

Abstract
AIM: To investigate the effect of carvedilol on angiogenesis and the underlying signaling pathways.
METHODS: The effect of carvedilol on angiogenesis was examined using a human umbilical vascular endothelial cell (HUVEC) model. The effect of carvedilol on cell viability was measured by CCK8 assay. Flow cytometry was used to assess the effect of carvedilol on cell cycle progression. Cell migration, transwell migration and tube formation assays were performed to analyze the effect of carvedilol on HUVEC function. Vascular endothelial growth factor (VEGF) induced activation of HUVECs, which were pretreated with different carvedilol concentrations or none. Western blot analysis detected the phosphorylation levels of three cell signaling pathway proteins, VEGFR-2, Src, and extracellular signal-regulated kinase (ERK). The specific Src inhibitor PP2 was used to assess the role of Src in the VEGF-induced angiogenic pathway.
RESULTS: Carvedilol inhibited HUVEC proliferation in a dose-dependent manner (IC50 = 38.5 mmol/L). The distribution of cells in the S phase decreased from 43.6% to 37.2%, 35.6% and 17.8% by 1, 5 and 10 μmol/L carvedilol for 24 h, respectively. Carvedilol (10 μmol/L) reduced VEGF-induced HUVEC migration from 67.54 ± 7.83 to 37.11 ± 3.533 (P < 0.001). Carvedilol concentrations of 5 μmol/L and 10 μmol/L reduced cell invasion from 196.3% ± 18.76% to 114.0% ± 12.20% and 51.68% ± 8.28%, respectively. VEGF-induced tube formation was also reduced significantly by 5 μmol/L and 10 μmol/L carvedilol from 286.0 ± 36.72 to 135.7 ± 18.13 (P < 0.05) and 80.27 ± 11.16 (P < 0.01) respectively. We investigated several intracellular protein levels to determine the reason for these reductions. Treatment with 10 μmol/L carvedilol reduced VEGF-induced tyrosine phosphorylation of VEGFR-2 from 175.5% ± 8.54% to 52.67% ± 5.33% (P < 0.01). Additionally, 10 μmol/L carvedilol reduced VEGF-induced ERK 1/2 phosphorylation from 181.9% ± 18.61% to 56.45% ± 7.64% (P < 0.01). The VEGF-induced increase in Src kinase activity was alleviated by carvedilol [decreased from 141.8% ± 15.37% to 53.57 ± 7.18% (P < 0.01) and 47.04% ± 9.74% (P < 0.01) at concentrations of 5 and 10 μmol/L, respectively]. Pretreatment of HUVECs with Src kinase inhibitor almost completely prevented the VEGF-induced ERK upregulation [decreased from 213.2% ± 27.68% to 90.96% ± 17.16% (P < 0.01)].
CONCLUSION: Carvedilol has an anti-angiogenic effect on HUVECs. This inhibitory effect is mediated by VEGF-induced Src-ERK signaling pathways.
Keywords: Carvedilol, Adrenergic β-antagonists, Angiogenesis, Liver cirrhosis, Drug utilization
Core tip: Carvedilol has been used for the treatment of portal hypertension for many years. In this study, carvedilol directly inhibited the proliferation and tube formation of cultured human umbilical vascular endothelial cells. Moreover, this study is the first to investigate the mechanism of the anti-angiogenic effect of carvedilol, which functions by inhibiting vascular endothelial growth factor-induced mitogen-activated protein kinase signaling pathways. These novel activities of carvedilol provide insight into the anti-angiogenic mechanisms involved, and highlight its potential therapeutic application against angiogenesis-dependent liver fibrosis.
INTRODUCTION
Angiogenesis, the growth of new blood vessels from pre-existing ones, is an essential process during embryonic and postnatal development[1]. Although angiogenesis may be considered beneficial for tissue growth and regeneration, as well as for the growth and repair of injured tissues, the same process is also currently believed to promote inflammatory and fibro-proliferative diseases and malignancies in different organs, including the chronically injured liver. Data from the literature during the last decade have unequivocally linked angiogenesis with liver fibrogenesis and chronic liver disease progression, suggesting that angiogenesis may favor fibrogenesis[2,3]. Experimental anti-angiogenic therapies, regardless of the specific drug or therapeutic strategy employed, have resulted in significant inhibition of fibrogenic progression, reductions in inflammatory infiltrates and α-smooth muscle actin-positive myofibroblasts, a decrease in portal pressure, and, with some drugs, a reduction in porto-systemic collateral vessel formation and splanchnic vascularization in portal hypertensive animal models or in cirrhotic animals[2-4].
