

Abstract
The conserved Notch pathway functions in diverse developmental and disease-related processes, requiring mechanisms to ensure appropriate target selection and gene activation in each context. To investigate the influence of chromatin organisation and dynamics on the response to Notch signalling, we partitioned Drosophila chromatin using histone modifications and established the preferred chromatin conditions for binding of Su(H), the Notch pathway transcription factor. By manipulating activity of a co-operating factor, Lozenge/Runx, we showed that it can help facilitate these conditions. While many histone modifications were unchanged by Su(H) binding or Notch activation, we detected rapid changes in acetylation of H3K56 at Notch-regulated enhancers. This modification extended over large regions, required the histone acetyl-transferase CBP and was independent of transcription. Such rapid changes in H3K56 acetylation appear to be a conserved indicator of enhancer activation as they also occurred at the mammalian Notch-regulated Hey1 gene and at Drosophila ecdysone-regulated genes. This intriguing example of a core histone modification increasing over short timescales may therefore underpin changes in chromatin accessibility needed to promote transcription following signalling activation.
Keywords: Notch, CSL, chromatin state, H3K56ac, CBP/Nejire
Introduction
Notch is the receptor in a highly conserved signalling pathway that is of major importance in many developmental and disease contexts (Bray, 2006; Miele et al, 2006; Kopan & Ilagan, 2009; Louvi & Artavanis-Tsakonas, 2012). Despite a relatively simple transduction pathway, the outcomes of Notch activation are diverse and such diversity is essential for animal development. It is also of considerable significance in cancers, since Notch activity can promote tumorigenesis in some tissues and suppress it in others (Radtke & Raj, 2003; Miele et al, 2006; Roy et al, 2007; Ntziachristos et al, 2014). However, the mechanisms underlying pathway specificity remain poorly understood. Likewise, little is known about the genomic changes that occur during the transition of Notch-regulated genes to activated states.
Although its effects are pleiotropic, Notch acts primarily through a single core transcription factor (Bray, 2006; Kopan & Ilagan, 2009) which is known generally as CSL (CBF1, Suppressor of Hairless, Lag1) and specifically as Suppressor of Hairless [Su(H)] in Drosophila. When Notch receptors are activated by ligands carried on an adjacent cell, they become susceptible to cleavage by Adam 10 and γ-secretase. Receptor cleavage results in the release of an intracellular fragment, NICD, which forms a complex with CSL and the co-activator Mastermind (Mam) (Kopan & Ilagan, 2009; Kovall & Blacklow, 2010). This complex recruits histone acetylases (HAT) of the p300 family and up-regulates expression of genes to which it is recruited (Borggrefe & Oswald, 2009), the best characterised being genes of the HES/HEY family (Bray & Bernard, 2010). These include the Drosophila Enhancer of split Complex [E(spl)-C], where 12 Notch-responsive genes are clustered in a 60-kb region.
Genome-wide studies in Drosophila, mouse and human cells have shown that CSL is bound at different target sites depending on the cell type (e.g. Krejcí et al, 2009; Jin et al, 2013; Terriente-Felix et al, 2013; Wang et al, 2014, 2012). Although it is evident that tissue-specific factors help to mediate this specificity, it is not yet clear how they do so. For example, in Drosophila blood cells, the Runx family transcription factor Lozenge (Lz) is necessary for activity of Notch-regulated enhancers and it helps promote binding of Su(H) to its target sites via an unknown mechanism (Terriente-Felix et al, 2013). Likewise, in mammalian T-lymphoblastic leukaemia cells, CSL-bound enhancers frequently overlapped with RUNX1 binding motifs, suggesting that similar mechanisms could confer specificity in these cells (Wang et al, 2014). As there is no evidence for direct interactions between CSL and Lz/Runx, it seems likely that recruitment involves indirect effects.
Unlike transcription factors, such as FoxA, which are capable of binding to their target motifs even when wrapped around the histone core (Sekiya et al, 2009; Zaret & Carroll, 2011), CSL appears to preferentially bind to motifs located outside the nucleosome (Lake et al, 2014). Thus, one way that variations in CSL binding could be specified is through changes in the organisation of the chromatin, directed by cell-specific pioneer transcription factors. It is evident that the structure of chromatin differs considerably in a manner that correlates with different histone modifications and with the presence of architectural proteins, such as HP1 (Kouzarides, 2007; de Wit & van Steensel, 2009). For example, specific histone tail modifications, H3K4 mono-methylation (H3K4me1) and H3K27 acetylation (H3K27ac), are associated with active enhancers (Creyghton et al, 2010; Calo & Wysocka, 2013; Smith & Shilatifard, 2014). Such modifications can be recognised by so-called Readers, which are often constituents of complexes that alter the organisation or sub-nuclear localisation of the modified locus (Lalonde et al, 2014; Swygert & Peterson, 2014). For example, several chromatin-remodelling complexes contain proteins with bromodomains that recognise acetylated histone motifs (Taverna et al, 2007; Filippakopoulos & Knapp, 2014). Additionally, modifications to the histone core, such as H3K56 acteylation (H3K56ac), have the potential to directly alter DNA interactions (Neumann et al, 2009; Simon et al, 2011). Thus, it is possible that particular combinations of histone modifications, engineered by tissue-specific regulators, could be a pre-requisite for CSL recruitment at Notch-regulated enhancers.
Changes to chromatin modifications and conformation may also be important for the induction of target gene expression following Notch activation. While there is a low level of CSL binding at Notch-regulated enhancers prior to pathway activation, its recruitment is greatly enhanced in the presence of NICD (Krejcí & Bray, 2007; Castel et al, 2013; Housden et al, 2013; Wang et al, 2014). Additional localised chromatin alterations may contribute to enhanced stability of the CSL/NICD complex on DNA. Furthermore, transitions in enhancer activity are also associated with changes in histone modifications. For example, the presence of H3K27ac at enhancers is thought to distinguish active from primed enhancers (Calo & Wysocka, 2013; Smith & Shilatifard, 2014) and has been detected following changes to Notch activity in myogenic and leukaemic cells (Castel et al, 2013; Wang et al, 2014). Exploring the mechanisms associated with Notch-induced enhancer activation is important in the context of emerging therapeutic strategies targeting chromatin regulators (Helin & Dhanak, 2013).
In order to investigate whether chromatin organisation can explain the cell-type specificity of Su(H) recruitment and how it relates to Notch-regulated enhancer activation, we took advantage of detailed profiling of histone modifications in Drosophila cells performed by modENCODE. Combining our new data on H3K56ac with modENCODE data on 23 different histone characteristics and DNase I hypersensitivity (Kharchenko et al, 2011), we derived models of the chromatin signatures in two different cell types and then related this to Su(H) binding profiles. We found that Su(H) occupancy is predominantly associated with two specific chromatin signatures and that the co-operating Lz/Runx transcription factor can help to confer characteristics of the preferred state. Su(H) and NICD also shape the chromatin at bound regions, in particular, H3K56ac is strongly increased across regulated loci following Notch activation. This modification to the H3 core appears to be dependent on CBP and occurs independently of transcription elongation from the transcribed loci. Thought to promote local DNA breathing (Neumann et al, 2009), we find that H3K56ac correlates with elevated transcription from the enhancer regions (intergenic RNAs). Analysis of H3K56ac after ecdysone stimulation indicates that changes to this histone modification are more generally applicable in Drosophila and our observation of similar Notch signalling-dependent changes at the mouse Hey1 enhancer indicates that this is a conserved mechanism.
Results
Relationship between chromatin states and Su(H) occupancy
Our initial goal was to determine which aspects of the chromatin environment, as defined by the presence or absence of particular histone modifications, could contribute to Su(H) binding and hence to the cell specificity of Notch-responsive genes. To achieve this, we generated a map of chromatin states within Drosophila BG3 (CNS) and Kc167 (blood) cells and also determined the positions where Su(H) was bound. Since chromatin states have not previously been derived for these cell types, we utilised an adaptation of a previously described Hidden Markov model (HMM) approach (Kharchenko et al, 2011) to generate genome-wide chromatin maps for the two cell types.
We used the available data sets for 23 histone modifications, DNase I hypersensitivity maps and new genome-wide H3K56ac data sets generated as part of this study (Fig1A, Supplementary Fig S1A). Although H3K56ac overlaps with key regulators of pluripotency in human cells (Xie et al, 2009), this modification has not been profiled in many transcriptional contexts nor included in previous chromatin models. In Drosophila cells, we found that H3K56ac was highly enriched at enhancers and around active transcription start sites (TSS), correlating most strongly with H3K9ac and H3K4me2 (Supplementary Fig S2). H3K56ac also showed strong relationships with the H3K27ac and H3K4me1 modifications known to be associated with enhancers (Supplementary Fig S2). A single data matrix was therefore created, combining the H3K56ac and modENCODE ChIP data with DNase I accessibility, and then, tied parameterisation was used to identify the maximum number of unique chromatin signatures that could be inferred before splitting a signature into two similar ones (see Supplementary Information for more details). This strategy was used to minimise the risk of over-fitting, one potential drawback of this type of maximum likelihood HMM. The fact that we recovered similar signatures to those obtained through a more complex Bayesian model (Supplementary Fig S1C) indicates the success of the strategy, as did with results from a leave-one-out analysis, which demonstrates the robustness of the signatures (Supplementary Fig S1F). The latter also highlights that some histone modifications have more dominant roles, while others are less discriminatory for the chromatin signatures.
