosin heavy chain in hypertrophied, failing ventricular myocardium. J Clin Invest. 1997;100:2315–2324. doi: 10.1172/JCI119770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariño G, Ugalde AP, Fernández AF, Osorio FG, Fueyo A, Freije JM, López-Otín C. Insulin-like growth factor 1 treatment extends longevity in a mouse model of human premature aging by restoring somatotroph axis function. Proc Natl Acad Sci USA. 2010;107:16268–16273. doi: 10.1073/pnas.1002696107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins RP, Finan JD, Guilak F, Lee DA. Mechanical regulation of nuclear structure and function. Annu Rev Biomed Eng. 2012;14:431–455. doi: 10.1146/annurev-bioeng-071910-124638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellad JA, Warren DT, Shanahan CM. Nesprins LINC the nucleus and cytoskeleton. Curr Opin Cell Biol. 2011;23:47–54. doi: 10.1016/j.ceb.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GT, Meinecke P, Kleijer WJ, Vijg J, Jaspers NG, Hoeijmakers JH. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–1043. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- Osorio FG, Navarro CL, Cadinanos J, Lopez-Mejia IC, Quiros PM, Bartoli C, Rivera J, Tazi J, Guzman G, Varela I, Depetris D, de CF, Cobo J, Andres V, De S-GA, Freije JM, Levy N, Lopez-Otin C. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci Transl Med. 2011;3:106ra107. doi: 10.1126/scitranslmed.3002847. [DOI] [PubMed] [Google Scholar]
- Osorio FG, Barcena C, Soria-Valles C, Ramsay AJ, de Carlos F, Cobo J, Fueyo A, Freije JM, Lopez-Otin C. Nuclear lamina defects cause ATM-dependent NF-kappaB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012;26:2311–2324. doi: 10.1101/gad.197954.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overall CM, Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2:657–672. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- Pendas AM, Zhou Z, Cadinanos J, Freije JM, Wang J, Hultenby K, Astudillo A, Wernerson A, Rodriguez F, Tryggvason K, Lopez-Otin C. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet. 2002;31:94–99. doi: 10.1038/ng871. [DOI] [PubMed] [Google Scholar]
- Postel R, Ketema M, Kuikman I, de Pereda JM, Sonnenberg A. Nesprin-3 augments peripheral nuclear localization of intermediate filaments in zebrafish. J Cell Sci. 2011;124:755–764. doi: 10.1242/jcs.081174. [DOI] [PubMed] [Google Scholar]
- Puente XS, Quesada V, Osorio FG, Cabanillas R, Cadinanos J, Fraile JM, Ordonez GR, Puente DA, Gutierrez-Fernandez A, Fanjul-Fernandez M, Levy N, Freije JM, Lopez-Otin C. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am J Hum Genet. 2011;88:650–656. doi: 10.1016/j.ajhg.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin AC, Hendry BM, Corcoran JP, Xu Q. An in vitro model for the pro-fibrotic effects of retinoids: mechanisms of action. Br J Pharmacol. 2013;170:1177–1189. doi: 10.1111/bph.12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell SJ, Kahn CR. Endocrine regulation of ageing. Nat Rev Mol Cell Biol. 2007;8:681–691. doi: 10.1038/nrm2234. [DOI] [PubMed] [Google Scholar]
- Sabeh F, Ota I, Holmbeck K, Birkedal-Hansen H, Soloway P, Balbin M, Lopez-Otin C, Shapiro S, Inada M, Krane S, Allen E, Chung D, Weiss SJ. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J Cell Biol. 2004;167:769–781. doi: 10.1083/jcb.200408028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha B, Zitnik G, Johnson S, Nguyen Q, Risques RA, Martin GM, Oshima J. DNA damage accumulation and TRF2 degradation in atypical Werner syndrome fibroblasts with LMNA mutations. Front Genet. 2013;4:129. doi: 10.3389/fgene.2013.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroen DJ, Brinckerhoff CE. Nuclear hormone receptors inhibit matrix metalloproteinase (MMP) gene expression through diverse mechanisms. Gene Expr. 1996;6:197–207. [PMC free article] [PubMed] [Google Scholar]
- Shimizu-Hirota R, Xiong W, Baxter BT, Kunkel SL, Maillard I, Chen XW, Sabeh F, Liu R, Li XY, Weiss SJ. MT1-MMP regulates the PI3Kdelta. Mi-2/NuRD-dependent control of macrophage immune function. Genes Dev. 2012;26:395–413. doi: 10.1101/gad.178749.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147:913–920. doi: 10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, Rehfeldt F, Speicher DW, Discher DE. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341:1240104. doi: 10.1126/science.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Rowe RG, Botvinick EL, Kurup A, Putnam AJ, Seiki M, Weaver VM, Keller ET, Goldstein S, Dai J, Begun D, Saunders T, Weiss SJ. MT1-MMP-dependent control of skeletal stem cell commitment via a beta1-Integrin/YAP/TAZ signaling axis. Dev Cell. 2013;25:402–416. doi: 10.1016/j.devcel.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123:966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugalde AP, Ramsay AJ, de la Rosa J, Varela I, Marino G, Cadinanos J, Lu J, Freije JM, Lopez-Otin C. Aging and chronic DNA damage response activate a regulatory pathway involving miR-29 and p53. EMBO J. 2011;30:2219–2232. doi: 10.1038/emboj.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdes-Mas R, Gutierrez-Fernandez A, Gomez J, Coto E, Astudillo A, Puente DA, Reguero JR, Alvarez V, Moris C, Leon D, Martin M, Puente XS, Lopez-Otin C. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat Commun. 2014;5:5326. doi: 10.1038/ncomms6326. [DOI] [PubMed] [Google Scholar]
- Varela I, Cadinanos J, Pendas AM, Gutierrez-Fernandez A, Folgueras AR, Sanchez LM, Zhou Z, Rodriguez FJ, Stewart CL, Vega JA, Tryggvason K, Freije JM, Lopez-Otin C. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 2005;437:564–568. doi: 10.1038/nature04019. [DOI] [PubMed] [Google Scholar]
- Varela I, Pereira S, Ugalde AP, Navarro CL, Suarez MF, Cau P, Cadinanos J, Osorio FG, Foray N, Cobo J, de Carlos F, Levy N, Freije JM, Lopez-Otin C. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat Med. 2008;14:767–772. doi: 10.1038/nm1786. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ross AC. Retinoic acid and the transcription factor MafB act together and differentially to regulate aggrecan and matrix metalloproteinase gene expression in neonatal chondrocytes. J Cell Biochem. 2013;114:471–479. doi: 10.1002/jcb.24387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Apte SS, Soininen R, Cao R, Baaklini GY, Rauser RW, Wang J, Cao Y, Tryggvason K. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc Natl Acad Sci USA. 2000;97:4052–4057. doi: 10.1073/pnas.060037197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Supplementary Figure Legends
Review Process File