During angiogenesis, multiple signaling pathways are activated by various factors such as vascular endothelial growth factor (VEGF), platelet-derived growth factor, angiopoietin, Notch, Wnt, transforming growth factor-β, and guanine nucleotide binding regulatory protein-coupled receptor agonists, with VEGF as a dominating player. VEGF-A is the primary mediator of angiogenesis and primarily activates the tyrosine kinase receptor VEGF receptor (VEGFR)-2[5]. The activity of Src, a membrane-associated nonreceptor tyrosine kinase, is central in signal transduction downstream of several growth factor receptors including VEGFR[6]. Its downstream transduction pathways include phosphatidylinositol-3-kinase, focal adhesion kinase, and mitogen-activated protein kinase (MAPK)[6]. The Ras/Raf/MEK/ERK signaling pathway, also known as the extracellular signal-regulated kinase (ERK) pathway, is the most classical pathway of MAPK pathways[7], and plays an important role in angiogenesis[8,9]
The β-adrenergic system interferes with several essential steps of neovascularization, thus providing novel therapeutic opportunities for the use of β-blockers in the treatment of angiogenesis-dependent diseases. For instance, propranolol was reported to inhibit the growth of capillary hemangiomas[10] and could inhibit human umbilical vascular endothelial cell (HUVEC) proliferation and migration, as well as the VEGF-induced activation of VEGFR-2 in these cells[11]. Alpha-blockers also exhibited anti-angiogenic effects. Doxazosin and terazosin inhibited the growth and tube formation of human vascular endothelium cells and suppressed angiogenesis in mice[12,13]. Prazosin has also been reported to exhibit anti-angiogenic activity and to differentially modulate apoptotic pathways depending on the cell type. Patients with cirrhosis exhibited increased levels of catecholamines with increasing liver disease severity[14]. Therefore, non-selective beta blockers (NSBBs) may be particularly potent for the inhibition of angiogenesis in cirrhosis. Carvedilol is a third-generation, nonselective-blocker that also possesses α1-adrenergic blocking[15], antioxidant[16], and calcium antagonist properties[17]. Similar to propranolol and nadolol, the current standard NSBBs for treating portal hypertension, carvedilol has been proposed for use in decreasing portal pressure. Because of its α1- and β-blocking effects, carvedilol has the potential to induce a more pronounced decrease in portal pressure.
The present study investigated whether carvedilol can modulate HUVEC functions that are essential for angiogenesis. We observed that carvedilol inhibited growth factor-induced proliferation, migration, and tube formation in vitro. We also found that carvedilol caused G1 cell cycle arrest of HUVECs and inhibited VEGF-induced MAPK signaling pathways. Taken together, these novel activities of carvedilol provided insight regarding the anti-angiogenic mechanisms involved, and highlighted its potential therapeutic application against angiogenesis-dependent liver fibrosis.
MATERIALS AND METHODS
Cell culture
HUVECs were obtained from ATCC (Manassas, VA, United States) and cultured in RPMI 1640 medium containing 10% fetal calf serum, 100 U/mL penicillin, and 100 mg/mL streptomycin at 37 °C in a humidified atmosphere of 5% CO2.
Recombinant human VEGF 165 was obtained from PeproTech (Rocky Hill, NJ, United States), and Matrigel was obtained from BD Biosciences (Bedford, MA, United States). Anti-phospho-VEGFR2 (Tyr1175), anti-total VEGFR, anti-phospho-p44/p42 MAPK (ERK 1/2), anti-total ERK 1/2, anti-phospho-Src family (Thr416), and anti-total Src family rabbit monoclonal antibodies were obtained from Cell Signaling Technology (Bedford, MA, United States). PP2 (Src family kinase inhibitor) was obtained from Selleckchem (United States).
Cell counting Kit-8 assay
HUVECs were seeded in 96-well plates at 5000 cells/well and allowed to adhere overnight. After the cells were treated with various concentrations of carvedilol dissolved in 1% FBS medium for 24 h, 10 μL of cholecystokinin-8 (Beyotime, Haimen, Jiangsu, China) was added to each well. The plates were incubated at 37 °C for 2 h, and then the optical density (OD) was measured at 450 nm using a scanning multi-well spectrophotometer (Bio-Rad Model 550, CA, United States). The cell inhibitory rate was calculated using the following equation: cell inhibitory rate = [(OD control group-OD experiment group)/OD control group] × 100%. All experiments were performed in triplicate and repeated three times.
Cell cycle analysis
HUVECs were seeded at an initial density of 5 × 104 cells/mL and incubated with or without the indicated concentration of carvedilol supplemented with 5% FBS for 24 h. At the end of treatment, the cells were collected by mild trypsin digestion, washed with ice-cold PBS and fixed in 70% (v/v) ethanol overnight at 4 °C. Then, the cells were stained with propidium iodide (PI) working solution with 20 μg/mL RNase A and 50 μg/mL PI for cellular DNA staining at room temperature for 1 h in the dark before analysis. The cell population fraction in each phase of the cell cycle was determined as a function of the DNA content using a BD Biosciences FACSCalibur flow cytometer equipped with FACSDiva 7.0 software.
Endothelial cell migration assay
HUVECs were allowed to grow to full confluence in 6-well plates and then starved with cell medium containing 1% FBS overnight to inactivate cell proliferation. The next day, scrape wounds were made, and the cells were treated with or without VEGF (50 ng/mL) and various concentrations of carvedilol for 24 h. Images were acquired at time 0 and 24 h after scraping. ImageJ software was used to analyze the areas of cell migration into the scrape wound.
Endothelial cell transwell migration assay
Transwells (8-μm pore size; Costar) were precoated with 0.1% gelatin for 30 min in a cell incubator. Then, the transwells were washed with PBS and assembled into 24-well plates. HUVECs (5 × 104 cells) suspended in 100 μL of serum-free medium plus various concentrations of carvedilol were seeded in the top chambers, while 600 μL of serum-free medium was added to the lower chamber. After 2 h, cell migration was initiated by adding VEGF-enriched medium to the lower chamber. The plate was incubated at 37 °C in 5% CO2/95% air for another 12 h. At the end of the incubation period, the cells were fixed in 4% (v/v) paraformaldehyde and stained with hematoxylin and eosin. The migrated cells in the lower surface were imaged and counted under a light microscope.
Tube formation assay
Matrigel was thawed at 4 °C overnight before the experiment. A 96-well plate was coated with cold Matrigel and incubated for 0.5-1 h at 37 °C. Carvedilol and the VEGF-treated cell suspensions (100 mmol/L) were added to the wells and were incubated at 37 °C with 5% CO2. The cells were monitored every 2 h under a microscope for 6-12 h, and tube formation was imaged at 8 h.