Figure 1.

Relationship between Su(H) binding and the chromatin state
- A E(spl)-C along with the histone modification data used to generate the chromatin signatures, where enrichment is shaded brown (highly enriched) to blue (depleted). Su(H) binding profile for BG3 cells (brown graph: fold enrichment, Log2 scale is −1.2 to 3.89, and blue shading indicates significant peaks 1% FDR) aligned with the chromatin map colour coded as in (B). Gene models are depicted in blue.
- B Summary of the 11 chromatin signatures derived from Hidden Markov model, showing enrichments for a subset of histone modifications, brown (highly enriched) to blue (depleted) (see Supplementary Fig S1 for full profiles and comparisons).
- C Numbers and extent of overlap in Su(H)-occupied regions between Kc and BG3 cells, the Su(H) motif was highly enriched in both data sets (P-values 4.2 × 10−10 and 2.12 × 10−14).
- D, E Distribution of Su(H)-bound regions according to chromatin type in BG3 (D) and Kc (E) cells; chromatin is colour coded according to the scheme in (B) and Supplementary Fig S1A. Grey bars indicate the proportion of the genome in each chromatin signature.
The 11 chromatin signatures we recovered showed extensive similarities with the chromatin states generated through a comparative analysis of metazoan chromatin (Fig1B and Supplementary Fig S1A–C; Ho et al, 2014). Despite differences in the approaches used, both identified similar types of regulatory chromatin, indicating that our simple method adequately captured the most frequent chromatin patterns. In particular, the active regulatory chromatin was partitioned into three types, which we refer to as enhancer (Enh; red), competent (Comp; green) and active intron (aIntr; purple) (Supplementary Fig S1A–E). Enh encompasses canonical modifications H3K4me1 and H3K27ac associated with enhancers and is also enriched for H3K56ac. In BG3 cells, it also corresponds to regions with regulatory activity, as identified by functional methods for discovering enhancers in BG3 cells (STARR-seq, Supplementary Fig S1A; Yanez-Cuna et al, 2014). Competent chromatin is marked by H3K4me1 but has low levels of the other enhancer modifications and frequently appears to represent enhancer regions that are not yet fully active, as supported by our subsequent experiments. The third regulatory chromatin, active intron, differs in its enrichment for H2B ubiquitination, one of the modifications important in differentiating these three types (Supplementary Fig S1F).
Having generated chromatin signature maps for BG3 and Kc cells, we then determined the genome-wide profile of Su(H) binding in the two cell types to determine the relationship between bound regions and chromatin states (e.g. see E(spl)-C, Fig1A). A comparable number of regions were bound by Su(H) in the two cell lines, of which 25–30% were bound in both cell types (Fig1C). Although relatively few peaks were identified in comparison to some other DNA-binding proteins, the low number is consistent with the observation that CSL/Su(H) exhibits low occupancy in the absence of NICD at many Notch-regulated enhancers (Krejcí & Bray, 2007; Castel et al, 2013; Housden et al, 2013; Wang et al, 2014) and the data were consistent across replicates (Supplementary Fig S3A). In both cell types, the mean peak width was similar (circa 500 bp; Supplementary Fig S3B), but occasionally, in regions of high occupancy at the E(spl) locus, these peaks overlapped to generate a super peak of several kb. By far the majority of the bound regions were located within Enh chromatin (red, Fig1D and E). The remainder were predominantly in another active region with TSS features (aTSS, orange; Fig1D and E) with a small proportion in Comp or Polycomb domains. The few peaks that mapped to other types of chromatin may reflect unusual binding events but could also arise from false positives in the chromatin assignments or in the ChIP data. Given the representation of each signature across the genome, there is clearly a highly significant preference for Enh and aTSS in the chromatin environment at Su(H)-bound loci.
To assess how well the chromatin signature predicted Su(H) occupancy, we considered four high-affinity Su(H) binding motifs and asked what proportion of these in each chromatin state were occupied. Of the small fraction of bound motifs in each cell type (Fig2A and B; 59/11,783 bound in BG3 cells and 89/11,783 bound in Kc cells), the majority were in Enh and aTSS states despite these housing a minority of the four motifs (Fig2B). In contrast, “Basal” (black) chromatin contained the largest proportion of motifs (3,520 motifs in BG3; 5,897 motifs in Kc), yet had negligible binding. These data indicate that > 91% of the Su(H) motifs are likely to be masked from binding due to the chromatin environment. Knowledge about the chromatin state in a given cell type can therefore help identify which motifs are more likely to be bound by Su(H), thus rendering the associated loci sensitive to Notch signalling.
Figure 2.

Differences in chromatin correlate with Su(H) binding at some, but not all, loci
- High-affinity motifs used in the analysis and numbers occupied by Su(H) in each cell type as indicated.
- Distribution of bound and unbound motifs according to chromatin type. Colour code indicates chromatin type, and the number of motifs in each state are indicated.
- Examples where Su(H) binding is concordant with chromatin. Each panel depicts a gene region with the chromatin map (colours as in Fig1B), Su(H) binding profiles for each cell type (fold enrichment Log2 with ranges −1.2 to 3.89 for BG3 and −0.86 to 2.09 for Kc167; significant 1% FDR peaks are shaded in blue) and positions of Su(H) motifs indicated. Gene models are depicted beneath each plot.
- Graph summarising the relationship between binding and chromatin in the two cell types. Brown bars, loci bound in both cell types; chromatin environment of bound motifs in Kc cells that were bound in Enh chromatin in BG3 cells. 100% are in Enh chromatin in both cell types. Green bars, motifs bound in Kc but not in BG3; chromatin environment of unbound motifs in BG3 cells that were bound in Enh chromatin in Kc cells. Blue bars, motifs bound in BG3 but not in Kc; chromatin environment of unbound motifs in Kc cells that were bound in Enh chromatin in BG3 cells. Some unbound motifs in each cell type are in less favourable chromatin but many remain in Enh even though not detectably bound.
Despite the considerable gains we observed by mapping motifs onto chromatin states, there remains a paradox, since only 40/545 Su(H) motifs present in “favoured” Enh chromatin were actually occupied in BG3 cells (Fig2B) and only 55/529 in Kc cells (Fig2B). Therefore, the majority of positions in the genome where the motif was in a favourable chromatin state, with accessible DNA and histone modifications linked to enhancer activity, were not stably bound by Su(H). This suggests that, while the chromatin environment is important, additional factors and/or as yet unknown chromatin characteristics determine where Su(H) binds.
We then examined whether the chromatin signatures could account for cell-type Su(H) binding, focussing again on the four high-affinity motifs (Fig2A). For motifs that were bound in both cell types, the situation was straightforward, as 100% of those present in Enh state in one cell type were also in Enh state in the other (Fig2D, brown). These included the Notch locus, where binding occurred at motifs that were in favourable chromatin state in both cell types (Fig2C, top). For those with differential binding in the two cell types, only a subset fit the predicted pattern. These included bigbrain (bib), where the motif was bound by Su(H) in BG3 cells and was within Enh (red) chromatin, but was unbound in Kc cells, where it was in basal chromatin (Fig2C middle). Likewise myc/diminutive (dm) was bound in Kc cells in the Enh (red) state, but not in BG3 cells where it was in Comp (green) (Fig2C). Overall, for approximately 30–40% of bound motifs that were in Enh3 in one cell type, the differential binding was correlated with a change in chromatin state (so that it was less favourable in the cells where the motif was unbound; Fig2D). For the remainder, there was no change in the chromatin state between the cell types to account for the differences in occupancy as they mapped to Enh chromatin in both (Fig2D). Thus, while Enh chromatin appears conducive to Su(H) binding, this condition is not sufficient to predict that binding will occur.
Roles of cooperating factors and Su(H) in conferring chromatin characteristics at enhancers
We have previously shown that cooperating transcription factors, such as Lz/Runx, are important for conferring specificity to the Notch response (Terriente-Felix et al, 2013). To ask whether Lz was capable of generating a favourable chromatin environment, we induced elevated Lz expression in Kc cells (where Lz levels are normally low (Terriente-Felix et al, 2013)) and in BG3 cells (where Lz was not detectably expressed) and then monitored the consequences at the pebbled locus (peb). Of three mapped Notch-responsive enhancers in peb, two (peb3 and peb2) were in unfavourable chromatin states (Comp or Pc) in both cell types and exhibited no Su(H) recruitment (Fig3A). The other, peb1, was in Enh state (red) in Kc cells, where low levels of Su(H) binding below the threshold of detection in genome-wide ChIP but apparent in specific ChIP-PCR (Fig3B), correlates with a mild (5×) induction of peb in these cells (Terriente-Felix et al, 2013).
