

Abstract
Nonfunctional neuroendocrine tumors of the pancreas (NF-PNETs) are a heterogeneous group of neoplasms. Although rare, the incidence of NF-PNETs is increasing significantly. The classification of PNETs has evolved over the past decades and is now based on a proliferation grading system. While most NF-PNETs are slow growing, tumors with more aggressive biology may become incurable once they progress to unresectable metastatic disease. Tumors of higher grade can be suspected preoperatively based on the presence of calcifications, hypoenhancement on arterial phase computed tomography, positron emission technology avidity and lack of octreotide scan uptake. Surgery is the only curative treatment and is recommended for most patients for whom complete resection is possible. Liver-directed therapies (thermal ablation, transarterial embolization) can be useful in controlling unresectable hepatic metastatic disease. In the presence of unresectable progressive disease, somatostatin analogues, everolimus and sunitinib can prolong progression-free survival. This article provides a comprehensive review of NF-PNETs with special emphasis on recent advances in diagnosis and management.
Keywords: Pancreas, Neuroendocrine tumor, Neuroendocrine carcinoma, Islet cell, Octreotide
Core tip: Pancreatic neuroendocrine tumors (PNETs) are a fascinating and diverse group of neoplasms. While the clinical features of functional PNETs are frequently discussed, the majority of PNETs are actually non-functional. Although typically slow growing, tumors with more aggressive biology may progress to unresectable metastatic disease. Surgery should be considered for all patients for whom complete resection is possible, while liver directed therapies are useful for managing hepatic metastases. For patients with progressive metastatic disease, strong evidence supports the use of somatostatin analogues, everolimus and sunitinib in prolonging survival. The purpose of this article is to provide a comprehensive review of NF-PNETs.
INTRODUCTION
Pancreatic neuroendocrine tumors (PNET) are a rare heterogeneous group of neoplasms that arise from progenitor islet cells. PNETs may be classified as either functional (F-PNET) or non-functional (NF-PNET), depending on their ability to secrete biologically active hormones and elicit characteristic symptomatology. NF-PNETs exhibit a wide range of malignant potential, ranging from slow-growing and non-infiltrative tumors to locally invasive and rapidly metastasizing ones, thereby making standardization of the diagnosis, surgical and medical management, follow-up surveillance and prognosis challenging. Fortunately, significant advances in diagnostic modalities, tumor localization and therapeutic options have been made over the past decade. This article provides a comprehensive review of NF-PNETs and an update on advances in their diagnosis and management.
EPIDEMIOLOGY
Although neuroendocrine neoplasms can occur nearly anywhere in the body, gastroenteropancreatic neuroendocrine tumors (GEP-NET) and pulmonary neuroendocrine tumors comprise the majority. PNETs comprise approximately 7% of all NETs[1]. However, compared to other pancreatic pathology, PNETs are relatively rare, comprising only 1%-2% of all pancreatic neoplasms. The incidence of PNETs increases significantly after the age of 40 with a peak incidence around age 65[1]. There is only a slight male predominance[2]. Between 60%-90% of all PNETs are non-functional and given their frequently asymptomatic nature the majority of patients present with distant metastasis[2,3].
By all measures, the incidence of PNETs is increasing. The Surveillance, Epidemiology and End Results (SEER) program has shown that the incidence has increased from 0.17 per 100000 people in 1973 to 0.47 per 100000 people in 2007[1]. Likewise, a six-fold increase in the incidence was found in Ontario, Canada between 1994 and 2009 (from 0.1 to 0.6 per 100000 persons)[4]. Autopsy studies would also suggest that the prevalence of PNETs is higher than previously suspected[5]. Interestingly, this trend of increasing incidence of PNETs seems to be true of all neoplasms of neuroendocrine origin[4,6] and may be partly related to increased incidental discovery due to more frequent use and improving sensitivity of cross-sectional imaging.
STAGING AND PROGNOSIS
In 2000, the World Health Organization (WHO) first established guidelines that distinguished between well-differentiated tumors with benign behavior (localized to pancreas, size < 2 cm, low mitotic rate and Ki-67, no angioinvasion or perineural invasion), tumors with uncertain behavior (limited to the pancreas, angioinvasion or perineural invasion, size ≥ 2 cm) and tumors with clearly malignant behavior (gross local invasion or distant metastases)[7]. In 2010, the WHO revised their previous grading system to a proliferation based grading system (Table 1). Based on mitotic counts and Ki-67 indices, well-differentiated tumors included those of low and intermediate grade while poorly differentiated tumors included high grade tumors. It was concluded that mitotic count and Ki-67 should be performed on all specimens and that the grade would reflect the higher value when discordant[8]. In fact, Ki-67 and differentiation has been found to be some of the most important factors in determining prognosis[9].
Table 1.
Current pancreatic neuroendocrine tumor classification and staging systems
| WHO 2010/ENETS grading | |||
| Grade | Differentiation | Ki-67 index (%) | Mitotic count/10 HPF |
| G1 (low) | Well | ≤ 2 | < 2 |
| G2 (intermediate) | Well | 3-20 | 2-20 |
| G3 (high) | Poorly | > 20 | > 20 |
| ENETS T staging1 | |||
| T stage | Description | ||
| TX | Cannot be assessed | ||
| T0 | No evidence of tumor | ||
| T1 | < 2 cm, limited to pancreas | ||
| T2 | 2-4 cm, limited to pancreas | ||
| T3 | > 4 cm, limited to pancreas | ||
| T4 | Involving adjacent organs or large blood vessels | ||
| AJCC T staging1 | |||
| T stage | Description | ||
| TX | Cannot be assessed | ||
| T0 | No evidence of tumor | ||
| T1 | ≤ 2 cm, limited to pancreas | ||
| T2 | > 2 cm, limited to pancreas | ||
| T3 | Involves adjacent organs | ||
| T4 | Involving celiac axis or superior mesenteric artery | ||
| Stage | ENETS staging | AJCC staging | |
| IA | T1 N0 M0 | T1 N0 M0 | |
| IB | T2 N0 M0 | ||
| IIA | T2 N0 M0 | T3 N0 M0 | |
| IIB | T3 N0 M0 | T1-3 N1 M0 | |
| IIIA | T4 N0 M0 | T4 N1 M0 | |
| IIIB | T1-4 N1 M0 | ||
| IV | T1-4 N0-1 M1 | T1-4 N0-1 M0 | |
Both AJCC and ENETS share common N and M staging: N0, no regional lymph node metastatic; N1, regional lymph node metastasis; M0, no distant metastasis; M1, distant metastasis. WHO: World Health Organization; ENETS: European neuroendocrine tumor society; AJCC: 7th edition American joint committee on cancer.
Based on results of a consensus conference in 2005, the European Neuroendocrine Tumour Society (ENETS) proposed a classification scheme for all foregut NETs that combined a TNM staging system with a histologic grading system[10]. The most commonly used staging system, however, is from the 7th edition American Joint Committee on Cancer (AJCC)[11]. Revised in 2010, this system applies to all neoplasms of the pancreas, both endocrine and exocrine, and is based on TNM staging (Table 1). Importantly, the AJCC system does not incorporate histological grading criteria.
Both the ENETS and the AJCC system have been validated and provide important prognostic information for patients with PNETs[12]. However, some have called into question whether the AJCC system provides adequate discriminatory value. Specifically, validation studies by Strosberg et al[13,14] showed no survival difference between stages I and II as well as stages III and IV. In addition, Rindi et al[15] studied a large international cohort of resected PNETs and found no significant differences in survival between stage II and III. A large range of outcomes was seen in patients of all stages, suggesting poor discriminatory ability, and they concluded that the ENETS staging system was superior[15]. Qadan et al[16] utilized SEER to demonstrate that no significant survival differences could be replicated between stages II and III or III and IV, and suggested a revised TNM staging system with potentially improved prognostic capabilities.
CLINICAL PRESENTATION
Unlike other solid tumors (including F-PNETs), NF-PNETs can remain asymptomatic before they reach a significant tumor burden. When they become symptomatic, their symptomatology is typically related to mass effect from the primary tumor or the metastasis. Many PNETs occur in the head of the pancreas where symptoms may include jaundice, abdominal pain, or weight loss. Other less frequent symptoms may include anorexia, nausea, intra-abdominal bleeding, or a palpable mass. Many will be asymptomatic and found incidentally on cross-sectional imaging performed for other indications. The vast majority of metastases occur in the liver, though other sites including bone, peritoneum, adrenal, brain and spleen have been reported[17]. Liver metastases more frequently occur with non-functional tumors and patients with symptoms. When liver metastases occur, most are multifocal and bilobar[17].