Western blot analysis
HUVECs were treated as described for the cell cycle analysis. Then, the cells were harvested with trypsin/EDTA (Clonetics) and lysed with RIPA buffer, which contains 1% PMSF and 1% protein phosphatase inhibitor mixture (Applygen, Beijing, China). The protein concentration of the samples was determined by the BCA assay (Biocolor Bioscience and Technology Company) according to the manufacturer’s protocol. Equal amounts of protein extracts (50 mg) were subjected to electrophoretic analysis using a 10% sodium dodecyl sulfate (SDS)/polyacrylamide gel and transferred to PVDF membranes (Millipore, MA, United States). The membranes were incubated overnight with phospho-VEGFR, VEGFR, phospho-Src, Src, phospho-ERK and ERK monoclonal antibodies. Then, the membranes were washed and incubated with species-specific horseradish peroxidase-labeled secondary antibodies (Zhongshan Golden Bridge Biotechnology, Beijing, China). Signal detection was achieved using an enhanced chemiluminescence system (ECL; Millipore, MA, United States) and autoradiographed using X-ray film. Western blot results were analyzed using MultiGauge Ver. 3.2 software (Fujifilm Life Science, Japan) and all bands were normalized to β-actin expression (AA128, Beyotime, Nantong, China).
Statistical analysis
To compare two or more groups with the control group, one way analysis of variance followed by Dunnett’s test was used. All statistical analyses were performed using SPSS version 17.0 (SPSS Inc., Chicago, IL, United States). The data are presented as the mean ± SE. P values < 0.05 were considered statistically significant.
RESULTS
Carvedilol inhibits HUVEC proliferation
HUVECs were treated with carvedilol for 24 h in a concentration range of 0.01 to 40 μmol/L. As shown in Figure 1, HUVEC proliferation was strongly inhibited by carvedilol in a dose-dependent manner. The half-maximal inhibition (IC50) was 38.5 μmol/L, which suggested a specific inhibitory effect of carvedilol on EC proliferation.
Figure 1.
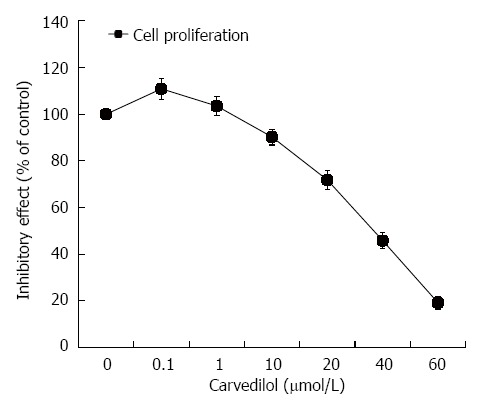
Effect of carvedilol on human umbilical vascular endothelial cell proliferation. Subconfluent cultures of human umbilical vascular endothelial cells were exposed to increasing concentrations of carvedilol (0.01-40 μmol/L), and the extent of cell proliferation was measured by cholecystokinin-8 assay. The values are the means of three independent experiments.
Carvedilol induces G1 phase arrest of ECs
To study the mechanism of carvedilol inhibition on cell proliferation, we performed cell cycle analysis of the carvedilol-treated HUVECs as shown in Figure 2. Exposing HUVECs to carvedilol resulted in prolongation of the G0/G1 phase (P < 0.05 vs control), which was associated with a reduction in S and M phases. These data suggested that carvedilol inhibited HUVEC proliferation by inhibiting cell cycle progression. Treatment with 1, 5 and 10 μmol/L carvedilol for 20 h effectively decreased the distribution of cells in the S phase from 43.6% to 37.2%, 35.6% and 17.8%, respectively (Figure 2B).
Figure 2.
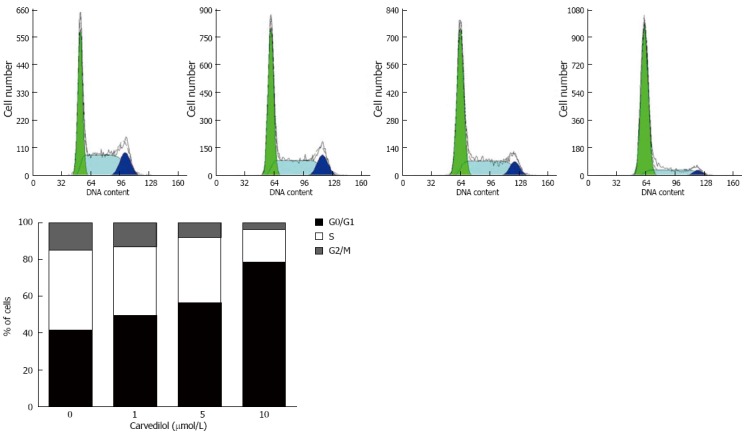
Effects of carvedilol on cell cycle progression in human umbilical vascular endothelial cells. Cells were exposed to either control medium (containing 5% FBS) or medium containing the indicated concentrations of carvedilol and were treated as indicated in the Materials and Methods section. Representatives cytometric profiles (upper) and the percentage of cells at G0/G1, S and G2/M phases (%) (lower). The DNA content was determined by measuring the fluorescence intensity of incorporated propidium iodide.