Figure 3.
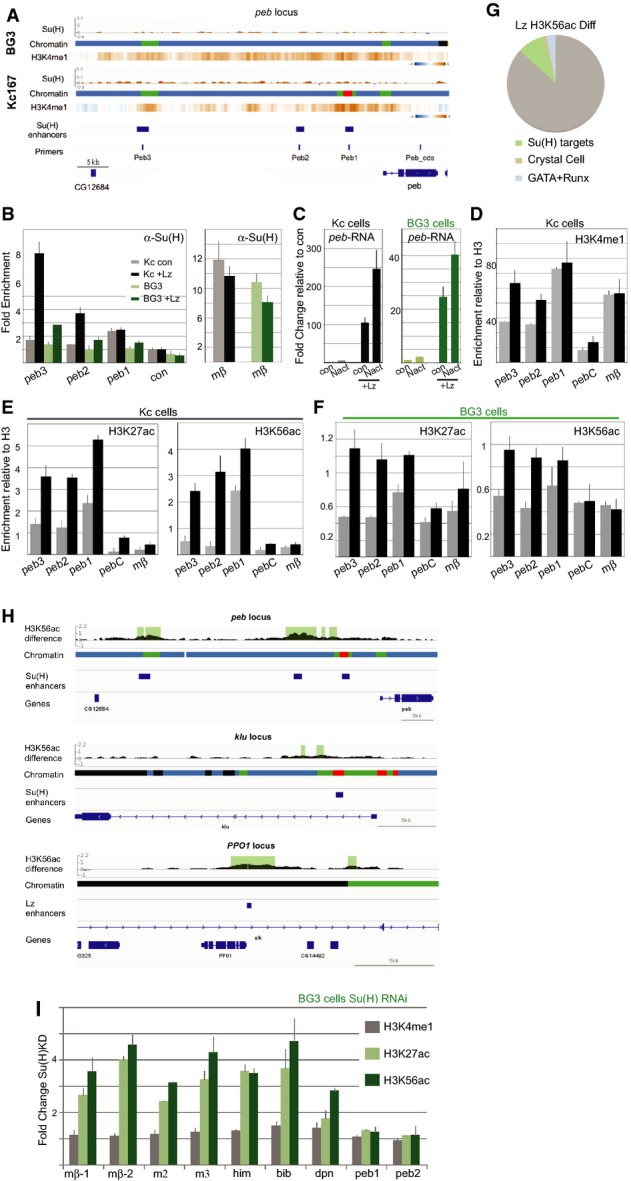
Roles of a cooperating factor and of Su(H) in conferring chromatin characteristics at bound enhancers
- A peb region with chromatin maps, Su(H) binding profiles and H3K4me1 heat maps for each cell type, and the known Su(H)/Notch-regulated peb enhancers are indicated, along with primers used.
- B Fold change in RNA levels in Kc and BG3 cells in the presence of Lz compared to control conditions (con: empty pMT) and to cells with Notch activation (Nact). Lz expression was induced for 3 days prior to the experiment, and RNA was analysed 30 min after exposing cells to control conditions or to EGTA to elicit Notch activation.
- C Bound Su(H) was captured by ChIP from Kc (grey/black) and BG3 (light/dark green) cells, with (black, dark green) or without (grey, light green) ectopic Lz, and the levels of the indicated enhancers analysed. Binding was significantly enriched at peb3 in BG3 and Kc cells and at peb2 in Kc cells (P < 0.05).
- D–F Enrichment of the indicated histone modifications detected by ChIP in Kc (D, E) and BG3 cells (F); control cells (grey; empty pMT), with ectopic Lz (black). peb regions analysed correspond to those depicted in (A) (primers), and enrichment was calculated relative to total H3. Changes in the modifications at peb3 and peb2 were significant in all cases (P < 0.05).
- G, H Differences in the profile of H3H56ac upon expression of Lz. (G) Percentage of regions with significant differences located in proximity to 224 Notch-/Su(H)-regulated genes (green, P = 1.6 × 10−18; Krejcí et al, 2009), to 31 known crystal cell-expressed genes (brown, P = 0.00057; Ferjoux et al, 2007) and to 269 genes with defined GATA-RUNX motif (blue, P = 0.0019; Ferjoux et al, 2007). (H) Regions with significant differences detected at Notch-regulated enhancers in peb and klu and at the Lz-regulated enhancer in PPO1 (Ferjoux et al, 2007). Gene regions with chromatin maps showing difference in H3K56ac, regions where the difference is significant (1% FDR) are shaded.
- I Fold change in enrichment of the indicated histone modifications at enhancers following RNAi treatment to deplete Su(H) compared to control (GFP RNAi). Increase in H3K27ac and H3K56ac occur at mβ-1, mβ-2, m2, m3, him, bib and dpn, which are bound by Su(H) in BG3 cells but not at peb2, peb3 which are not bound by Su(H) in BG3 cells.
Data information: All graphs depict average results from ≥ 3 experiments, error bars are standard error of the mean, and significance was determined by t-test for graphed results and hypergeometric test for pie chart.
Production of Lz in each cell type strongly induced the expression of peb mRNA, which was further enhanced by Notch activation (Fig3C). The changes in RNA expression correlated with de novo recruitment of Su(H) at peb3 and peb2 enhancers (Fig3B) and with an increase in Enh-associated histone modifications (Fig3D–F). Notably, H3K4me1, H3K27ac and H3K56ac were all increased at peb3 and peb2 in both cell types. Although detectable in both, the effects of Lz were much greater in Kc cells than in BG3 cells, suggesting that pre-existing factors facilitate its actions. Furthermore, peb1 appeared the least responsive in BG3 cells, despite manifesting an active conformation in Kc cells. Nevertheless, the results demonstrate that Lz expression can elicit changes in histone modifications to convert the chromatin at two target enhancers towards the active Enh state, which correlates with their ability to recruit Su(H).
To assess whether Lz has more widespread effects, we profiled the changes in H3K56ac following Lz expression in Kc cells. These results confirmed that the acetylation was increased at several other loci in addition to peb enhancers, with 459 regions showing significant change (1% FDR). Notably these included significant relationship with Notch-regulated genes (Fig3G), as exemplified by peb and klu (Fig3H), as well as known crystal cell and Lz-regulated genes (Fig3G) such as PPO1/Bc (Fig3H; Ferjoux et al, 2007). Few (3%) of the changes occurred in regions of Enh chromatin. Around 26% mapped to Comp chromatin (as for peb) and 14% mapped to Basal (as for PPO1), supporting the model that Lz expression facilitates their conversion to a more active chromatin state.
Since Su(H) is present at many enhancers in un-activated cells, it could potentially also contribute to the chromatin landscape, as suggested by reports that the mammalian homologue acts as a “bookmark” remaining on chromatin at mitosis (Lake et al, 2014). To investigate this possibility, we analysed the consequences of depleting Su(H) levels by RNAi treatment on Enh modifications at several target enhancers in BG3 cells. Under conditions where < 20% Su(H) mRNA remained, H3K4me1 was unchanged, but both H3K27ac and H3K56ac were markedly increased (Fig3I). These changes were specifically detected at the enhancers where Su(H) was bound in BG3 cells (e.g. Him, bib, dpn; Fig3I) not at those that were unbound (e.g. peb1, peb2; Fig3I). Analysis of target gene mRNA changes under similar conditions showed that a number of these loci were de-repressed (e.g. E(spl)mβ-HLH, bib, Supplementary Table S1). This is consistent with a model where co-repressors associated with Su(H) recruit histone deacetylases (HDACs) that are important in suppressing expression from target loci in the absence of Notch signalling. Thus, it appears that, although Su(H)-bound regions exhibit characteristics of Enh chromatin, these regions often have lower than average levels of H3K27ac and H3K56ac due to the presence of Su(H) (Supplementary Fig S4A). This does not appear to be the case for all bound regions, potentially indicative of different modes of regulation.
Changes in histone modifications following Notch activation include robust increase in H3K56 acetylation
Although CSL resides at some target enhancers prior to Notch activation, several studies have shown that CSL binding, as detected by ChIP, increases substantially following Notch activation and that CSL is detected de novo at many loci (Krejcí & Bray, 2007; Castel et al, 2013; Housden et al, 2013; Wang et al, 2014). To address whether such de novo binding occurs at sites located in different chromatin contexts, we analysed the profile of Su(H) binding 30 min after eliciting Notch activation in BG3 cells. To achieve this temporal control, we used the calcium chelator EGTA, which disrupts the extracellular domain of Notch making it accessible to the activating proteases (e.g. Gupta-Rossi et al, 2001; Krejcí and Bray, 2007; Ilagan et al, 2011). As anticipated, a large increase in the number of Su(H)-bound sites was detected under these conditions (Fig4A; 388 activation-only peaks). We therefore asked whether de novo-bound regions differed in their chromatin state from those where Su(H) was detected prior to activation. This was generally not the case: the distribution was broadly similar to control un-stimulated cells with the majority (70%) of occupied sites mapping to Enh chromatin (Fig4B) which increased the fraction of bound high-affinity motifs in Enh to 16% (89/545). However, for a small fraction of loci (10%), the bound regions occurred in the chromatin state indicative of Polycomb regulation (Pc, blue). The majority of these had bivalent characteristics, with low levels of enhancer-associated modifications (such as H3K4me1) in addition to the H3K27me3 Polycomb-related modification.