F-PNETs present with symptoms caused by the specific hormone produced. Common F-NETs include insulinoma, which presents with hypoglycemia, and gastrinoma, which presents with peptic ulcer disease, gastroesophageal reflux disease or secretory diarrhea. Less common F-NETs include VIPomas, glucagonomas, and somatostainomas. These tumors are summarized in Table 2 but are not discussed further in this review. NF-PNETs either do not produce hormones, produce hormones at a low enough level to not cause symptoms, or are associated with hormones that do not cause symptoms, such as pancreatic polypeptide, chromogranin A, ghrelin, calcitonin or neurotensin.
Table 2.
Clinical features of functional pancreatic neuroendocrine tumors
| Tumor | Percentage | Secreted hormone | Malignant | Clinical features | Biochemical evaluation |
| Insulinoma | 40%-60% | Insulin | < 10% | Hypoglycemia | Insulin, pro-insulin, C-peptide, 72 h fasting insulin/glucose ratio |
| Gastrinoma | 20%-50% | Gastrin | 60%-90% | PUD, GERD, diarrhea | Fasting gastrin (off PPI), secretin stimulation test |
| Glucagonoma | Rare | Glucagon | 50%-80% | Necrolytic migratory erythema, diabetes, venous thrombosis, depression | Glucagon |
| Somatostatinoma | Rare | Somatostatin | > 70% | Diabetes, hypochlorhydria, cholelithiasis, diarrhea | Somatostatin (not widely available) |
| VIPoma | Rare | Vasoactive Intestinal Peptide | 40%-70% | Watery diarrhea, hypokalemia, achlorhydria | VIP |
PUD: Peptic ulcer disease; GERD: Gastroesophageal reflux disease; PPI: Proton pump inhibitor.
While most NF-NETs are sporadic, approximately 10% of NETs will be associated with an inherited genetic syndrome[18]. Multiple endocrine neoplasia type 1 (MEN1) is an inherited autosomal dominant disease characterized by hyperparathyroidism (nearly 100%), PNETs (up to 75%) and pituitary tumors (less than 50%)[19]. NF-PNETs are the most common pancreatic neoplasms in MEN1, followed by gastrinomas, and then insulinomas. Patients with MEN1 frequently present with multiple PNETs throughout the pancreas[20]. Von Hippel-Lindau (VHL) disease is an autosomal dominant disorder that is associated with pancreatic tumors or cysts. In order of frequency, patients develop pancreatic cysts, NF-PNETs (10%-20% of patients), cystadenomas, hemangioblastomas and adenocarcinoma; F-PNETs are rare[20]. PNETs in neurofibromatosis type 1 (NF1) are relatively rare (0%-10%) but are almost exclusively duodenal somatostatinomas in the periampullary region[21]. Other functional and non-functional PNETs may rarely occur[22-24]. PNETs associated with tuberous sclerosis (TS) are relatively uncommon and may be either functional or non-functional[25].
DIAGNOSIS
Patients with PNETs require a thorough evaluation for symptoms classically associated with functional tumors as well as symptoms directly related to the primary or metastatic tumor. Past medical and family history should be carefully reviewed. A comprehensive physical examination should be undertaken. Ultimately, the diagnosis of PNETs depends on comprehensive biochemical and radiographic evaluation.
Biochemical
Neuroendocrine markers are important not only for confirming diagnosis, but also as screening tools for future surveillance. The most commonly utilized neuroendocrine markers include chromogranin A (CgA), pancreatic polypeptide (PPP), pancreastatin, and neuron-specific enolase (NSE). CgA is a glycoprotein used commonly as a tumor marker in histopathology but also has elevated circulating levels in patients with both functional and non-functional PNETs[26,27]. However, falsely elevated levels can be observed in patients with chronic renal insufficiency, liver failure and with proton-pump inhibitor (PPI) use[28,29]. Recent recommendations by the North American Neuroendocrine Tumor Society[30] and a Canadian national expert group[29] have recommended utilizing CgA in the diagnosis and surveillance of advanced PNETs. PPP may be elevated in up to 63% of PNETs[31] and has a specificity of 84% when used during surveillance[32]. Pancreastatin may provide additional diagnostic utility, especially in patients on PPIs or with normal CgA levels[33,34]. Laboratory evaluation should also include tests to rule out F-NETs, including insulinoma and gastrinoma, if suspected (Table 2). Screening for MEN1 with serum parathyroid hormone and calcium levels should be performed in appropriate patients (e.g., diagnosis at young age, multifocal tumors, and/or with relevant personal or family history).
Localization
Cross-sectional imaging should be performed in all patients suspected of having a PNET. Computed tomography (CT) remains the initial imaging modality of choice given its good sensitivity, specificity and availability. PNETs typically are well-circumscribed lesions that appear hyperenhancing on contrast-enhanced scans. In fact, there is some evidence that hypoenhancement on arterial phase imaging is associated with more aggressive tumors and worse prognosis (Figure 1)[35]. Similarly, the presence of calcifications within these tumors on CT is associated with higher grade and the presence of lymph node metastases (Figure 2)[36]. Magnetic resonance imaging (MRI) is an alternative modality with the advantage of less radiation exposure. PNETs should be low signal intensity on T1 weighted images and high signal intensity on T2 weighted images. In addition, MRI may be more sensitive than CT for detecting smaller pancreatic lesions and liver metastases[37,38]. While ultrasound has a limited role in the diagnosis of PNETs, intraoperative ultrasound (IOUS) is very sensitive in identifying small PNETs[39], and endoscopic ultrasound (EUS) is a valuable technique for detection, localization and diagnosis through fine needle aspiration of identified lesions[40].
Figure 1.
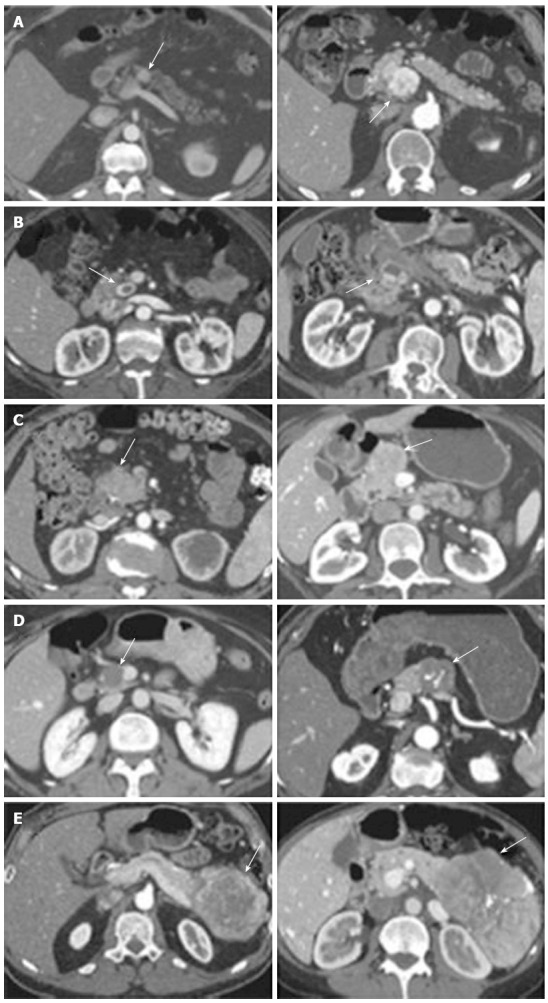
Representative images of the 5 types of pancreatic neuroendocrine tumor enhancement pattern on arterial phase computed tomography. Two images are shown for each type. A: Hyperenhancing, solid; B: Cystic with hyperenhancing rim; C: Isoenhancing or no mass visualized; D: Homogeneously hypoenhancing; E: Heterogeneous but mostly hypoenhancing with some peripheral enhancement. Groups D and E had worse survival after resection compared with groups A, B, and C (From Worhunsky et al[35]. Pancreatic neuroendocrine tumours: hypoenhancement on arterial phase computed tomography predicts biological aggressiveness. HPB 2014; 16: 304-311). Arrows indicate PNET.
Figure 2.