Inhibitory effects of Carvedilol on chemotactic motility in HUVECs
To assess the anti-angiogenic action of carvedilol in vitro, we investigated the inhibitory effects of carvedilol on the chemotactic motility of HUVECs by wound-healing migration assay (Figure 3) and Transwell assay (Figure 4). We found that 10 μmol/L carvedilol could reduce VEGF-induced HUVEC migration from 67.54% ± 7.831% to 37.11% ± 3.53% (P < 0.01) (Figure 3). Cell invasion was also reduced significantly by 5 μmol/L and 10 μmol/L carvedilol [from 196.3% ± 18.76% to 114.0% ± 12.20% (P < 0.05) and 51.68% ± 8.28% (P < 0.01), respectively] (Figure 5). These data showed that carvedilol can suppress HUVEC chemotactic motility in a dose-dependent manner.
Figure 3.
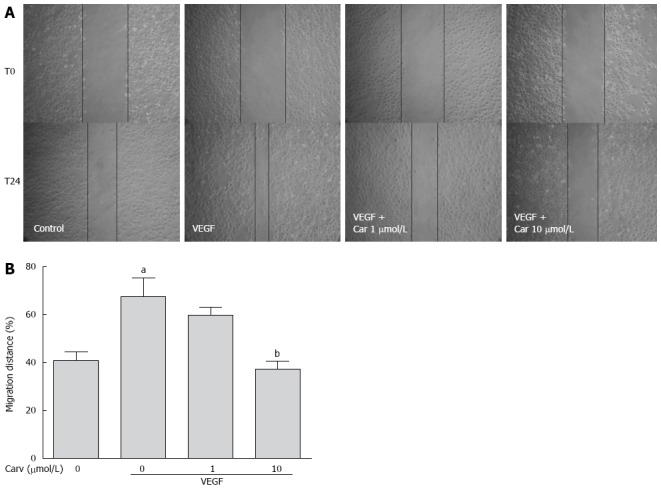
Effect of carvedilol on cell migration. A: HUVECs were scraped and then treated with or without VEGF (50 ng/mL) and various concentrations of carvedilol. Images were taken at 0 and 24 h after scraping; B: Cell migration across the scraped area was determined using ImageJ software. (n = 12, aP < 0.05 vs control, bP < 0.01 vs VEGF alone). HUVEC: Human umbilical vascular endothelial cell; VEGF: Vascular endothelial growth factor.
Figure 4.
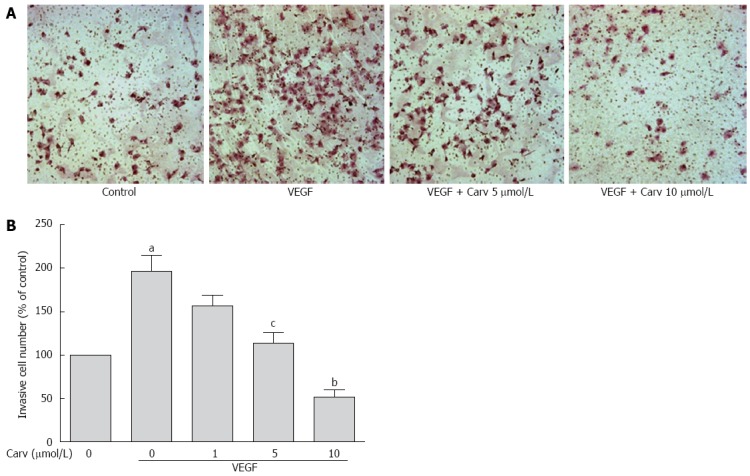
Effect of carvedilol on cell invasion. A: HUVECs were seeded in the upper chamber of a Transwell and treated with various concentrations of carvedilol. The bottom chamber was filled with VEGF-enriched media. After 12 h, the HUVECs that migrated through the membrane were stained by hematoxylin and eosin; B: Cell numbers were quantified (n = 12, aP < 0.05 vs control, cP < 0.05, bP < 0.01 vs VEGF alone).
Figure 5.
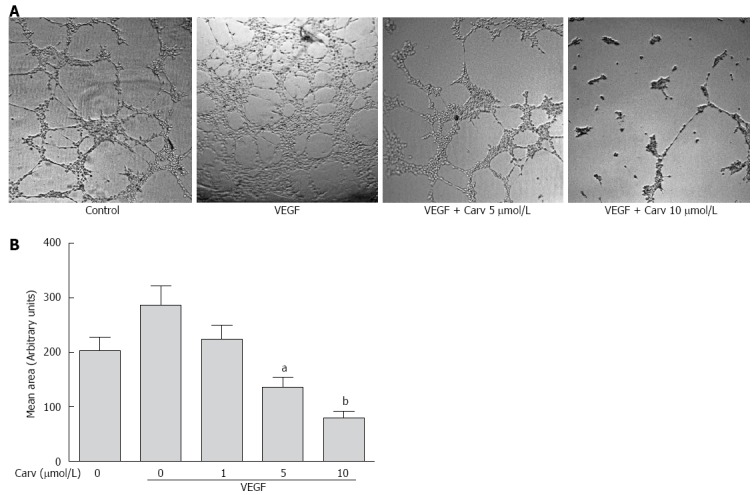
Carvedilol inhibited the vascular endothelial growth factor-induced tube formation of human umbilical vascular endothelial cells. A: HUVECs were placed in Matrigel with or without VEGF (50 ng/mL) in the presence or absence of different concentrations of carvedilol. After 8 h, tubular structures were imaged (magnification, × 100); B: Quantification of the area of cell alignment was determined using ImageJ software (n = 3, aP < 0.05, bP < 0.01 vs VEGF alone).