Figure 4.

Changes in histone modifications after Notch activation: rapid and robust increase in H3K56 acetylation
- AVenn diagram summarising the relationship between Su(H)-bound regions in control and Notch-activated (EGTA-treated for 30 mins) cells.
- B Distribution of de novo Su(H) peaks, detectable only in activated cells, according to chromatin state.
- C, D Representative experiment comparing the enrichment of the indicated histone modifications at several loci in control and Notch-activated (30 min) cells. Details of loci analysed are provided in the Supplementary Information.
- E Changes in enrichment of the indicated modifications at selected loci 30 min after Notch activation compared to control-treated cells.
- F Treatment with a γ-secretase inhibitor (GSI: 10 nM Compound E) prevents the increase in mRNA levels in Notch-activated cells.
- G Treatment with a γ-secretase inhibitor (GSI: 10 nM Compound E, conditions as in F) prevents the increase in H3K56ac in Notch-activated cells.
Data information: Graphs in (E–G) depict average results from ≥ 3 experiments, error bars are standard error of the mean, and differences in (F, G) were all significant (P ≤ 0.05, t-test).
To investigate whether Notch activation could induce changes in chromatin at the regulated loci, we examined a wide range of histone modifications at Su(H)-bound regions before and after Notch activation by EGTA (Fig4C–E). The majority of those tested were unchanged by Notch activity. Notable exceptions were H2B ubiquitination (H2Bub), H3K27ac and H3K56ac which all increased following Notch activation, while several loci showed reduced H3K4me1. Of the changes observed, the increase in H3K56ac was the most unexpected as this residue, located within the histone core, has not previously been shown to exhibit dynamic changes and has largely been linked to DNA replication or damage. In contrast, recent studies in mammalian cells have reported changes in H3K27 acetylation with longer-term differences in Notch activity (Wang et al, 2014). Our results indicate that both the H3K27ac and H3K56ac are acute effects, occurring within minutes, and are likely related to NICD recruitment directly. To confirm whether the increases in H3K56ac is also directly dependent on Notch activity, we assayed H3K56ac in the presence of a γ-secretase inhibitor (GSI) to prevent the presenilin-mediated release of NICD. Treating cells with GSI (10 nM Compound E) was sufficient to block the up-regulation of target genes (Fig4F). The same conditions eliminated the increase in H3K56ac, indicating the likelihood that it is Notch dependent (Fig4G). Furthermore, similar changes were detected in cells where NICD expression was induced using an alternate strategy (see below).
Since there have been no previous reports of dynamic changes in H3K56ac, we assayed the specificity of the antibody by probing a histone peptide array containing many different modifications (Supplementary Fig S5A). This confirmed that the antibody was specific for the H3K56ac peptide, in agreement with studies in other cells (Das et al, 2014). Further confirming our observations, we showed that the ChIP enrichment was eliminated in the presence of competing K56Ac peptide (Supplementary Fig S5B). Finally, we obtained similar results using an independent anti-H3K56ac antibody (Supplementary Fig S5C). Taken altogether, these data support the conclusion that the levels of H3K56ac change substantially at target loci upon Notch activation.
Increased H3K56 acetylation after Notch activation spans large domains at regulated loci and is associated with intergenic RNA
To gain a more complete picture of H3K56ac changes following Notch activation, we compared genome-wide ChIP profiles from control and Notch-activated Kc and BG3 cells. This comparison revealed a significant increase in H3K56ac in Notch-activated cells at several specific loci (e.g. Figs5 and 6A). The majority have Su(H) bound, are known Notch targets and/or were detectably Notch regulated in these cell types (Supplementary Table S1). Strikingly, increased H3K56ac was not restricted to the locality of the Su(H)-bound enhancers but was spread across relatively broad regions. At E(spl)-C, the range of increased H3K56ac in BG3 cells spanned the whole locus, while the Su(H) binding was restricted to specific enhancers (Fig5). The boundaries of the H3K56ac domain corresponded with chromatin state boundaries, and the extent of spreading was best predicted by the presence of H3K4me1 (Supplementary Fig S4B). For example, in Kc cells, H3K56ac covered a more restricted portion of E(spl)-C, exhibiting little up-regulation over distal m6/m7/m8 regions where H3K4me1 was less enriched (Fig5). Up-regulation of H3K56ac in Kc cells across a similar region of the complex was also detected following a 2-h induction of NICD under the control of a metallothionein promoter (Fig5), along with significant changes at 656 other regions that included peb and klu enhancers as well as other Notch-regulated genes (e.g. Supplementary Table S1). The differences in the H3K56ac profiles over the E(spl)-C in the BG3 and Kc cell types correlated with differences in responding gene activities, as more distal genes, such as E(spl)m6-BFM were poorly up-regulated in Kc cells (Terriente-Felix et al, 2013); Supplementary Table S1).
Figure 5.

Increase in H3K56 acetylation at E(spl)-C in BG3 and Kc cells extends across the 60-kb locus
E(spl)-C genomic region. For each cell type, the enrichment profiles for H3K56ac ChIP from control and Notch-activated (EGTA-treated) cells are plotted in brown (Log2, scales are: −3.39 to 4.35 in BG3 and −2.92 to 5.19 in Kc167), with the differences plotted below in dark green and the regions of significant difference shaded (1% FDR see Supplementary Methods). For Kc cells, differences in H3K56ac in NICD-expressing cells (after 2-h induction) versus controls are also plotted in the same way. Chromatin maps, colour coded as in Fig1B; Su(H) binding profiles for each cell type and enrichment maps for H3K4me1 from ModENCODE are shown as indicated. For BG3, Su(H) binding profile in Notch-activated cells and the consequences on chromatin state from the changes in H3K56ac are also shown. For Su(H) ChIP profiles, Log2 scales are −1.205 to 3.89 BG3 controls, −1.145 to 4.03 BG3 Nact and −0.862 to 2.09 Kc controls and shaded regions indicate significant peaks (1% FDR). Gene models are depicted in dark blue.
Figure 6.

Selective increase in H3K56 acetylation is dependent on CBP-HAT activity, independent of transcription elongation and occurs at Hey1 enhancer in mammalian cells
- Him-Her gene region showing that Notch activity induces changes in H3K56ac in BG3 cells but not in Kc Cells. Graphs and details as in Fig5A. Nact chromatin shows the results from running the HMM using the data from the Notch-activated BG3 cells, and the Su(H)-bound region has gained an Enh (red) signature.
- Fold change in expression of intergenic RNAs at E(spl) locus after Notch treatment (30 mins EGTA) in the presence and absence of the indicated inhibitors. Upper panel summarises the primers used to detect non-coding RNAs and their relationship to Su(H) binding profile (plotted as in Fig5). Graphs summarise the expression of intergenic RNAs and coding RNAs under the conditions indicated.
- Effects of indicated inhibitors (see Supplementary Table S2) on E(spl)mβ-HLH and E(spl)m3-HLH RNA levels following Notch activation. RNA levels from untreated (-) and Notch-activated (+; EGTA-treated) cells were normalised to rp49.
- Effects of the indicated inhibitors (see Supplementary Table S2) on the fold change in H3K56ac in Nact cells compared to control, measured by the enrichment for the indicated loci in anti-H3K56ac ChIP.
- Hey1 locus in mouse cells with previously documented CSL/RBPJ and p300 profiles (Castel et al, 2013) and positions of primers used in (D).
- Enrichment for H3K56ac at the indicated positions relative to control region (Hey1-L4; Kr9/14-con) in control cells (grey; exposed to Fc and DAPT) or in Notch-activated cells, (blue: exposed to Fc-DLL1) after 2.5 h (light shading) or 6 h (dark shading).
Data information: Graphs in (B–D and F) depict average results from ≥ 3 experiments, and error bars are standard error of the mean. In (B, C), differences between each treatment and control condition (DMSO) were all significant (P ≤ 0.05, t-test). In (D), all CBP-HAT C646 values were significantly different from DMSO (P ≤ 0.05, t-test), but none of those with CBP-BRM SGC or DBR were significant (P > 0.05, t-test). In (F), Hey1, Pr, L2, L1 and R1 were all significantly different from control condition (P ≤ 0.05, t-test), while none of the Kr9/14 levels were significantly different (P > 0.05, t-test).