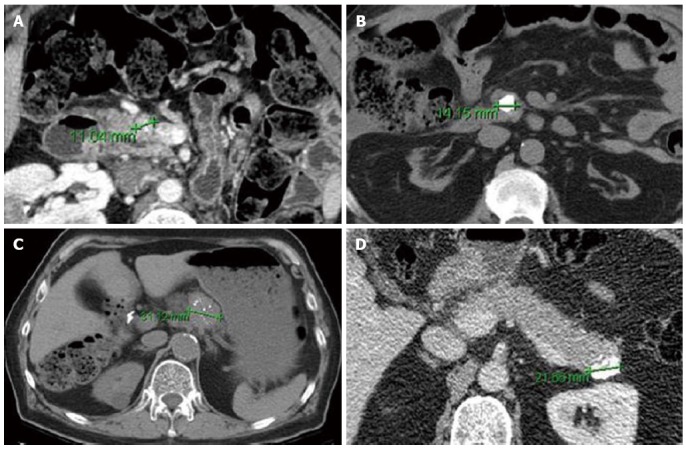
Axial computed tomography images of Pancreatic neuroendocrine tumor with punctate (A, C) and dense/coarse calcifications (B, D). Despite their small size, all lesions were associated with either lymph node metastasis (A-C) or intermediate (G2) grade (B-D) on pathologic evaluation (From Poultsides et al[36]. Pancreatic Neuroendocrine Tumors: Radiographic Calcifications Correlate with Grade and Metastasis. Ann Surg Onc 2012; 19: 2295-2303).
Somatostatin receptor scintigraphy (SRS), also known as an octreotide scan, is a whole body functional imaging study that uses 111indium labeled pentetreotide, a somatostatin analogue. Advantages include identification of unknown metastatic sites and providing important information on functional expression of somatostatin receptors which may guide systemic therapy decisions[41]. Although less available at most institutions compared to SRS, newer functional imaging studies utilizing 68Gallium labeled 1,4,7,10-tetraazacyclododecane-N,N’,N’’,N’’’-tetraacetic acid-d-Phe(1)-Tyr(3)-octreotide ((68)Ga-DOTA-TOC) show promising results that may be superior to conventional SRS[42,43]. Although standard positron emission technology (PET) with 18flourodeoxyglucose is not typically useful in the diagnosis of NF-PNETs, PET with newer radiolabeled tracers may prove more advantageous[44,45]. In general, well differentiated tumors are positive on Octreotide scan and negative on PET scan, with the opposite being true for poorly differentiated grade 3 tumors[46].
SURGICAL MANAGEMENT
Primary
Surgery remains the only curative treatment for NF-PNETs and is the mainstay of treatment in most cases. Appropriate candidates who undergo surgery have a significant survival advantage compared with those who do not. Hill et al[47] demonstrated a median survival difference of 114 mo vs 35 mo for patients who underwent resection compared to those who did not but were recommended to, across all patients with localized, regional and metastatic disease.
The exact surgical management must be individualized for each patient based on their particular tumor and staging. In general, most NF-PNETs should be resected. However, given the increase in incidentally discovered asymptomatic NF-PNETs, there is growing interest in the role of observation for patients with small indolent tumors. Lee et al[48] retrospectively analyzed 77 patients who underwent nonoperative observation of small, sporadic NF-PNETs without evidence of local invasion or metastasis. Median initial size was 1.0 cm and there was no documented disease specific progression or mortality during a median follow-up of 45 mo. In addition, Bettini et al[49] found that of 51 patients with incidentally diagnosed NF-PNETs < 2 cm, only 6% were malignant and there were no disease specific deaths on long term follow-up. Other population-based analyses have attempted to investigate this question but have been limited by methodological concerns[50-52]. Until better data are available, the ENETS guidelines states that intensive observation could be considered for NF-PNETs < 2 cm but risks and benefits must be carefully weighed in each patient[53].
Small low grade PNETs may safely undergo enucleation regardless of location in the pancreas, provided they are far away from the pancreatic duct and the integrity of this structure can be maintained during enucleation[54]. Enucleation may be performed in an open, laparoscopic or robotic fashion; the technique does not have an appreciable impact on morbidity, mortality, length of hospital stay or survival[55]. For larger or more aggressive NF-PNETs, formal resection is recommended. Tumors in the head of the pancreas typically require pancreaticoduodenectomy (PD) while body and tail lesions may be resected via distal pancreatectomy with or without splenic preservation. Distal pancreatectomy can often be performed via minimally invasive techniques which are associated with decreased morbidity, operative blood loss and hospital length of stay with similar rates of negative margins[56]. Minimally invasive PD has been slow to gain popularity given its greater learning curve and longer operating times. However, recent evidence suggests that it is a feasible option at select centers with potential benefits in morbidity and perhaps oncologic outcomes[57,58].
Several reports now have stressed the prognostic importance of lymph node involvement in patients with NF-PNETs[59-61]. Krampitz et al[61] found that positive lymph nodes were associated with a shorter time interval to the development of liver metastases, and in long term follow-up, a shorter disease specific survival. Similarly, Hashim et al[60] found that lymph node positivity was associated with PNETs of greater size, location in the head, high Ki-67 and with lymphovascular invasion. Furthermore, positive lymph nodes were associated with decreased median disease free survival. These data support the use of routine lymphadenectomy during resection for PNETs. Controversy exists over which lesions may forego lymphadenectomy during simple enucleation. Curran et al[59] analyzed the SEER database and found no lymph node metastases in any low grade PNETs < 1 cm. In contrast, Gratian et al[50] found that among tumors < 0.5 cm in the national cancer database, 33% presented with regional lymph node metastases and 11% with distant metastases. Formal resection with adjacent lymphadenectomy, as opposed to enucleation, is the procedure of choice for PNETs greater than 2 cm, of higher grade, or with radiographic calcifications.
Several authors have described the role of aggressive extended resections for advanced PNETs[62-65]. For example, Norton et al[62] describe acceptable morbidity, low mortality, and excellent overall survival rates, albeit high recurrence rates, in patients with advanced PNETs. Norton et al[63] also described good outcomes for patients with PNETs with major vessel involvement undergoing simultaneous vascular reconstruction. Surgeons at experienced hepatopancreaticobiliary centers may follow standard oncologic principles, including multivisceral and vascular resections, in order to accomplish R0 resections.
Liver metastases
All patients with liver metastases from PNETs should be considered for surgical intervention. Although resection can be associated with high recurrence rates, it does improve progression free survival as well as symptom control[66-71]. Saxena et al[66] performed a meta-analysis of 1469 GEP-NETs metastatic to the liver and found 3, 5, and 10 year overall survival rates of 83%, 70.5%, and 42%, respectively, following hepatic resection. Predictors of poor outcomes included poor histologic grade, incomplete resection and extrahepatic disease[66]. When patients are not candidates for resection, alternative methods, such as thermal ablation or hepatic artery embolization, are helpful strategies that improve local control and palliate symptoms[67,69,71,72]. Insufficient data exists to recommend one liver-directed strategy over another[73]. Liver transplantation has been described for well selected patients with metastatic GEP-NET[74]. However, liver transplantation for neuroendocrine liver metastases of pancreatic origin is associated with worse overall outcomes and is not typically recommended[75].
In the setting of metastatic disease, controversy remains regarding the role of surgery for the primary tumor[53,76,77]. Capurso et al[77] performed a systematic review on this topic and found improved overall survival in patients undergoing resection of the primary in 2 of 3 retrospective cohort studies identified. One potential benefit of removal of the primary tumor is allowing providers to focus treatment on the liver metastatic sites with hepatic artery therapies. Primary tumors that are symptomatic should generally undergo resection for palliation of symptoms[78].
SYSTEMIC THERAPY
The goal of systemic therapy is to prolong survival in patients with recurrence or relapse as well as improve quality of life by controlling symptoms. Currently, there is no evidence to support the use of various systemic modalities in an adjuvant fashion following complete surgical resection of PNETs.
Somatostatin analogues
Nearly 80% of NF-NETs express somatostatin receptors, making them a suitable target for therapy with somatostatin analogues. In addition to their favorable safety profile and effectiveness in controlling symptoms, recent evidence has suggested improvements in oncologic outcomes as well. The PROMID trial[79] was a placebo-controlled double blinded randomized controlled trial (RCT) of long acting release (LAR) octreotide in patients with metastatic well differentiated midgut NETs. Median progression free survival was 14.3 mo in patients receiving octreotide vs 6 mo in the placebo group. More recently, the CLARINET trial randomized 204 patients with enteropancreatic NETs to receive long acting lanreotide or placebo and found significantly prolonged progression free survival in the lanreotide group (65.1% vs 33.0% at 24 mo); this finding was confirmed in a subset of patients with PNETs (Figure 3)[80].
Figure 3.