Inhibitory effects of carvedilol on tube formation in HUVECs
Tube formation in HUVECs incubated on Matrigel-coated plates represents the initial step in angiogenesis. To examine whether carvedilol can directly inhibit angiogenesis, the effect of carvedilol on tube formation was investigated using an in vitro angiogenesis assay. When HUVECs were seeded on growth factor-reduced Matrigel, robust tubular-like structures were formed in the presence of VEGF. However, treatment with 5 and 10 μmol/L carvedilol significantly suppressed VEGF-induced tube formation from 286.0 ± 36.72 to 135.7 ± 18.13 (P < 0.05) and 80.27 ± 11.16 (P < 0.01). These results indicated that carvedilol could block VEGF-induced angiogenesis in vitro by inhibiting EC motility and tube formation.
Carvedilol suppresses VEGF-induced VEGFR2 and ERK phosphorylation
The biological action of VEGF in ECs is mediated primarily via VEGFR-2. VEGFR-2 is dimerized and auto-phosphorylated after binding with VEGF. Therefore, we examined whether carvedilol could have an effect on VEGF-induced tyrosine phosphorylation of VEGFR-2. Quiescent HUVECs were pretreated with various concentrations of carvedilol for 24 h and then stimulated with 50 ng/mL VEGF for 2 h. We observed that VEGF-induced tyrosine phosphorylation of VEGFR-2 was reduced by carvedilol in a dose-dependent manner [from 175.5% ± 8.54% to 129.6% ± 14.83% (P < 0.05), 103.5% ± 14.32% (P < 0.01) and 52.67% ± 5.33% (P < 0.01) at concentrations of 1, 5 and 10 μmol/L, respectively] (Figure 6A).
Figure 6.
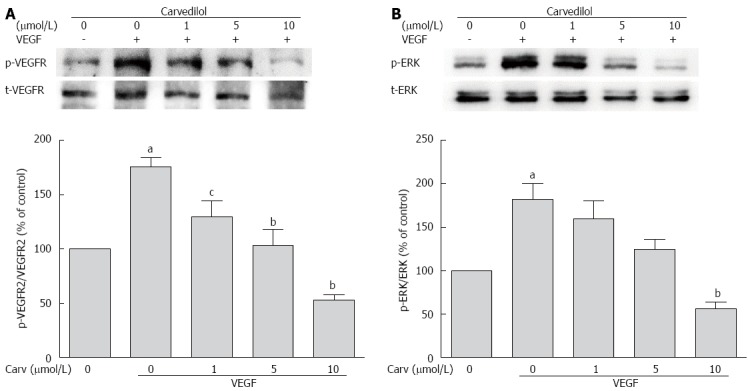
Effect of carvedilol on vascular endothelial growth factor angiogenic signaling. HUVECs were treated with VEGF (50 ng/mL) for 2 h in the presence or absence of different concentrations of carvedilol. Cell lysates were processed as indicated in the Materials and Methods. p-VEGFR2/VEGFR2 (A) and p-ERK/ERK (B) levels (n = 3, aP < 0.05 vs control, cP < 0.05, bP < 0.01 vs VEGF alone). ERK: Extracellular signal-regulated kinase.
Quiescent HUVECs were treated as mentioned above. Compared with the control group, the relative protein expression of phospho-ERK increased in the VEGF-treated group. Treatment with 1, 5, and 10 μmol/L carvedilol also reduced VEGF-induced ERK 1/2 phosphorylation (Figure 6B) in a dose-dependent manner [from 181.9% ± 18.61% to 159.8% ± 20.52%, 124.3% ± 11.48% and 56.45% ± 7.64% (P < 0.01), respectively].
Role of Src kinase in carvedilol-induced inhibition of tyrosine phosphorylation of ERK1/2
Because VEGFR-2 activation results in the activation of the Src family kinase[18], we studied whether Src kinase mediates tyrosine phosphorylation signaling from VEGFR to ERK. First, we tested whether carvedilol had an effect on VEGF-induced tyrosine phosphorylation of Src. We observed an increase in Src kinase activity in cells treated with VEGF for 30 min, and this increase was attenuated by carvedilol [decreased from 141.8% ± 15.37% to 110.3% ± 10.40%, 53.57% ± 7.18% (P < 0.01) and 47.04% ± 9.74% (P < 0.01) at concentrations of 1, 5, and 10 μmol/L, respectively] (Figure 7A). Then, the experiments illustrated in Figure 7B were conducted. Pretreating HUVECs with PP2 (10 μmol/L), a Src kinase inhibitor, almost completely prevented the VEGF-induced upregulation of ERK levels [decreased from 213.2% ± 27.68% to 90.96% ± 17.16% (P < 0.01)]. Evidently, Src activation may be a preceding step in the VEGF-induced upregulation of ERK and may play an important role in angiogenesis. These findings suggest that Src may serve as a mediating factor for signaling from VEGFR to ERK during carvedilol-induced inhibition of angiogenesis.
Figure 7.