Other loci exhibited similar cell-specific broad increases in H3K56ac. At the Him locus, the increase in H3K56ac after Notch activation occurred only in BG3 cells, where Him is Notch regulated, and encompassed the divergently transcribed Him and Her genes up to the boundaries of neighbouring genes (Fig6A). Strikingly, when the Hidden Markov Models were re-run including the profile of H3K56ac from Notch-activated cells, the Him enhancer was reclassified as Enh (red) from Comp (green; Fig6A). Similarly, bib also exhibited an increase in H3K56ac following Notch activation in BG3 cells, where the modified region spread across the intronic peak of Su(H) binding into the intergenic region (data not shown). Thus, not only were changes in H3K56ac detected rapidly after Notch activation, they also extended through large territories associated with the regulated loci.
Since H3K56 hyper-acetylation is reported to facilitate divergent transcription (Marquardt et al, 2014), we assessed whether any intergenic transcription of non-coding RNAs was detectable within the E(spl)-C and whether this increased following Notch activation. Probing several of the regions flanking Su(H)-bound enhancers, we detected low levels of intergenic RNAs whose expression increased substantially in the Notch-activated cells (Fig6B). This increase was attenuated when the cells were treated with the γ-secretase inhibitor (GSI, CompE), indicating that it was dependent on the activating cleavage (Fig6B). Thus, it appears that Notch activation results in increased transcription of intergenic enhancer RNAs (e-RNA) as well as an increase in H3K56ac.
Increased H3K56 acetylation is CBP dependent, transcription independent and also occurs at mammalian Hey1
Assays of total levels of H3K56ac in Drosophila and mammalian cells have demonstrated that this modification requires CBP/p300 histone acetylases (Das et al, 2009). Since p300 is recruited to target loci by a complex containing NICD and Mastermind (Oswald et al, 2001; Fryer et al, 2002; Wallberg et al, 2002), it is a prime candidate to mediate modifications associated with Notch activation. To ascertain whether the single Drosophila homologue of CBP/p300 (Nejire) is necessary for the H3K56ac increase following Notch activation, we treated cells with C646, an inhibitor that is specific for the CBP-HAT domain (Bowers et al, 2010). A brief 30-min treatment with C646, which was sufficient to prevent the up-regulation of E(spl)m3-HLH and E(spl)mβ-HLH mRNAs (Fig6B and C) and of intergenic RNAs (Fig6B), fully suppressed the increase in H3K56ac at target enhancers from E(spl)-C and from other loci (Fig6D). A second inhibitor, curcumin, which inhibits CBP as well as other acetylases, similarly blocked both mRNA up-regulation and the increase in H3K56ac (Supplementary Fig S6A and B). Likewise, depletion of CBP in Kc cells and in vivo (using RNAi) led to reduced H3K56ac and compromised expression of a Notch-regulated gene (cut; Supplementary Fig S6C–F). In contrast, neither the changes in mRNA expression nor H3K56ac levels were prevented by an inhibitor that interferes with another functional domain in CBP (ICG-001; Supplementary Fig S6A and B). Together, the results from these inhibitors suggest that the change in H3K56ac following Notch activation is dependent on the catalytic HAT activity of CBP.
When free histone dimers undergo H3K56ac modification, they are associated with the chaperones CAF-1 and Asf1. Under these circumstances, the bromodomain of CBP is necessary for the interaction with Asf-1, which brings the enzyme in proximity to its lysine substrate (Das et al, 2014). To investigate whether a similar mechanism could be involved at Notch-regulated enhancers, we tested the consequences of treating cells with two CBP bromodomain inhibitors (I-CBP112, SGC-CBP30) (Gallenkamp et al, 2014; Hay et al, 2014). Of the two, only SGC-CBP30 inhibited Notch-dependent transcription from E(spl)-C genes at the concentrations tested, resulting in a 4- to 5-fold reduction in the induced mRNA levels and a similar reduction in the levels of intergenic RNAs (Fig6B and C). Under the same conditions, SGC-CBP30 had no effect on H3K56ac levels following Notch activation (Fig6D), suggesting that the Notch-induced modification occurs independently of the CBP bromodomain, although the inhibitory effects on mRNA induction imply that the domain could have some role in Notch-regulated transcription.
A second question was whether the change in H3K56ac is a consequence of increased transcription at the regulated loci, since the modification also occurs at TSS. To address this, cells were treated with DRB, a potent inhibitor of the kinase subunit in P-TEF, which prevents entry into transcription elongation (Marshall & Price, 1995). Despite the fact that the drug effectively eliminated mRNA expression from the regulated genes (Fig6C) and strongly reduced the up-regulation of intergenic RNAs (Fig6B), it did not affect H3K56ac levels (Fig6D), arguing that the modification occurs independently of transcription elongation. Similar results were obtained with flavopiridol, which also blocks transcription elongation. Flavopiridol prevented mRNA up-regulation without affecting the increase in H3K56ac (Supplementary Fig S6). These results suggest that H3K56ac precedes the effects on mRNA transcription and fit with the fact that some genes with increased H3K56ac were not detectably up-regulated under the conditions used (Supplementary Table S1). Taken together, the results from the inhibitor experiments argue that H3K56ac modification in response to Notch at regulated loci requires CBP-HAT activity but is largely independent of both mRNA and e-RNA transcription elongation.
Finally, we asked whether such changes in H3K56ac also occur at Notch-regulated enhancers in mammalian cells, using mouse C2C12 cells where the binding profile of mouse CSL (also known as RBPJ) has been described (Castel et al, 2013). Those studies revealed that binding was dynamic at Notch-inducible enhancers, such as Hey1, and constitutive at other loci, such as Krt9/14, whose expression was unchanged following Notch activation (Castel et al, 2013). C2C12 cells were exposed to the Notch ligand (Dll1) for 2.5 and 6 h and the consequences on H3K56ac at the Hey1 and Krt9/14 enhancers analysed. Hey1 was selected because the enhancer is well separated from the gene body (Fig6E) and the RNA levels were induced 3× compared to controls. A robust increase in H3K56ac was detected at the Hey1 enhancer, but not at the constitutive Krt9/14 locus where the levels of this modification were already comparatively high (Fig6F). Thus, mammalian cells exhibit an increase in H3K56ac at the Notch-inducible Hey1 enhancer, similar to that seen in Drosophila cells.
Increased H3K56 acetylation also occurs in response to ecdysone
Since H3K56ac is detected widely at enhancer regions, the modification may be acquired in response to other signals besides Notch. The steroid hormone ecdysone acts through the nuclear ecdysone receptor (EcR; Thummel, 1995) and, as with NICD/CSL, hormone binding converts the receptor from a repressing to an activating complex (Tsai et al, 1999). We therefore assessed whether the response to ecdysone is accompanied by a similar change in H3K56ac, using data on EcR binding and EcR-responsive enhancers (Gauhar et al, 2009; Shlyueva et al, 2014) to select appropriate regions. For example, distal ecdysone-responsive enhancers are located between Dip-B and pri/tal (Chanut-Delalande et al, 2014; Shlyueva et al, 2014) and EcR binding to Eip78C has been mapped to a regulatory enhancer in a large intron (Gauhar et al, 2009; Fig7A). Strikingly, large changes in H3K56ac were detected around these target enhancers within 1 h of ecdysone exposure and remained consistent over longer periods of stimulation (Fig7B). Similar large-scale changes also occurred at Hr4 (Fig7C), correlating with the robust increases in RNA levels (Fig7D). Rapid increases in H3K56ac are therefore a characteristic of ecdysone-activated enhancers in addition to those regulated by Notch.
Figure 7.

- Eip78C and DlpB-pri/tal gene regions showing chromatin maps and H3K56ac in un-stimulated cells, along with positions of primers used in (B). Ecdysone-responsive enhancers for DlpB-pri/tal are those identified in S2 cells (Shlyueva et al, 2014), EcR DamID peaks, regions bound by EcR in Kc cells (Gauhar et al, 2009; Chanut-Delalande et al, 2014).
- Fold change in H3K56ac at the indicated regions following 1-h (grey) or 3-h (black) treatment with 20-hydroxy-ecdysone.
- Fold change in H3K56ac across Hr4, under the same conditions as in (B) (see Supplementary Methods for primers).
- Fold change in RNA levels for the indicated loci following 20-hydroxy-ecdysone treatment as in (B).
Data information: Graphs in (B–D) depict average results from ≥ 3 experiments, and error bars are standard error of the mean. In (B), pri/tal-bef, pri/tal-1, pri/tal-2 and Eip78C-EcR were significantly different from control treatment (P ≤ 0.05, t-test). In (C), primers 2, 3, 4 and 5 showed significant differences from controls in both conditions (P ≤ 0.05, t-test). In (D), pri/tal, Dip-B, Eip-78, Hr4 and Eip75 mRNA levels were significantly increased (P ≤ 0.05, t-test).