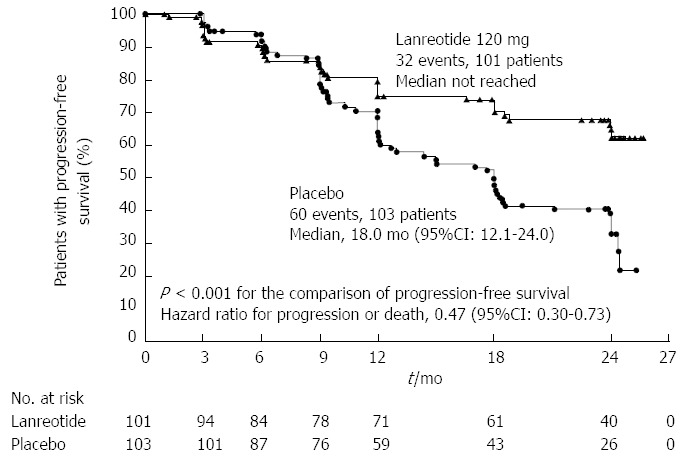
Results of the Clarinet trial which randomized patients with enteropancreatic neuroendocrine tumors to lanreotide vs placebo. From: Caplin et al[80]. Lanreotide in Metastatic Enteropancreatic Neuroendocrine Tumors. N Engl J Med 2014; 371: 224-233.
Radionucleide therapy
Peptide receptor radionuclide therapy (PRRT) also makes use of PNETs’ octreotide receptor expression by coupling radionuclides to somatostatin analogues. Typical radionuclides include 90yttrium and 177lutetium. Response rates range only between 10%-40% with toxicity (primarily bone marrow and renal) rates in a similar range so PRRT should be reserved for cases not responsive to less toxic therapies[81-83]. Furthermore, having been pioneered at the Erasmus Medical Center in the Netherlands, PRRT is still only available at select centers in Europe and North America and randomized data are lacking.
Chemotherapy
Indolent and well differentiated NETs are typically resistant to traditional systemic chemotherapy which is therefore reserved for patients with high grade, poorly differentiated tumors or with rapidly progressive unresectable disease[84]. However, NETS of pancreatic origin, generally respond better to chemotherapy than other GEP-NETs. Streptozocin was one of the first agents shown to have activity patients with metastatic PNETs, either as monotherapy or in combination with doxorubicin or fluorouracil[85-87]. Currently, platinum based therapy remains the standard of care for patients with high grade metastatic PNETs. Various combinations exist but the most common regimen utilized consists of cisplatin and etoposide. Nevertheless, data supporting the use of this regimen is limited and more evidence is needed to clarify its role as aggressive first line therapy[88].
More recent research has focused on the use of oral temozolamide with or without capecitabine given its ease of administration and favorable side effect profile. A retrospective review of 30 patients with well or moderately differentiated PNETs treated with this regimen demonstrated a 70% response rate and a median PFS of 18 mo[89]. Additional research is becoming available regarding the safety and efficacy of this regimen[90,91].
Targeted therapy
Increasingly, PNETs have been found to be responsive to targeted therapies. The purpose of these molecular agents is to stabilize disease progression in metastatic unresectable cases[92]. Much focus has been placed on Everolimus, an oral mTOR (mammalian target of rapamycin) inhibitor. Previously, Everolimus had been found to have clinical benefit in patients who progressed while on systemic cytotoxic chemotherapy[93]. The RADIANT-3 trial was an international, multisite, RCT comparing daily Everolimus to placebo in patients with low or moderate grade NF-PNETs. Although response rates were low, PFS was longer in the Everolimus group (11.0 mo vs 4.6 mo) (Figure 4)[94]. Similarly, the RADIANT-2 trial evaluated Everolimus in conjunction with long acting octreotide and found improved PFS in the Everolimus plus octreotide LAR group vs octreotide LAR alone(16.4 mo vs 11.3 mo)[95]. Common adverse effects included stomatitis, rash and diarrhea. Some have suggested this regimen should be first line therapy for most NETs[96].
Figure 4.
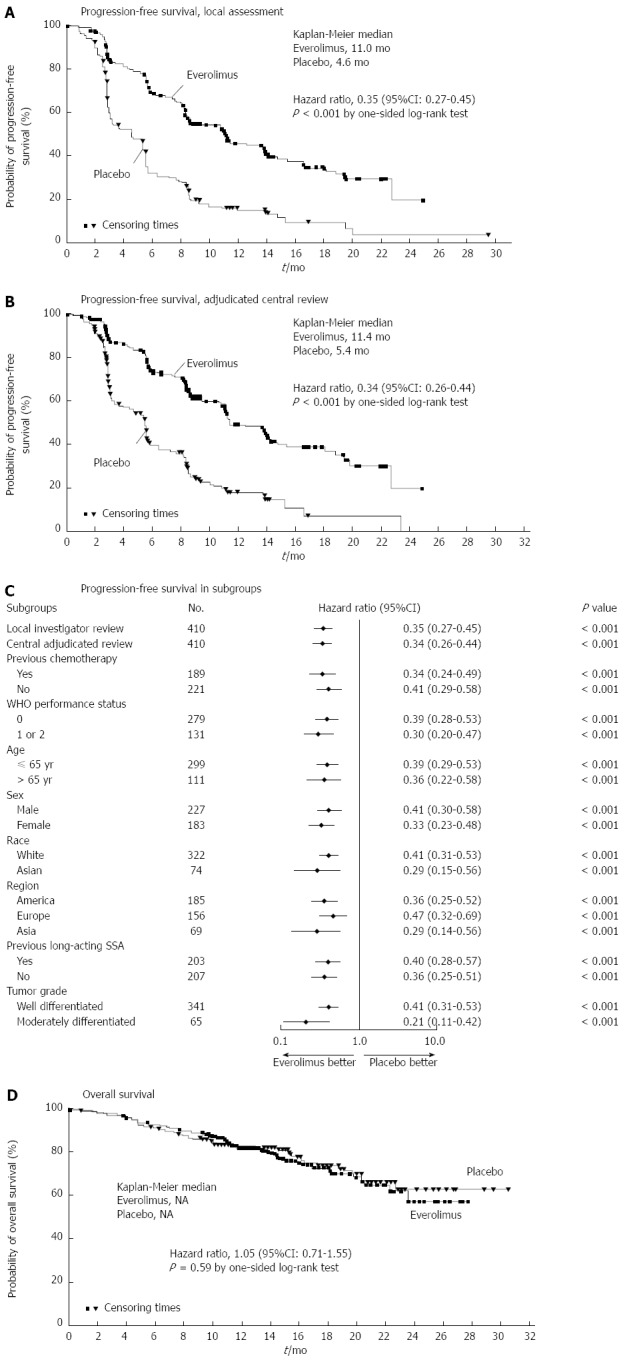
Results of the RADIANT-3 trial which randomized patients with nonfunctional neuroendocrine tumors of the pancreas to Everolimus vs placebo. From Yao et al[94]. Everolimus for Advanced Pancreatic Neuroendocrine Tumors. N Engl J Med 2011; 364: 514-523.
Sunitinib is an oral, small-molecule, tyrosine kinase inhibitor with activity against vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) both of which are expressed abundantly in PNETs[97]. A placebo controlled, double blind, RCT of daily sunitinib in patients with well-differentiated PNETs with documented disease progression found improved PFS in patients receiving Sunitinib (11.4 mo vs 5.5 mo) (Figure 5)[98]. Finally, newer therapies that target the mTOR (e.g., temsirolimus) and VEGF (e.g., bevacizumab) pathways are currently being investigated and hold promise both as single agents and in combination[92,99].
Figure 5.
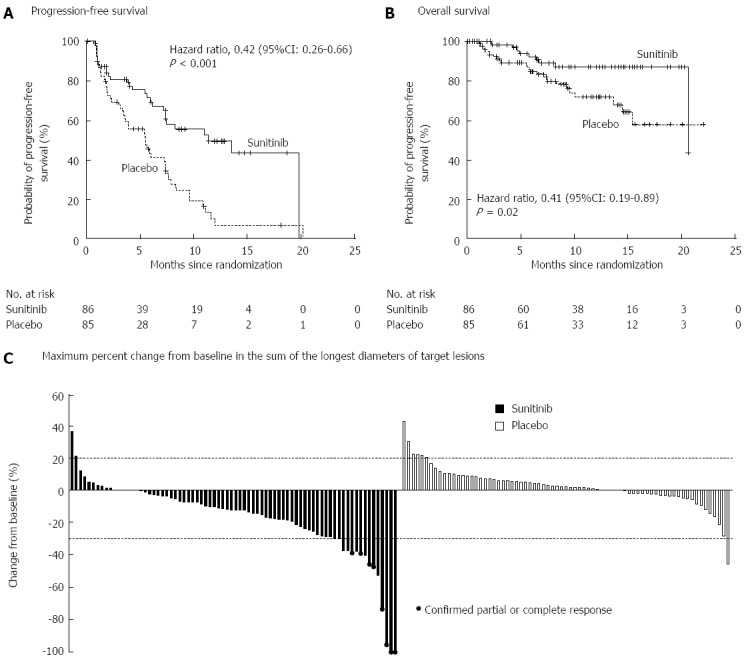
Results of a randomized controlled trial of Sunitinib vs placebo for well-differentiated pancreatic neuroendocrine tumors demonstrating (A) progression free survival and (B) overall survival. From: Raymond et al[98]. Sunitinib Malate for the Treatment of Pancreatic Neuroendocrine Tumors. N Engl J Med 2011; 364: 501-513.