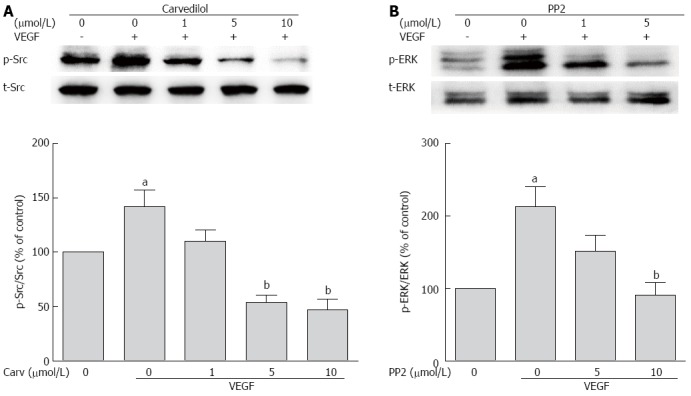
Role of Src in angiogenic signaling. A: HUVECs were treated with VEGF (50 ng/mL) for 2 h in the presence or absence of different concentrations of PP2. Cell lysates were subjected to SDS-PAGE for Src phosphorylation; B: HUVECs were treated with VEGF (50 ng/mL) for 2 h in the presence or absence of different concentrations of PP2. Cell lysates were processed as indicated in the Materials and Methods (n = 3, aP < 0.05 vs control, bP < 0.01 vs VEGF alone).
DISCUSSION
Our present study is the first to demonstrate that carvedilol inhibits VEGF-induced angiogenesis by blocking the VEGF/VEGFR-Src-ERK mediated pathway. The data obtained here provide a novel mechanism to account for the inhibitory effect of carvedilol on VEGF-mediated angiogenesis.
VEGF is a focus of applications of current concepts in the development of new anti-cancer and anti-angiogenetic therapies[19,20], and is capable of provoking angiogenesis by interacting with its two receptor tyrosine kinases [VEGFR-1; Flt-1) and VEGFR-2 (Flk-1)] expressed on ECs[21,22]. This study demonstrated that carvedilol significantly suppressed the VEGF-induced phosphorylation of Tyr1175 of VEGFR-2, suggesting that carvedilol may exert anti-angiogenic behavior by targeting VEGFR-2, and could impair its associated downstream signal transduction cascade. VEGFR-2 appears to mediate the majority of the known cellular responses to VEGFA, and the largest class of neoangiogenesis blocking drugs are the multiple receptor tyrosine kinase inhibitors that target VEGFR-2, such as sorafenib, vatalanib, and TSU-68[23-25]. The present results showed that carvedilol inhibited the phosphorylation of VEGFR-2, and thus blocked the subsequent activation of downstream signals. However, carvedilol did not inhibit VEGFR-2 activation completely. Thus, other signaling pathways may be involved in VEGFR-2 and ERK activation in response to carvedilol; however, this possibility requires further investigation. The reduction in phosphorylated VEGFR-2 may explain the reason for the observed decrease in the VEGF-induced phosphorylation of ERK1⁄2, the major downstream target of VEGFR-2, following treatment with carvedilol. Various agents have been shown to block VEGFA-induced angiogenesis by inhibiting the activation of the MAPK signal cascade[26-28]. VEGF-mediated survival[29,30] and protection against receptor-mediated apoptosis were Src-dependent[31]. Thus, we further investigated the effect of carvedilol on Src, and showed that Src plays an important role in the signaling process. However, the mechanism by which carvedilol decreases VEGFR-2 phosphorylation requires further investigation.
Historically, an interest in angiogenesis has been associated with studies regarding cancer and factors involved in tumor progression[32]. Recent data regarding chronic liver diseases have demonstrated that angiogenesis may contribute to the progression of fibrosis during the wound healing process in chronic liver damage[2,3,33-35]. Anti-angiogenic therapy has been found to be efficient in the prevention of fibrosis in experimental models of chronic liver diseases; blocking angiogenesis could be a promising therapeutic option in patients with advanced fibrosis[36-38]. Carvedilol is a cardiovascular drug that is licensed for the treatment of chronic heart failure and is widely used to reduce portal pressure in patients with cirrhosis. In the present study, we extended the current knowledge regarding the role of carvedilol in anti-angiogenesis. Catecholamines exhibited pro-carcinogenic effects[39], and NSBBs antagonized catecholamine-driven cell migration and tumor angiogenesis, invasiveness and proliferation in gastric, breast and pancreatic cancers[40-42]. A number of studies have recently highlighted the potential anti-angiogenic properties of β-blockers[10,43]. Carvedilol has also been shown to inhibit the proliferation of various human cell lines such as osteosarcoma MG63[44] and rat C6 glioma cells[45]. One report indicated that carvedilol could inhibit tube formation similar to other β-blockers[40]; however, no detailed mechanism had been explored. Here, we demonstrated that carvedilol directly inhibited the proliferation and formation of capillary-like tube structures in cultured HUVECs. Importantly, we provided compelling evidence that activation of the VEGFR/Src/ERK1/2 signaling pathway was involved in the carvedilol-induced inhibition of angiogenesis in HUVECs. Interestingly, carvedilol was reported previously to have anti-inflammatory and pro-angiogenic effects in a canine model of chronic ischemic cardiomyopathy[40]. Our study did not contradict the results of that study because the model and inducing factors are completely different. Taken together, these studies suggest that carvedilol may have a two-way regulatory effect on angiogenesis.
In addition to the effect of carvedilol on angiogenesis, carvedilol can induce human hepatoma cell death[46]. Because most cases of hepatocellular carcinoma (HCC) develop in patients with chronic liver disease (70%-90%)[47,48], finding a drug that would ameliorate either liver cirrhosis or HCC may be beneficial for the other disease. One limitation of our study was the lack of in vivo experiments; however, carvedilol was reported previously to have antifibrotic effects on chronic carbon tetrachloride-induced liver damage[49], suggesting that carvedilol may ameliorate liver fibrosis-related angiogenesis. We are currently examining this possibility. If carvedilol can be demonstrated to have anti-angiogenic and anti-neoplastic effects, then patients with chronic liver disease could benefit greatly from this treatment.