Discussion
Signalling pathways such as Notch have diverse functions depending on the context in which they are activated and on the specific subsets of genes that are regulated in each context. This specificity necessitates mechanisms that enable Su(H) to recognise and bind to appropriate enhancers and effect relevant gene expression changes. By utilising the comprehensive collection of chromatin modifications gathered by the modENCODE project (Kharchenko et al, 2011), we have generated maps of chromatin states in two Drosophila cell types and related those to the loci that are bound by Su(H). In doing so, we also analysed the profile of H3K56ac across the genome and found that this core histone modification is present at enhancers, and at transcription start sites, similar to the reported distribution in mammalian ES cells (Xie et al, 2009). Significantly, the inclusion of H3K56ac-binding data in the computational model helped to discriminate the active enhancers. Even more striking was the robust increase in this core nucleosome modification in response to Notch activation. Such changes were also detected in mammalian cells and at ecdysone-regulated genes in Drosophila, arguing that H3K56ac is likely to be a widespread modification associated with enhancer activation.
Unlike the modifications to exposed histone tails, which primarily provide docking sites for further chromatin modifying proteins, H3K56ac can directly alter nucleosomal DNA accessibility by increasing DNA breathing and unwrapping rate (Neumann et al, 2009; North et al, 2012). As a consequence, this modification can influence transcription factor (TF) occupancy within the nucleosome (Shimko et al, 2011; Tan et al, 2013) and it has been argued that H3K56ac drives chromatin towards the disassembled state during transcriptional activation (Williams et al, 2008). As the increase in H3K56ac appears to precede transcription elongation, it fits with the latter model. Furthermore, as mammalian CSL has been found to bind preferentially to motifs at the nucleosome exit point (Lake et al, 2014), H3K56ac may enhance recruitment, giving a feed-forward benefit that could potentially explain the increase in occupancy following Notch activation. In addition, H3K56ac facilitates divergent transcription by promoting rapid nucleosome turnover (Marquardt et al, 2014) and also promotes small RNA production in neurospora (Zhang et al, 2014), which is consistent with our detection of intergenic enhancer-templated RNAs in the modified regions following Notch activation.
The increase in H3K56ac appears to require CBP-HAT activity, which is also essential for catalysing this modification on free histones (Das et al, 2009). It is plausible therefore that the increase in H3K56ac could occur through the incorporation of pre-modified nucleosomes. The modification of histone dimers requires interaction with the chaperones CAF1 and ASF1, and while genetic evidence that the chaperone subunit dCAF-1-p105 can help promote Notch signalling (Yu et al, 2013) favours such a model, our results suggest this is less likely. First, we find that CBP is required at the time of activation, making it improbable that the increase in H3K56ac is a consequence of loading pre-modified histones. Second, an inhibitor of the CBP bromodomain, which plays an important role in enabling H3K56ac on histone dimers via its interaction with chaperones (Das et al, 2014), had no effect on the increase in H3K56ac. Thus, it seems more likely that the modification occurs at the time of enhancer activation, although it may nevertheless involve nucleosome exchange. For example, SWI/SNF nucleosome remodellers have been found to act in combination with H3K56ac to promote nucleosome turnover and gene activity in yeasts (Xu et al, 2005; Watanabe et al, 2014). At several loci where we detected changes in H3K56ac, the modification extended broadly from the site of Su(H)/NICD binding, correlating with domains that already possessed H3K4me1. Along with data from other studies of enhancer activation (Calo & Wysocka, 2013; Smith & Shilatifard, 2014), and the observation that levels of H3K56ac are affected by mutation of H3K4 (Guan et al, 2013), this suggests that H3K4me1 is likely to be one of the earliest modifications, prefiguring sites of active enhancer. It may also facilitate the spread of H3K56ac across the regulated regions.
Our analysis of the relationship between chromatin states and regions occupied by Su(H) suggests that the pre-existing chromatin environment is likely to make an important contribution to recruitment. First, Su(H)-occupied motifs were almost exclusively located in highly accessible chromatin, with modifications such as H3K4me1 characteristic of enhancer states. Second, expression of the cooperating transcription factor Lz converted enhancers towards this preferred chromatin state where additional Su(H) was recruited. By having a preference for a particular chromatin signature, the vast majority (> 91%) of potential Su(H) binding motifs will be masked by unfavourable chromatin. Indeed, the small fraction of sites that do not fit with this pattern may reflect false positives in the ChIP data or in chromatin assignment. The greater paradox is that only 7–10% of CSL motifs within the favourable Enh chromatin were bound. Furthermore, many of the positions that were differentially bound in two cell types existed in Enh chromatin in both cell types examined. These observations suggest that additional factors restrict CSL binding to a subset of sites located within favourable chromatin. Such factors might include currently unknown histone modifications, protein–protein interactions, 3D organisation and/or DNA sequence properties around the CSL motif.
Once bound, Su(H) itself also helps to shape the local chromatin environment. Depleting cells of Su(H) resulted in an increase in local histone acetylation (H3K27ac, H3K56ac), suggesting that, in the absence of NICD, Su(H) helps to suppress enhancer activity through its association with co-repressors. Thus, a model emerges in which Su(H) is recruited to regions that have already acquired regulatory competence and that it keeps these in a transitional state with low levels of H3K56ac. As there is considerable variability between enhancers, this suggests that each attains an activity that reflects the balance between the transcription factors promoting enhancer activity and those, such as Su(H), that can antagonise it. In those instances where Su(H)-corepressor complexes win out, then the enhancer is suppressed until the complimentary activity of NICD converts it from a transitional to an active state, a conversion that is associated with a large-scale increase in H3K56ac.
The extent that the principles we have observed here will be of general relevance for other signalling pathways remains to be established, although it seems likely that their target gene specificity will be similarly dependant on the pre-existing chromatin substrate. However, it is possible that the inferred transitional enhancer states may be particularly relevant for those pathways/contexts where there is a fine-scale switch between repression and activation, as occurs for Notch and ecdysone signalling. Nevertheless, the correlation of H3K56ac with H3K4me1 suggests that H3K56ac is likely to be of widespread importance in enhancer activation. Whether this will be mediated through its direct effects on DNA–histone core interactions or through intermediate bromodomain containing proteins that link to the core transcription machinery, such as Brd4, remains to be determined.
Materials and Methods
Computational methods
Details of the datasets, normalisation and training for the Hidden Markov Models are provided in Supplementary Methods along with a summary of the algorithm used to detect differences in H3K56ac and of motif enrichment analysis. The BED files containing the chromatin maps are available on GitHub at https://github.com/rstojnic/notch-chromatin as are the data and scripts used.
Cell culture, Notch activation and inhibitor treatments
Kc cells were cultured at 25°C in Shields and Sang M3 insect medium (Sigma, S3652), supplemented with 5% FBS (Sigma, F9665), 1 g/l yeast extract (Oxoid, LP0021), 2.5 g/l bacto-peptone (BD Biosciences, 211677) and 1× Antibiotic-Antimycotic (Gibco, 15240-062). BG3 cells were cultured at 25°C in Shields and Sang M3 insect medium, supplemented with 10% FBS (Sigma, F4135), 10 mg/l insulin (Sigma, I9278) and also 1× Antibiotic-Antimycotic. For Notch activation, Kc cells were treated with 4 mM EGTA in PBS and BG3 cells were treated with 4 mM EGTA in HBSS (Invitrogen, 14170) for 30 min unless otherwise stated. EGTA destabilises the Notch negative regulatory region, exposing the site for Adam10, and consequently results in γ-secretase cleavage and release of NICD (e.g. Gupta-Rossi et al, 2001; Krejcí & Bray, 2007; Ilagan et al, 2011). To demonstrate Notch-dependent effects of this treatment, cells were incubated with γ-secretase inhibitor (10 nm Compound E, see Supplementary Table S3). Kc cells were treated with 5 μM ecdysone (Sigma, H5142) for 1 or 3 h before RNA or chromatin isolation. For inhibitor treatment, BG3 cells were pre-treated with inhibitors, as listed in Supplementary Table S3, and then incubated with EGTA or HBSS plus inhibitors for further 30 min. Conditions for culturing and Dll1 treatment of C2C12 cells were as described previously (Castel et al, 2013). 1 × 106 cells were plated onto culture dishes pre-coated with Delta-like 1 fused to the Fc fragment of human IgG (Dll1-Fc) or with Fc fragment of human IgG (Control-Fc). 20 μM DAPT (Calbiochem, #565784) was included in control-Fc incubations.