CONCLUSION
Nonfunctional neuroendocrine tumors of the pancreas are a heterogeneous group of neoplasms that are generally slow growing, however, they may become incurable when they progress to unresectable metastatic disease. Tumors of higher grade can be suspected preoperatively based on the presence of calcifications, hypoenhancement on arterial phase computed tomography, PET avidity and lack or octreotide scan uptake. Surgery is the only curative treatment and is recommended for most patients for whom complete resection is possible. Liver-directed therapies (thermal ablation, transarterial embolization) can be useful in controlling unresectable hepatic metastatic disease. In the presence of unresectable progressive disease, level 1 evidence suggests that somatostatin analogues, everolimus and sunitinib can prolong progression-free survival.
Footnotes
Conflict-of-interest statement: The authors have no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 11, 2015
First decision: June 2, 2015
Article in press: July 8, 2015
P- Reviewer: Fujino Y, Sakata N, Sumi S, Teo AKK S- Editor: Ji FF L- Editor: A E- Editor: Zhang DN
References
- 1.Lawrence B, Gustafsson BI, Chan A, Svejda B, Kidd M, Modlin IM. The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am. 2011;40:1–18, vii. doi: 10.1016/j.ecl.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 2.Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol. 2008;19:1727–1733. doi: 10.1093/annonc/mdn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Metz DC, Jensen RT. Gastrointestinal neuroendocrine tumors: pancreatic endocrine tumors. Gastroenterology. 2008;135:1469–1492. doi: 10.1053/j.gastro.2008.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hallet J, Law CH, Cukier M, Saskin R, Liu N, Singh S. Exploring the rising incidence of neuroendocrine tumors: a population-based analysis of epidemiology, metastatic presentation, and outcomes. Cancer. 2015;121:589–597. doi: 10.1002/cncr.29099. [DOI] [PubMed] [Google Scholar]
- 5.Kimura W, Kuroda A, Morioka Y. Clinical pathology of endocrine tumors of the pancreas. Analysis of autopsy cases. Dig Dis Sci. 1991;36:933–942. doi: 10.1007/BF01297144. [DOI] [PubMed] [Google Scholar]
- 6.Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, Abdalla EK, Fleming JB, Vauthey JN, Rashid A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063–3072. doi: 10.1200/JCO.2007.15.4377. [DOI] [PubMed] [Google Scholar]
- 7.Solcia E, Kloppel G, Alhman H. Histological Typing of Endocrine Tumors: WHO International Histological Classification of Tumors. 2nd ed. Berlin: Springer; 2000. [Google Scholar]
- 8.Rindi G, Arnold R, Bosman F. Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In: WHO Classification of Tumours of the Digestive System., editor. 4th ed. Lyon: IARC Press; 2010. [Google Scholar]
- 9.Martin-Perez E, Capdevila J, Castellano D, Jimenez-Fonseca P, Salazar R, Beguiristain-Gomez A, Alonso-Orduña V, Martinez Del Prado P, Villabona-Artero C, Diaz-Perez JA, et al. Prognostic factors and long-term outcome of pancreatic neuroendocrine neoplasms: Ki-67 index shows a greater impact on survival than disease stage. The large experience of the Spanish National Tumor Registry (RGETNE) Neuroendocrinology. 2013;98:156–168. doi: 10.1159/000355152. [DOI] [PubMed] [Google Scholar]
- 10.Rindi G, Klöppel G, Alhman H, Caplin M, Couvelard A, de Herder WW, Erikssson B, Falchetti A, Falconi M, Komminoth P, et al. TNM staging of foregut (neuro)endocrine tumors: a consensus proposal including a grading system. Virchows Arch. 2006;449:395–401. doi: 10.1007/s00428-006-0250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edge S, Byrd D, Compton C. AJCC Cancer Staging Manual. 7th ed. New York: Springer; 2010. [Google Scholar]
- 12.Ellison TA, Wolfgang CL, Shi C, Cameron JL, Murakami P, Mun LJ, Singhi AD, Cornish TC, Olino K, Meriden Z, et al. A single institution’s 26-year experience with nonfunctional pancreatic neuroendocrine tumors: a validation of current staging systems and a new prognostic nomogram. Ann Surg. 2014;259:204–212. doi: 10.1097/SLA.0b013e31828f3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strosberg JR, Cheema A, Weber J, Han G, Coppola D, Kvols LK. Prognostic validity of a novel American Joint Committee on Cancer Staging Classification for pancreatic neuroendocrine tumors. J Clin Oncol. 2011;29:3044–3049. doi: 10.1200/JCO.2011.35.1817. [DOI] [PubMed] [Google Scholar]
- 14.Strosberg JR, Cheema A, Weber JM, Ghayouri M, Han G, Hodul PJ, Kvols LK. Relapse-free survival in patients with nonmetastatic, surgically resected pancreatic neuroendocrine tumors: an analysis of the AJCC and ENETS staging classifications. Ann Surg. 2012;256:321–325. doi: 10.1097/SLA.0b013e31824e6108. [DOI] [PubMed] [Google Scholar]
- 15.Rindi G, Falconi M, Klersy C, Albarello L, Boninsegna L, Buchler MW, Capella C, Caplin M, Couvelard A, Doglioni C, et al. TNM staging of neoplasms of the endocrine pancreas: results from a large international cohort study. J Natl Cancer Inst. 2012;104:764–777. doi: 10.1093/jnci/djs208. [DOI] [PubMed] [Google Scholar]
- 16.Qadan M, Ma Y, Visser BC, Kunz PL, Fisher GA, Norton JA, Poultsides GA. Reassessment of the current American Joint Committee on Cancer staging system for pancreatic neuroendocrine tumors. J Am Coll Surg. 2014;218:188–195. doi: 10.1016/j.jamcollsurg.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Zerbi A, Falconi M, Rindi G, Delle Fave G, Tomassetti P, Pasquali C, Capitanio V, Boninsegna L, Di Carlo V. Clinicopathological features of pancreatic endocrine tumors: a prospective multicenter study in Italy of 297 sporadic cases. Am J Gastroenterol. 2010;105:1421–1429. doi: 10.1038/ajg.2009.747. [DOI] [PubMed] [Google Scholar]
- 18.Kuo JH, Lee JA, Chabot JA. Nonfunctional pancreatic neuroendocrine tumors. Surg Clin North Am. 2014;94:689–708. doi: 10.1016/j.suc.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 19.Krampitz GW, Norton JA. Pancreatic neuroendocrine tumors. Curr Probl Surg. 2013;50:509–545. doi: 10.1067/j.cpsurg.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 20.Jensen RT, Berna MJ, Bingham DB, Norton JA. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer. 2008;113:1807–1843. doi: 10.1002/cncr.23648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mao C, Shah A, Hanson DJ, Howard JM. Von Recklinghausen’s disease associated with duodenal somatostatinoma: contrast of duodenal versus pancreatic somatostatinomas. J Surg Oncol. 1995;59:67–73. doi: 10.1002/jso.2930590116. [DOI] [PubMed] [Google Scholar]
- 22.Lee WS, Koh YS, Kim JC, Park CH, Joo YE, Kim HS, Cho CK, Choi SK, Rew JS, Kim SJ. Zollinger-Ellison syndrome associated with neurofibromatosis type 1: a case report. BMC Cancer. 2005;5:85. doi: 10.1186/1471-2407-5-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perren A, Wiesli P, Schmid S, Montani M, Schmitt A, Schmid C, Moch H, Komminoth P. Pancreatic endocrine tumors are a rare manifestation of the neurofibromatosis type 1 phenotype: molecular analysis of a malignant insulinoma in a NF-1 patient. Am J Surg Pathol. 2006;30:1047–1051. doi: 10.1097/00000478-200608000-00018. [DOI] [PubMed] [Google Scholar]