In conclusion, the results from the present study indicate that carvedilol can exert an anti-angiogenic effect by inhibiting the activation of the cSrc/ERK 1/2 signaling pathway. The findings from the present in vitro studies strongly suggest the potential application of carvedilol in the treatment of disorders associated with localized angiogenesis. If this hypothesis holds true, then the use of carvedilol in chronic liver diseases could be extended beyond the prevention of variceal bleeding; its multiple effects including anti-angiogenesis, anti-fibrosis, and even anti-cancer would reinforce the recommendation of this drug as first-line treatment in chronic liver disease patients.
COMMENTS
Background
Angiogenesis is an important process during embryonic and postnatal development. Data from the literature in the last decade have suggested that angiogenesis may favor fibrogenesis. Beta-blockers were reported to suppress angiogenesis. Carvedilol is a third-generation nonselective β-blocker that has been used for treating portal hypertension. This study investigated whether carvedilol could modulate human umbilical vascular endothelial cell functions that are essential for angiogenesis.
Research frontiers
Blocking angiogenesis could be a promising therapeutic option in patients with advanced fibrosis. Carvedilol is widely used to reduce portal pressure in patients with cirrhosis. In this study, the authors extended the current knowledge regarding the role of carvedilol in anti-angiogenesis.
Innovations and breakthroughs
This study is the first to demonstrate that carvedilol inhibits vascular endothelial growth factor (VEGF)-induced angiogenesis by blocking the VEGF/VEGFR-Src-ERK-mediated pathway. The data obtained here provide a novel mechanism to account for the inhibitory effect of carvedilol on VEGF-mediated angiogenesis.
Terminology
Angiogenesis is the growth of new blood vessels from pre-existing ones, it may fuel inflammatory and fibro-proliferative diseases including chronically injured liver. VEGF-A is the primary mediator of angiogenesis and primarily activates the tyrosine kinase receptor VEGF receptor (VEGFR)-2. Beta-blockers are a class of drugs that could block the action of endogenous catecholamines, epinephrine (adrenaline) and norepinephrine (noradrenaline), in particular on adrenergic β-receptors.
Applications
These results strongly suggest the potential application of carvedilol in the treatment of disorders associated with localized angiogenesis. The use of carvedilol in chronic liver diseases could be extended beyond the prevention of variceal bleeding; its multiple effects, including anti-angiogenesis, anti-fibrosis, and even anti-neoplastic, would reinforce the recommendation of this drug as first-line treatment in chronic liver disease patients.
Peer-review
This study investigated the effect of carvedilol on angiogenesis in vitro. The results suggest that carvedilol could inhibit angiogenesis by blocking the VEGF-induced VEGFR-Src-Erk pathway. Thus, these findings suggest potential new uses for carvedilol.
Footnotes
Supported by National Natural Science Foundation of China, No. 81370590.
Conflict-of-interest statement: The authors have no conflict of interest related to the manuscript.
Data sharing statement: No additional unpublished data are available.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: March 31, 2015
First decision: April 23, 2015
Article in press: July 9, 2015
P- Reviewer: Gorrell MD, Ruiz-Margain A, Vezali E S- Editor: Ma YJ L- Editor: Cant MR E- Editor: Zhang DN
References
- 1.Marone G, Granata F. Angiogenesis, lymphangiogenesis and clinical implications. Preface. Chem Immunol Allergy. 2014;99:XI–XII. doi: 10.1159/000352074. [DOI] [PubMed] [Google Scholar]
- 2.Fernández M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J. Angiogenesis in liver disease. J Hepatol. 2009;50:604–620. doi: 10.1016/j.jhep.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 3.Valfrè di Bonzo L, Novo E, Cannito S, Busletta C, Paternostro C, Povero D, Parola M. Angiogenesis and liver fibrogenesis. Histol Histopathol. 2009;24:1323–1341. doi: 10.14670/HH-24.1323. [DOI] [PubMed] [Google Scholar]
- 4.Rosmorduc O, Housset C. Hypoxia: a link between fibrogenesis, angiogenesis, and carcinogenesis in liver disease. Semin Liver Dis. 2010;30:258–270. doi: 10.1055/s-0030-1255355. [DOI] [PubMed] [Google Scholar]
- 5.Varricchi G, Granata F, Loffredo S, Genovese A, Marone G. Angiogenesis and lymphangiogenesis in inflammatory skin disorders. J Am Acad Dermatol. 2015;73:144–153. doi: 10.1016/j.jaad.2015.03.041. [DOI] [PubMed] [Google Scholar]
- 6.Mayer EL, Krop IE. Advances in targeting SRC in the treatment of breast cancer and other solid malignancies. Clin Cancer Res. 2010;16:3526–3532. doi: 10.1158/1078-0432.CCR-09-1834. [DOI] [PubMed] [Google Scholar]