Chromatin immunoprecipitation, ChIP array and RNA isolation
ChIP experiments, RNA isolation and real-time PCR were basically performed as described (Krejcí & Bray, 2007) with the following modifications. 1% formaldehyde was used for ChIP cross-linking, and the DNA was purified after proteinase K treatment using columns (Qiagen, 28106). 6–10 × 106 cells were used as a starting material for Su(H) ChIP arrays, and the resulting DNA, along with 10 ng of input, was amplified using the WGA2 (Sigma) for two rounds of amplification (14 and 6 PCR cycles). For H3K56ac ChIP arrays, 5 × 106 cells and 14 amplification cycles were used. All antibodies used for ChIP are listed in Supplementary Table S4. For whole-genome analysis, 1 μg double-stranded ChIP or input DNA was labelled with either Cy3- or Cy5-random primers using the Nimblegen Dual Colour kit. Both ChIP and input were co-hybridised to NimbleGen D. melanogaster ChIP-chip 2.1 M whole-genome tiling arrays in the Nimblegen hybridisation station at 42°C for 16 h and then washed according to the NimbleGen Wash Buffer kit instructions. The arrays were scanned at 5-μm resolution with a GenePix 4000B (Axon) dual laser scanner at individually optimised PMT gain settings and images processed with NimbleScan software (Roche-Nimblegen) with Loess spatial correction. The data were then normalised using quantile normalisation across the replicate arrays in R. Window smoothing and peak calling were performed using the Bioconductor package Ringo (Toedling et al, 2007) with a winHalfSize of 600 bp and min.probes = 5. Probe levels were then assigned P-values based on the normalNull method, corrected for multiple testing using the Hochberg–Benjamini algorithm and then condensed into regions using distCutOff of 300 bp. Data from genome-wide ChIP experiments have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO, www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession number GSE66227.
In RNA isolation, DNase treatments (Ambion, AM1906) were performed before RNA reverse transcription. To assess non-coding/intergenic transcription, 5 μg RNA was reverse transcribed with random primers and Superscript III. The product was then diluted 1:5, and 2 μl was used in each real-time PCR. “No reverse transcription” control was performed in parallel to confirm the absence of genomic DNA. All primers used in real-time PCR are listed in Supplementary Table S5.
Lz overexpression, NICD expression and Su(H) RNAi in cells lines
Kc and BG3 cells were transfected with pMT-puro plasmid, pMT-puro-Lz or pMT-puro-NICD construct and then grown under permanent selection with 2 μg/ml puromycin (Sigma). Expression of Lz and NICD were induced by adding 500 μM CuSO4 (Sigma) to cell culture medium for 3 days or for 2 h, respectively. pMT-puro was a gift from D. Sabatini (Center for Cancer Research, MIT, Cambridge, USA), and Lz cDNA plasmid was a gift from M. Haenlin (Centre de Biologie du Développement, Toulouse, France). For Su(H) RNAi, BG3 cells were transfected with 20 μg dsRNA in Fugene 6 (Promega, E2691) in 10-cm plates according to the standard protocol and then incubated for 72 h.
Acknowledgments
We thank members of the Bray and Adryan laboratories for helpful discussions, Kat Millen for the images shown in Supplementary Fig S5, Marc Haenlin and David Sabatini for plasmids, and Philippos Mourikis for advice on C2C12 experiments and for helpful comments on the manuscript. This work was supported by a BBSRC project grant [BB/J008842/1] to SJB, BA and SR and by a MRC programme grant [G0800034] to SJB. JL is the recipient of a scholarship from the China Scholarship Council Cambridge.
Author contributions
LS, RS, JL, SJB, BA, SR and ST conceived and designed the experiments. LS, JL, GC-M, BF and HS performed the experiments. RS, JL, BF and SJB analysed the data. RS, BF, HS and ST contributed reagents/materials/analysis tools. SJB, RS, JL, LS, SR and BA wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Information
Review Process File
References
- Borggrefe T, Oswald F. The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci. 2009;66:1631–1646. doi: 10.1007/s00018-009-8668-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, Crump NT, Hazzalin CA, Liszczak G, Yuan H, Larocca C, Saldanha SA, Abagyan R, Sun Y, Meyers DJ, Marmorstein R, Mahadevan LC, Alani RM, Cole PA. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471–482. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- Bray S, Bernard F. Notch targets and their regulation. Curr Top Dev Biol. 2010;92:253–275. doi: 10.1016/S0070-2153(10)92008-5. [DOI] [PubMed] [Google Scholar]
- Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel D, Mourikis P, Bartels SJ, Brinkman AB, Tajbakhsh S, Stunnenberg HG. Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev. 2013;27:1059–1071. doi: 10.1101/gad.211912.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanut-Delalande H, Hashimoto Y, Pelissier-Monier A, Spokony R, Dib A, Kondo T, Bohere J, Niimi K, Latapie Y, Inagaki S, Dubois L, Valenti P, Polesello C, Kobayashi S, Moussian B, White KP, Plaza S, Kageyama Y, Payre F. Pri peptides are mediators of ecdysone for the temporal control of development. Nat Cell Biol. 2014;16:1035–1044. doi: 10.1038/ncb3052. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature. 2009;459:113–117. doi: 10.1038/nature07861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das C, Roy S, Namjoshi S, Malarkey CS, Jones DN, Kutateladze TG, Churchill ME, Tyler JK. Binding of the histone chaperone ASF1 to the CBP bromodomain promotes histone acetylation. Proc Natl Acad Sci U S A. 2014;111:E1072–E1081. doi: 10.1073/pnas.1319122111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferjoux G, Auge B, Boyer K, Haenlin M, Waltzer L. A GATA/RUNX cis-regulatory module couples Drosophila blood cell commitment and differentiation into crystal cells. Dev Biol. 2007;305:726–734. doi: 10.1016/j.ydbio.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Fryer CJ, Lamar E, Turbachova I, Kintner C, Jones KA. Mastermind mediates chromatin-specific transcription and turnover of the Notch enhancer complex. Genes Dev. 2002;16:1397–1411. doi: 10.1101/gad.991602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallenkamp D, Gelato KA, Haendler B, Weinmann H. Bromodomains and their pharmacological inhibitors. ChemMedChem. 2014;9:438–464. doi: 10.1002/cmdc.201300434. [DOI] [PubMed] [Google Scholar]
- Gauhar Z, Sun LV, Hua S, Mason CE, Fuchs F, Li TR, Boutros M, White KP. Genomic mapping of binding regions for the Ecdysone receptor protein complex. Genome Res. 2009;19:1006–1013. doi: 10.1101/gr.081349.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X, Rastogi N, Parthun MR, Freitas MA. Discovery of histone modification crosstalk networks by stable isotope labeling of amino acids in cell culture mass spectrometry (SILAC MS) Mol Cell Proteomics. 2013;12:2048–2059. doi: 10.1074/mcp.M112.026716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta-Rossi N, Le Bail O, Gonen H, Brou C, Logeat F, Six E, Ciechanover A, Israel A. Functional interaction between SEL-10, an F-box protein, and the nuclear form of activated Notch1 receptor. J Biol Chem. 2001;276:34371–34378. doi: 10.1074/jbc.M101343200. [DOI] [PubMed] [Google Scholar]
- Hay DA, Fedorov O, Martin S, Singleton DC, Tallant C, Wells C, Picaud S, Philpott M, Monteiro OP, Rogers CM, Conway SJ, Rooney TP, Tumber A, Yapp C, Filippakopoulos P, Bunnage ME, Muller S, Knapp S, Schofield CJ, Brennan PE. Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J Am Chem Soc. 2014;136:9308–9319. doi: 10.1021/ja412434f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502:480–488. doi: 10.1038/nature12751. [DOI] [PubMed] [Google Scholar]
- Ho JW, Jung YL, Liu T, Alver BH, Lee S, Ikegami K, Sohn KA, Minoda A, Tolstorukov MY, Appert A, Parker SC, Gu T, Kundaje A, Riddle NC, Bishop E, Egelhofer TA, Hu SS, Alekseyenko AA, Rechtsteiner A, Asker D, et al. Comparative analysis of metazoan chromatin organization. Nature. 2014;512:449–452. doi: 10.1038/nature13415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housden BE, Fu AQ, Krejcí A, Bernard F, Fischer B, Tavare S, Russell S, Bray SJ. Transcriptional dynamics elicited by a short pulse of notch activation involves feed-forward regulation by E(spl)/Hes genes. PLoS Genet. 2013;9:e1003162. doi: 10.1371/journal.pgen.1003162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilagan MX, Lim S, Fulbright M, Piwnica-Worms D, Kopan R. Real-time imaging of notch activation with a luciferase complementation-based reporter. Sci Signal. 2011;4:rs7. doi: 10.1126/scisignal.2001656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Mutvei AP, Chivukula IV, Andersson ER, Ramskold D, Sandberg R, Lee KL, Kronqvist P, Mamaeva V, Ostling P, Mpindi JP, Kallioniemi O, Screpanti I, Poellinger L, Sahlgren C, Lendahl U. Non-canonical Notch signaling activates IL-6/JAK/STAT signaling in breast tumor cells and is controlled by p53 and IKKalpha/IKKbeta. Oncogene. 2013;32:4892–4902. doi: 10.1038/onc.2012.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, Linder-Basso D, Plachetka A, Shanower G, Tolstorukov MY, Luquette LJ, Xi R, Jung YL, Park RW, Bishop EP, Canfield TK, et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011;471:480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Kovall RA, Blacklow SC. Mechanistic insights into Notch receptor signaling from structural and biochemical studies. Curr Top Dev Biol. 2010;92:31–71. doi: 10.1016/S0070-2153(10)92002-4. [DOI] [PubMed] [Google Scholar]