- 24.Fujisawa T, Osuga T, Maeda M, Sakamoto N, Maeda T, Sakaguchi K, Onishi Y, Toyoda M, Maeda H, Miyamoto K, et al. Malignant endocrine tumor of the pancreas associated with von Recklinghausen’s disease. J Gastroenterol. 2002;37:59–67. doi: 10.1007/s535-002-8135-x. [DOI] [PubMed] [Google Scholar]
- 25.Dworakowska D, Grossman AB. Are neuroendocrine tumours a feature of tuberous sclerosis? A systematic review. Endocr Relat Cancer. 2009;16:45–58. doi: 10.1677/ERC-08-0142. [DOI] [PubMed] [Google Scholar]
- 26.Modlin IM, Gustafsson BI, Moss SF, Pavel M, Tsolakis AV, Kidd M. Chromogranin A--biological function and clinical utility in neuro endocrine tumor disease. Ann Surg Oncol. 2010;17:2427–2443. doi: 10.1245/s10434-010-1006-3. [DOI] [PubMed] [Google Scholar]
- 27.Campana D, Nori F, Piscitelli L, Morselli-Labate AM, Pezzilli R, Corinaldesi R, Tomassetti P. Chromogranin A: is it a useful marker of neuroendocrine tumors? J Clin Oncol. 2007;25:1967–1973. doi: 10.1200/JCO.2006.10.1535. [DOI] [PubMed] [Google Scholar]
- 28.Giusti M, Sidoti M, Augeri C, Rabitti C, Minuto F. Effect of short-term treatment with low dosages of the proton-pump inhibitor omeprazole on serum chromogranin A levels in man. Eur J Endocrinol. 2004;150:299–303. doi: 10.1530/eje.0.1500299. [DOI] [PubMed] [Google Scholar]
- 29.Singh S, Dey C, Kennecke H, Kocha W, Maroun J, Metrakos P, Mukhtar T, Pasieka J, Rayson D, Rowsell C, et al. Consensus Recommendations for the Diagnosis and Management of Pancreatic Neuroendocrine Tumors: Guidelines from a Canadian National Expert Group. Ann Surg Oncol. 2015;22:2685–2699. doi: 10.1245/s10434-014-4145-0. [DOI] [PubMed] [Google Scholar]
- 30.Kunz PL, Reidy-Lagunes D, Anthony LB, Bertino EM, Brendtro K, Chan JA, Chen H, Jensen RT, Kim MK, Klimstra DS, et al. Consensus guidelines for the management and treatment of neuroendocrine tumors. Pancreas. 2013;42:557–577. doi: 10.1097/MPA.0b013e31828e34a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panzuto F, Severi C, Cannizzaro R, Falconi M, Angeletti S, Pasquali A, Corleto VD, Annibale B, Buonadonna A, Pederzoli P, et al. Utility of combined use of plasma levels of chromogranin A and pancreatic polypeptide in the diagnosis of gastrointestinal and pancreatic endocrine tumors. J Endocrinol Invest. 2004;27:6–11. doi: 10.1007/BF03350903. [DOI] [PubMed] [Google Scholar]
- 32.Walter T, Chardon L, Chopin-laly X, Raverot V, Caffin AG, Chayvialle JA, Scoazec JY, Lombard-Bohas C. Is the combination of chromogranin A and pancreatic polypeptide serum determinations of interest in the diagnosis and follow-up of gastro-entero-pancreatic neuroendocrine tumours? Eur J Cancer. 2012;48:1766–1773. doi: 10.1016/j.ejca.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 33.Raines D, Chester M, Diebold AE, Mamikunian P, Anthony CT, Mamikunian G, Woltering EA. A prospective evaluation of the effect of chronic proton pump inhibitor use on plasma biomarker levels in humans. Pancreas. 2012;41:508–511. doi: 10.1097/MPA.0b013e318243a0b6. [DOI] [PubMed] [Google Scholar]
- 34.Rustagi S, Warner RR, Divino CM. Serum pancreastatin: the next predictive neuroendocrine tumor marker. J Surg Oncol. 2013;108:126–128. doi: 10.1002/jso.23359. [DOI] [PubMed] [Google Scholar]
- 35.Worhunsky DJ, Krampitz GW, Poullos PD, Visser BC, Kunz PL, Fisher GA, Norton JA, Poultsides GA. Pancreatic neuroendocrine tumours: hypoenhancement on arterial phase computed tomography predicts biological aggressiveness. HPB (Oxford) 2014;16:304–311. doi: 10.1111/hpb.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poultsides GA, Huang LC, Chen Y, Visser BC, Pai RK, Jeffrey RB, Park WG, Chen AM, Kunz PL, Fisher GA, et al. Pancreatic neuroendocrine tumors: radiographic calcifications correlate with grade and metastasis. Ann Surg Oncol. 2012;19:2295–2303. doi: 10.1245/s10434-012-2305-7. [DOI] [PubMed] [Google Scholar]
- 37.Sundin A, Vullierme MP, Kaltsas G, Plöckinger U. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: radiological examinations. Neuroendocrinology. 2009;90:167–183. doi: 10.1159/000184855. [DOI] [PubMed] [Google Scholar]
- 38.Dromain C, de Baere T, Lumbroso J, Caillet H, Laplanche A, Boige V, Ducreux M, Duvillard P, Elias D, Schlumberger M, et al. Detection of liver metastases from endocrine tumors: a prospective comparison of somatostatin receptor scintigraphy, computed tomography, and magnetic resonance imaging. J Clin Oncol. 2005;23:70–78. doi: 10.1200/JCO.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 39.Hiramoto JS, Feldstein VA, LaBerge JM, Norton JA. Intraoperative ultrasound and preoperative localization detects all occult insulinomas; discussion 1025-6. Arch Surg. 2001;136:1020–1025. doi: 10.1001/archsurg.136.9.1020. [DOI] [PubMed] [Google Scholar]
- 40.Rösch T, Lightdale CJ, Botet JF, Boyce GA, Sivak MV, Yasuda K, Heyder N, Palazzo L, Dancygier H, Schusdziarra V. Localization of pancreatic endocrine tumors by endoscopic ultrasonography. N Engl J Med. 1992;326:1721–1726. doi: 10.1056/NEJM199206253262601. [DOI] [PubMed] [Google Scholar]
- 41.Schillaci O, Spanu A, Scopinaro F, Falchi A, Corleto V, Danieli R, Marongiu P, Pisu N, Madeddu G, Delle Fave G, et al. Somatostatin receptor scintigraphy with 111In-pentetreotide in non-functioning gastroenteropancreatic neuroendocrine tumors. Int J Oncol. 2003;23:1687–1695. [PubMed] [Google Scholar]
- 42.Hofman MS, Kong G, Neels OC, Eu P, Hong E, Hicks RJ. High management impact of Ga-68 DOTATATE (GaTate) PET/CT for imaging neuroendocrine and other somatostatin expressing tumours. J Med Imaging Radiat Oncol. 2012;56:40–47. doi: 10.1111/j.1754-9485.2011.02327.x. [DOI] [PubMed] [Google Scholar]
- 43.Gabriel M, Decristoforo C, Kendler D, Dobrozemsky G, Heute D, Uprimny C, Kovacs P, Von Guggenberg E, Bale R, Virgolini IJ. 68Ga-DOTA-Tyr3-octreotide PET in neuroendocrine tumors: comparison with somatostatin receptor scintigraphy and CT. J Nucl Med. 2007;48:508–518. doi: 10.2967/jnumed.106.035667. [DOI] [PubMed] [Google Scholar]
- 44.Koopmans KP, Neels OC, Kema IP, Elsinga PH, Sluiter WJ, Vanghillewe K, Brouwers AH, Jager PL, de Vries EG. Improved staging of patients with carcinoid and islet cell tumors with 18F-dihydroxy-phenyl-alanine and 11C-5-hydroxy-tryptophan positron emission tomography. J Clin Oncol. 2008;26:1489–1495. doi: 10.1200/JCO.2007.15.1126. [DOI] [PubMed] [Google Scholar]
- 45.Koopmans KP, de Vries EG, Kema IP, Elsinga PH, Neels OC, Sluiter WJ, van der Horst-Schrivers AN, Jager PL. Staging of carcinoid tumours with 18F-DOPA PET: a prospective, diagnostic accuracy study. Lancet Oncol. 2006;7:728–734. doi: 10.1016/S1470-2045(06)70801-4. [DOI] [PubMed] [Google Scholar]
- 46.Squires MH, Volkan Adsay N, Schuster DM, Russell MC, Cardona K, Delman KA, Winer JH, Altinel D, Sarmiento JM, El-Rayes B, et al. Octreoscan Versus FDG-PET for Neuroendocrine Tumor Staging: A Biological Approach. Ann Surg Oncol. 2015;22:2295–2301. doi: 10.1245/s10434-015-4471-x. [DOI] [PubMed] [Google Scholar]
- 47.Hill JS, McPhee JT, McDade TP, Zhou Z, Sullivan ME, Whalen GF, Tseng JF. Pancreatic neuroendocrine tumors: the impact of surgical resection on survival. Cancer. 2009;115:741–751. doi: 10.1002/cncr.24065. [DOI] [PubMed] [Google Scholar]
- 48.Lee LC, Grant CS, Salomao DR, Fletcher JG, Takahashi N, Fidler JL, Levy MJ, Huebner M. Small, nonfunctioning, asymptomatic pancreatic neuroendocrine tumors (PNETs): role for nonoperative management. Surgery. 2012;152:965–974. doi: 10.1016/j.surg.2012.08.038. [DOI] [PubMed] [Google Scholar]
- 49.Bettini R, Partelli S, Boninsegna L, Capelli P, Crippa S, Pederzoli P, Scarpa A, Falconi M. Tumor size correlates with malignancy in nonfunctioning pancreatic endocrine tumor. Surgery. 2011;150:75–82. doi: 10.1016/j.surg.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 50.Gratian L, Pura J, Dinan M, Roman S, Reed S, Sosa JA. Impact of extent of surgery on survival in patients with small nonfunctional pancreatic neuroendocrine tumors in the United States. Ann Surg Oncol. 2014;21:3515–3521. doi: 10.1245/s10434-014-3769-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharpe SM, In H, Winchester DJ, Talamonti MS, Baker MS. Surgical resection provides an overall survival benefit for patients with small pancreatic neuroendocrine tumors. J Gastrointest Surg. 2015;19:117–23; discussion 123. doi: 10.1007/s11605-014-2615-0. [DOI] [PubMed] [Google Scholar]
- 52.Kuo EJ, Salem RR. Population-level analysis of pancreatic neuroendocrine tumors 2 cm or less in size. Ann Surg Oncol. 2013;20:2815–2821. doi: 10.1245/s10434-013-3005-7. [DOI] [PubMed] [Google Scholar]
- 53.Falconi M, Bartsch DK, Eriksson B, Klöppel G, Lopes JM, O’Connor JM, Salazar R, Taal BG, Vullierme MP, O’Toole D. ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms of the digestive system: well-differentiated pancreatic non-functioning tumors. Neuroendocrinology. 2012;95:120–134. doi: 10.1159/000335587. [DOI] [PubMed] [Google Scholar]
- 54.Crippa S, Bassi C, Salvia R, Falconi M, Butturini G, Pederzoli P. Enucleation of pancreatic neoplasms. Br J Surg. 2007;94:1254–1259. doi: 10.1002/bjs.5833. [DOI] [PubMed] [Google Scholar]
- 55.Pitt SC, Pitt HA, Baker MS, Christians K, Touzios JG, Kiely JM, Weber SM, Wilson SD, Howard TJ, Talamonti MS, et al. Small pancreatic and periampullary neuroendocrine tumors: resect or enucleate? J Gastrointest Surg. 2009;13:1692–1698. doi: 10.1007/s11605-009-0946-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Venkat R, Edil BH, Schulick RD, Lidor AO, Makary MA, Wolfgang CL. Laparoscopic distal pancreatectomy is associated with significantly less overall morbidity compared to the open technique: a systematic review and meta-analysis. Ann Surg. 2012;255:1048–1059. doi: 10.1097/SLA.0b013e318251ee09. [DOI] [PubMed] [Google Scholar]
- 57.Correa-Gallego C, Dinkelspiel HE, Sulimanoff I, Fisher S, Viñuela EF, Kingham TP, Fong Y, DeMatteo RP, D’Angelica MI, Jarnagin WR, et al. Minimally-invasive vs open pancreaticoduodenectomy: systematic review and meta-analysis. J Am Coll Surg. 2014;218:129–139. doi: 10.1016/j.jamcollsurg.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 58.Croome KP, Farnell MB, Que FG, Reid-Lombardo KM, Truty MJ, Nagorney DM, Kendrick ML. Total laparoscopic pancreaticoduodenectomy for pancreatic ductal adenocarcinoma: oncologic advantages over open approaches? Ann Surg. 2014;260:633–68; discussion 633-68;. doi: 10.1097/SLA.0000000000000937. [DOI] [PubMed] [Google Scholar]
- 59.Curran T, Pockaj BA, Gray RJ, Halfdanarson TR, Wasif N. Importance of lymph node involvement in pancreatic neuroendocrine tumors: impact on survival and implications for surgical resection. J Gastrointest Surg. 2015;19:152–60; discussion 160. doi: 10.1007/s11605-014-2624-z. [DOI] [PubMed] [Google Scholar]
- 60.Hashim YM, Trinkaus KM, Linehan DC, Strasberg SS, Fields RC, Cao D, Hawkins WG. Regional lymphadenectomy is indicated in the surgical treatment of pancreatic neuroendocrine tumors (PNETs) Ann Surg. 2014;259:197–203. doi: 10.1097/SLA.0000000000000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krampitz GW, Norton JA, Poultsides GA, Visser BC, Sun L, Jensen RT. Lymph nodes and survival in pancreatic neuroendocrine tumors. Arch Surg. 2012;147:820–827. doi: 10.1001/archsurg.2012.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Norton JA, Kivlen M, Li M, Schneider D, Chuter T, Jensen RT. Morbidity and mortality of aggressive resection in patients with advanced neuroendocrine tumors. Arch Surg. 2003;138:859–866. doi: 10.1001/archsurg.138.8.859. [DOI] [PubMed] [Google Scholar]
- 63.Norton JA, Harris EJ, Chen Y, Visser BC, Poultsides GA, Kunz PC, Fisher GA, Jensen RT. Pancreatic endocrine tumors with major vascular abutment, involvement, or encasement and indication for resection. Arch Surg. 2011;146:724–732. doi: 10.1001/archsurg.2011.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Teh SH, Deveney C, Sheppard BC. Aggressive pancreatic resection for primary pancreatic neuroendocrine tumor: is it justifiable? Am J Surg. 2007;193:610–63; discussion 613. doi: 10.1016/j.amjsurg.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 65.Abu Hilal M, McPhail MJ, Zeidan BA, Jones CE, Johnson CD, Pearce NW. Aggressive multi-visceral pancreatic resections for locally advanced neuroendocrine tumours. Is it worth it? JOP. 2009;10:276–279. [PubMed] [Google Scholar]
- 66.Saxena A, Chua TC, Perera M, Chu F, Morris DL. Surgical resection of hepatic metastases from neuroendocrine neoplasms: a systematic review. Surg Oncol. 2012;21:e131–e141. doi: 10.1016/j.suronc.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 67.Chamberlain RS, Canes D, Brown KT, Saltz L, Jarnagin W, Fong Y, Blumgart LH. Hepatic neuroendocrine metastases: does intervention alter outcomes? J Am Coll Surg. 2000;190:432–445. doi: 10.1016/s1072-7515(00)00222-2. [DOI] [PubMed] [Google Scholar]
- 68.Cusati D, Zhang L, Harmsen WS, Hu A, Farnell MB, Nagorney DM, Donohue JH, Que FG, Reid-Lombardo KM, Kendrick ML. Metastatic nonfunctioning pancreatic neuroendocrine carcinoma to liver: surgical treatment and outcomes. J Am Coll Surg. 2012;215:117–24; discussion 124-5. doi: 10.1016/j.jamcollsurg.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 69.Musunuru S, Chen H, Rajpal S, Stephani N, McDermott JC, Holen K, Rikkers LF, Weber SM. Metastatic neuroendocrine hepatic tumors: resection improves survival. Arch Surg. 2006;141:1000–104; discussion 1005. doi: 10.1001/archsurg.141.10.1000. [DOI] [PubMed] [Google Scholar]
- 70.Birnbaum DJ, Turrini O, Vigano L, Russolillo N, Autret A, Moutardier V, Capussotti L, Le Treut YP, Delpero JR, Hardwigsen J. Surgical management of advanced pancreatic neuroendocrine tumors: short-term and long-term results from an international multi-institutional study. Ann Surg Oncol. 2015;22:1000–1007. doi: 10.1245/s10434-014-4016-8. [DOI] [PubMed] [Google Scholar]
- 71.Touzios JG, Kiely JM, Pitt SC, Rilling WS, Quebbeman EJ, Wilson SD, Pitt HA. Neuroendocrine hepatic metastases: does aggressive management improve survival? Ann Surg. 2005;241:776–83; discussion 783-5. doi: 10.1097/01.sla.0000161981.58631.ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mayo SC, de Jong MC, Bloomston M, Pulitano C, Clary BM, Reddy SK, Clark Gamblin T, Celinski SA, Kooby DA, Staley CA, et al. Surgery versus intra-arterial therapy for neuroendocrine liver metastasis: a multicenter international analysis. Ann Surg Oncol. 2011;18:3657–3665. doi: 10.1245/s10434-011-1832-y. [DOI] [PubMed] [Google Scholar]