- Krejcí A, Bray S. Notch activation stimulates transient and selective binding of Su(H)/CSL to target enhancers. Genes Dev. 2007;21:1322–1327. doi: 10.1101/gad.424607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejcí A, Bernard F, Housden BE, Collins S, Bray SJ. Direct response to Notch activation: signaling crosstalk and incoherent logic. Sci Signal. 2009;2:ra1. doi: 10.1126/scisignal.2000140. [DOI] [PubMed] [Google Scholar]
- Lake RJ, Tsai PF, Choi I, Won KJ, Fan HY. RBPJ, the major transcriptional effector of Notch signaling, remains associated with chromatin throughout mitosis, suggesting a role in mitotic bookmarking. PLoS Genet. 2014;10:e1004204. doi: 10.1371/journal.pgen.1004204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde ME, Cheng X, Cote J. Histone target selection within chromatin: an exemplary case of teamwork. Genes Dev. 2014;28:1029–1041. doi: 10.1101/gad.236331.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvi A, Artavanis-Tsakonas S. Notch and disease: a growing field. Semin Cell Dev Biol. 2012;23:473–480. doi: 10.1016/j.semcdb.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt S, Escalante-Chong R, Pho N, Wang J, Churchman LS, Springer M, Buratowski S. A chromatin-based mechanism for limiting divergent noncoding transcription. Cell. 2014;157:1712–1723. doi: 10.1016/j.cell.2014.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall NF, Price DH. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem. 1995;270:12335–12338. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]
- Miele L, Golde T, Osborne B. Notch signaling in cancer. Curr Mol Med. 2006;6:905–918. doi: 10.2174/156652406779010830. [DOI] [PubMed] [Google Scholar]
- Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, Chin JW. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Mol Cell. 2009;36:153–163. doi: 10.1016/j.molcel.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North JA, Shimko JC, Javaid S, Mooney AM, Shoffner MA, Rose SD, Bundschuh R, Fishel R, Ottesen JJ, Poirier MG. Regulation of the nucleosome unwrapping rate controls DNA accessibility. Nucleic Acids Res. 2012;40:10215–10227. doi: 10.1093/nar/gks747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntziachristos P, Lim JS, Sage J, Aifantis I. From fly wings to targeted cancer therapies: a centennial for notch signaling. Cancer Cell. 2014;25:318–334. doi: 10.1016/j.ccr.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald F, Tauber B, Dobner T, Bourteele S, Kostezka U, Adler G, Liptay S, Schmid RM. p300 acts as a transcriptional coactivator for mammalian Notch-1. Mol Cell Biol. 2001;21:7761–7774. doi: 10.1128/MCB.21.22.7761-7774.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke F, Raj K. The role of Notch in tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer. 2003;3:756–767. doi: 10.1038/nrc1186. [DOI] [PubMed] [Google Scholar]
- Roy M, Pear WS, Aster JC. The multifaceted role of Notch in cancer. Curr Opin Genet Dev. 2007;17:52–59. doi: 10.1016/j.gde.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Sekiya T, Muthurajan UM, Luger K, Tulin AV, Zaret KS. Nucleosome-binding affinity as a primary determinant of the nuclear mobility of the pioneer transcription factor FoxA. Genes Dev. 2009;23:804–809. doi: 10.1101/gad.1775509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimko JC, North JA, Bruns AN, Poirier MG, Ottesen JJ. Preparation of fully synthetic histone H3 reveals that acetyl-lysine 56 facilitates protein binding within nucleosomes. J Mol Biol. 2011;408:187–204. doi: 10.1016/j.jmb.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyueva D, Stelzer C, Gerlach D, Yanez-Cuna JO, Rath M, Boryn LM, Arnold CD, Stark A. Hormone-responsive enhancer-activity maps reveal predictive motifs, indirect repression, and targeting of closed chromatin. Mol Cell. 2014;54:180–192. doi: 10.1016/j.molcel.2014.02.026. [DOI] [PubMed] [Google Scholar]
- Simon M, North JA, Shimko JC, Forties RA, Ferdinand MB, Manohar M, Zhang M, Fishel R, Ottesen JJ, Poirier MG. Histone fold modifications control nucleosome unwrapping and disassembly. Proc Natl Acad Sci U S A. 2011;108:12711–12716. doi: 10.1073/pnas.1106264108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E, Shilatifard A. Enhancer biology and enhanceropathies. Nat Struct Mol Biol. 2014;21:210–219. doi: 10.1038/nsmb.2784. [DOI] [PubMed] [Google Scholar]
- Swygert SG, Peterson CL. Chromatin dynamics: interplay between remodeling enzymes and histone modifications. Biochim Biophys Acta. 2014;1839:728–736. doi: 10.1016/j.bbagrm.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Xue Y, Song C, Grunstein M. Acetylated histone H3K56 interacts with Oct4 to promote mouse embryonic stem cell pluripotency. Proc Natl Acad Sci U S A. 2013;110:11493–11498. doi: 10.1073/pnas.1309914110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terriente-Felix A, Li J, Collins S, Mulligan A, Reekie I, Bernard F, Krejcí A, Bray S. Notch cooperates with Lozenge/Runx to lock haemocytes into a differentiation programme. Development. 2013;140:926–937. doi: 10.1242/dev.086785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel CS. From embryogenesis to metamorphosis: the regulation and function of Drosophila nuclear receptor superfamily members. Cell. 1995;83:871–877. doi: 10.1016/0092-8674(95)90203-1. [DOI] [PubMed] [Google Scholar]
- Toedling J, Skylar O, Krueger T, Fischer JJ, Sperling S, Huber W. Ringo–an R/Bioconductor package for analyzing ChIP-chip readouts. BMC Bioinformatics. 2007;8:221. doi: 10.1186/1471-2105-8-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CC, Kao HY, Yao TP, McKeown M, Evans RM. SMRTER, a Drosophila nuclear receptor coregulator, reveals that EcR-mediated repression is critical for development. Mol Cell. 1999;4:175–186. doi: 10.1016/s1097-2765(00)80365-2. [DOI] [PubMed] [Google Scholar]
- Wallberg AE, Pedersen K, Lendahl U, Roeder RG. p300 and PCAF act cooperatively to mediate transcriptional activation from chromatin templates by Notch intracellular domains in vitro. Mol Cell Biol. 2002;22:7812–7819. doi: 10.1128/MCB.22.22.7812-7819.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zou J, Zhao B, Johannsen E, Ashworth T, Wong H, Pear WS, Schug J, Blacklow SC, Arnett KL, Bernstein BE, Kieff E, Aster JC. Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc Natl Acad Sci U S A. 2012;108:14908–14913. doi: 10.1073/pnas.1109023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zang C, Taing L, Arnett KL, Wong YJ, Pear WS, Blacklow SC, Liu XS, Aster JC. NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc Natl Acad Sci U S A. 2014;111:705–710. doi: 10.1073/pnas.1315023111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Radman-Livaja M, Rando OJ, Peterson CL. A histone acetylation switch regulates H2A.Z deposition by the SWR-C remodeling enzyme. Science. 2014;340:195–199. doi: 10.1126/science.1229758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SK, Truong D, Tyler JK. Acetylation in the globular core of histone H3 on lysine-56 promotes chromatin disassembly during transcriptional activation. Proc Natl Acad Sci U S A. 2008;105:9000–9005. doi: 10.1073/pnas.0800057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E, van Steensel B. Chromatin domains in higher eukaryotes: insights from genome-wide mapping studies. Chromosoma. 2009;118:25–36. doi: 10.1007/s00412-008-0186-0. [DOI] [PubMed] [Google Scholar]
- Xie W, Song C, Young NL, Sperling AS, Xu F, Sridharan R, Conway AE, Garcia BA, Plath K, Clark AT, Grunstein M. Histone H3 lysine 56 acetylation is linked to the core transcriptional network in human embryonic stem cells. Mol Cell. 2009;33:417–427. doi: 10.1016/j.molcel.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Zhang K, Grunstein M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell. 2005;121:375–385. doi: 10.1016/j.cell.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Yanez-Cuna JO, Arnold CD, Stampfel G, Boryn LM, Gerlach D, Rath M, Stark A. Dissection of thousands of cell type-specific enhancers identifies dinucleotide repeat motifs as general enhancer features. Genome Res. 2014;24:1147–1156. doi: 10.1101/gr.169243.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Wu H, Chen H, Wang R, Liang X, Liu J, Li C, Deng WM, Jiao R. CAF-1 promotes Notch signaling through epigenetic control of target gene expression during Drosophila development. Development. 2013;140:3635–3644. doi: 10.1242/dev.094599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yang Q, Sun G, Chen S, He Q, Li S, Liu Y. Histone H3K56 acetylation is required for quelling-induced small RNA production through its role in homologous recombination. J Biol Chem. 2014;289:9365–9371. doi: 10.1074/jbc.M113.528521. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Review Process File