- 73.Lesurtel M, Nagorney DM, Mazzaferro V, Jensen RT, Poston GJ. When should a liver resection be performed in patients with liver metastases from neuroendocrine tumours? A systematic review with practice recommendations. HPB (Oxford) 2015;17:17–22. doi: 10.1111/hpb.12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Le Treut YP, Grégoire E, Belghiti J, Boillot O, Soubrane O, Mantion G, Cherqui D, Castaing D, Ruszniewski P, Wolf P, et al. Predictors of long-term survival after liver transplantation for metastatic endocrine tumors: an 85-case French multicentric report. Am J Transplant. 2008;8:1205–1213. doi: 10.1111/j.1600-6143.2008.02233.x. [DOI] [PubMed] [Google Scholar]
- 75.Gregoire E, Le Treut YP. Liver transplantation for primary or secondary endocrine tumors. Transpl Int. 2010;23:704–711. doi: 10.1111/j.1432-2277.2010.01110.x. [DOI] [PubMed] [Google Scholar]
- 76.Bettini R, Mantovani W, Boninsegna L, Crippa S, Capelli P, Bassi C, Scarpa A, Pederzoli P, Falconi M. Primary tumour resection in metastatic nonfunctioning pancreatic endocrine carcinomas. Dig Liver Dis. 2009;41:49–55. doi: 10.1016/j.dld.2008.03.015. [DOI] [PubMed] [Google Scholar]
- 77.Capurso G, Bettini R, Rinzivillo M, Boninsegna L, Delle Fave G, Falconi M. Role of resection of the primary pancreatic neuroendocrine tumour only in patients with unresectable metastatic liver disease: a systematic review. Neuroendocrinology. 2011;93:223–229. doi: 10.1159/000324770. [DOI] [PubMed] [Google Scholar]
- 78.Hung JS, Chang MC, Lee PH, Tien YW. Is surgery indicated for patients with symptomatic nonfunctioning pancreatic neuroendocrine tumor and unresectable hepatic metastases? World J Surg. 2007;31:2392–2397. doi: 10.1007/s00268-007-9264-3. [DOI] [PubMed] [Google Scholar]
- 79.Rinke A, Müller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, Mayer C, Aminossadati B, Pape UF, Bläker M, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27:4656–4663. doi: 10.1200/JCO.2009.22.8510. [DOI] [PubMed] [Google Scholar]
- 80.Caplin ME, Pavel M, Ćwikła JB, Phan AT, Raderer M, Sedláčková E, Cadiot G, Wolin EM, Capdevila J, Wall L, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371:224–233. doi: 10.1056/NEJMoa1316158. [DOI] [PubMed] [Google Scholar]
- 81.Kwekkeboom DJ, de Herder WW, Kam BL, van Eijck CH, van Essen M, Kooij PP, Feelders RA, van Aken MO, Krenning EP. Treatment with the radiolabeled somatostatin analog [177 Lu-DOTA 0,Tyr3]octreotate: toxicity, efficacy, and survival. J Clin Oncol. 2008;26:2124–2130. doi: 10.1200/JCO.2007.15.2553. [DOI] [PubMed] [Google Scholar]
- 82.Kwekkeboom DJ, Krenning EP, Lebtahi R, Komminoth P, Kos-Kudła B, de Herder WW, Plöckinger U. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: peptide receptor radionuclide therapy with radiolabeled somatostatin analogs. Neuroendocrinology. 2009;90:220–226. doi: 10.1159/000225951. [DOI] [PubMed] [Google Scholar]
- 83.Villard L, Romer A, Marincek N, Brunner P, Koller MT, Schindler C, Ng QK, Mäcke HR, Müller-Brand J, Rochlitz C, et al. Cohort study of somatostatin-based radiopeptide therapy with [(90)Y-DOTA]-TOC versus [(90)Y-DOTA]-TOC plus [(177)Lu-DOTA]-TOC in neuroendocrine cancers. J Clin Oncol. 2012;30:1100–1106. doi: 10.1200/JCO.2011.37.2151. [DOI] [PubMed] [Google Scholar]
- 84.Strosberg JR. Systemic treatment of gastroenteropancreatic neuroendocrine tumors (GEP-NETS): current approaches and future options. Endocr Pract. 2014;20:167–175. doi: 10.4158/EP13262.RA. [DOI] [PubMed] [Google Scholar]
- 85.Moertel CG, Hanley JA, Johnson LA. Streptozocin alone compared with streptozocin plus fluorouracil in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1980;303:1189–1194. doi: 10.1056/NEJM198011203032101. [DOI] [PubMed] [Google Scholar]
- 86.Moertel CG, Lefkopoulo M, Lipsitz S, Hahn RG, Klaassen D. Streptozocin-doxorubicin, streptozocin-fluorouracil or chlorozotocin in the treatment of advanced islet-cell carcinoma. N Engl J Med. 1992;326:519–523. doi: 10.1056/NEJM199202203260804. [DOI] [PubMed] [Google Scholar]
- 87.Kouvaraki MA, Ajani JA, Hoff P, Wolff R, Evans DB, Lozano R, Yao JC. Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J Clin Oncol. 2004;22:4762–4771. doi: 10.1200/JCO.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 88.Fazio N, Spada F, Giovannini M. Chemotherapy in gastroenteropancreatic (GEP) neuroendocrine carcinomas (NEC): a critical view. Cancer Treat Rev. 2013;39:270–274. doi: 10.1016/j.ctrv.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 89.Strosberg JR, Fine RL, Choi J, Nasir A, Coppola D, Chen DT, Helm J, Kvols L. First-line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinomas. Cancer. 2011;117:268–275. doi: 10.1002/cncr.25425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fine RL, Gulati AP, Krantz BA, Moss RA, Schreibman S, Tsushima DA, Mowatt KB, Dinnen RD, Mao Y, Stevens PD, et al. Capecitabine and temozolomide (CAPTEM) for metastatic, well-differentiated neuroendocrine cancers: The Pancreas Center at Columbia University experience. Cancer Chemother Pharmacol. 2013;71:663–670. doi: 10.1007/s00280-012-2055-z. [DOI] [PubMed] [Google Scholar]
- 91.Tejani MA, Saif MW. Pancreatic neuroendocrine tumors: does chemotherapy work? JOP. 2014;15:132–134. doi: 10.6092/1590-8577/2301. [DOI] [PubMed] [Google Scholar]
- 92.Khagi S, Saif MW. Pancreatic neuroendocrine tumors: targeting the molecular basis of disease. Curr Opin Oncol. 2015;27:38–43. doi: 10.1097/CCO.0000000000000146. [DOI] [PubMed] [Google Scholar]
- 93.Yao JC, Lombard-Bohas C, Baudin E, Kvols LK, Rougier P, Ruszniewski P, Hoosen S, St Peter J, Haas T, Lebwohl D, et al. Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: a phase II trial. J Clin Oncol. 2010;28:69–76. doi: 10.1200/JCO.2009.24.2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514–523. doi: 10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pavel ME, Hainsworth JD, Baudin E, Peeters M, Hörsch D, Winkler RE, Klimovsky J, Lebwohl D, Jehl V, Wolin EM, et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase 3 study. Lancet. 2011;378:2005–2012. doi: 10.1016/S0140-6736(11)61742-X. [DOI] [PubMed] [Google Scholar]
- 96.Bajetta E, Catena L, Fazio N, Pusceddu S, Biondani P, Blanco G, Ricci S, Aieta M, Pucci F, Valente M, et al. Everolimus in combination with octreotide long-acting repeatable in a first-line setting for patients with neuroendocrine tumors: an ITMO group study. Cancer. 2014;120:2457–2463. doi: 10.1002/cncr.28726. [DOI] [PubMed] [Google Scholar]
- 97.Faivre S, Demetri G, Sargent W, Raymond E. Molecular basis for sunitinib efficacy and future clinical development. Nat Rev Drug Discov. 2007;6:734–745. doi: 10.1038/nrd2380. [DOI] [PubMed] [Google Scholar]
- 98.Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, Valle J, Metrakos P, Smith D, Vinik A, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501–513. doi: 10.1056/NEJMoa1003825. [DOI] [PubMed] [Google Scholar]
- 99.Hobday TJ, Qin R, Reidy-Lagunes D, Moore MJ, Strosberg J, Kaubisch A, Shah M, Kindler HL, Lenz HJ, Chen H, et al. Multicenter Phase II Trial of Temsirolimus and Bevacizumab in Pancreatic Neuroendocrine Tumors. J Clin Oncol. 2015;33:1551–1556. doi: 10.1200/JCO.2014.56.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]