- 7.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 8.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang F, Li J, Zhu J, Wang D, Chen S, Bai X. Hydroxysafflor yellow A inhibits angiogenesis of hepatocellular carcinoma via blocking ERK/MAPK and NF-κB signaling pathway in H22 tumor-bearing mice. Eur J Pharmacol. 2015;754:105–114. doi: 10.1016/j.ejphar.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 10.Léauté-Labrèze C, Dumas de la Roque E, Hubiche T, Boralevi F, Thambo JB, Taïeb A. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008;358:2649–2651. doi: 10.1056/NEJMc0708819. [DOI] [PubMed] [Google Scholar]
- 11.Lamy S, Lachambre MP, Lord-Dufour S, Béliveau R. Propranolol suppresses angiogenesis in vitro: inhibition of proliferation, migration, and differentiation of endothelial cells. Vascul Pharmacol. 2010;53:200–208. doi: 10.1016/j.vph.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 12.Pan SL, Guh JH, Huang YW, Chern JW, Chou JY, Teng CM. Identification of apoptotic and antiangiogenic activities of terazosin in human prostate cancer and endothelial cells. J Urol. 2003;169:724–729. doi: 10.1097/01.ju.0000037731.83941.db. [DOI] [PubMed] [Google Scholar]
- 13.Keledjian K, Garrison JB, Kyprianou N. Doxazosin inhibits human vascular endothelial cell adhesion, migration, and invasion. J Cell Biochem. 2005;94:374–388. doi: 10.1002/jcb.20240. [DOI] [PubMed] [Google Scholar]
- 14.Dietrich P, Moleda L, Kees F, Müller M, Straub RH, Hellerbrand C, Wiest R. Dysbalance in sympathetic neurotransmitter release and action in cirrhotic rats: impact of exogenous neuropeptide Y. J Hepatol. 2013;58:254–261. doi: 10.1016/j.jhep.2012.09.027. [DOI] [PubMed] [Google Scholar]
- 15.Bristow MR, Larrabee P, Müller-Beckmann B, Minobe W, Roden R, Skerl L, Klein J, Handwerger D, Port JD. Effects of carvedilol on adrenergic receptor pharmacology in human ventricular myocardium and lymphocytes. Clin Investig. 1992;70 Suppl 1:S105–S113. doi: 10.1007/BF00207620. [DOI] [PubMed] [Google Scholar]
- 16.Feuerstein GZ, Ruffolo RR. Carvedilol, a novel multiple action antihypertensive agent with antioxidant activity and the potential for myocardial and vascular protection. Eur Heart J. 1995;16 Suppl F:38–42. doi: 10.1093/eurheartj/16.suppl_f.38. [DOI] [PubMed] [Google Scholar]
- 17.Nichols AJ, Gellai M, Ruffolo RR. Studies on the mechanism of arterial vasodilation produced by the novel antihypertensive agent, carvedilol. Fundam Clin Pharmacol. 1991;5:25–38. doi: 10.1111/j.1472-8206.1991.tb00698.x. [DOI] [PubMed] [Google Scholar]
- 18.Schlessinger J. New roles for Src kinases in control of cell survival and angiogenesis. Cell. 2000;100:293–296. doi: 10.1016/s0092-8674(00)80664-9. [DOI] [PubMed] [Google Scholar]
- 19.Plate KH, Breier G, Weich HA, Mennel HD, Risau W. Vascular endothelial growth factor and glioma angiogenesis: coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms. Int J Cancer. 1994;59:520–529. doi: 10.1002/ijc.2910590415. [DOI] [PubMed] [Google Scholar]
- 20.Lu J, Zhang K, Nam S, Anderson RA, Jove R, Wen W. Novel angiogenesis inhibitory activity in cinnamon extract blocks VEGFR2 kinase and downstream signaling. Carcinogenesis. 2010;31:481–488. doi: 10.1093/carcin/bgp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Z, Neiva KG, Lingen MW, Ellis LM, Nör JE. VEGF-dependent tumor angiogenesis requires inverse and reciprocal regulation of VEGFR1 and VEGFR2. Cell Death Differ. 2010;17:499–512. doi: 10.1038/cdd.2009.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meissner M, Hrgovic I, Doll M, Kaufmann R. PPARδ agonists suppress angiogenesis in a VEGFR2-dependent manner. Arch Dermatol Res. 2011;303:41–47. doi: 10.1007/s00403-010-1091-y. [DOI] [PubMed] [Google Scholar]
- 23.Ibrahim N, Yu Y, Walsh WR, Yang JL. Molecular targeted therapies for cancer: sorafenib mono-therapy and its combination with other therapies (review) Oncol Rep. 2012;27:1303–1311. doi: 10.3892/or.2012.1675. [DOI] [PubMed] [Google Scholar]
- 24.Brave SR, Odedra R, James NH, Smith NR, Marshall GB, Acheson KL, Baker D, Howard Z, Jackson L, Ratcliffe K, et al. Vandetanib inhibits both VEGFR-2 and EGFR signalling at clinically relevant drug levels in preclinical models of human cancer. Int J Oncol. 2011;39:271–278. doi: 10.3892/ijo.2011.1022. [DOI] [PubMed] [Google Scholar]
- 25.Shin SJ, Jung M, Jeung HC, Kim HR, Rha SY, Roh JK, Chung HC, Ahn JB. A phase I pharmacokinetic study of TSU-68 (a multiple tyrosine kinase inhibitor of VEGFR-2, FGF and PDFG) in combination with S-1 and oxaliplatin in metastatic colorectal cancer patients previously treated with chemotherapy. Invest New Drugs. 2012;30:1501–1510. doi: 10.1007/s10637-011-9683-8. [DOI] [PubMed] [Google Scholar]
- 26.Pang X, Yi T, Yi Z, Cho SG, Qu W, Pinkaew D, Fujise K, Liu M. Morelloflavone, a biflavonoid, inhibits tumor angiogenesis by targeting rho GTPases and extracellular signal-regulated kinase signaling pathways. Cancer Res. 2009;69:518–525. doi: 10.1158/0008-5472.CAN-08-2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Asami Y, Kakeya H, Komi Y, Kojima S, Nishikawa K, Beebe K, Neckers L, Osada H. Azaspirene, a fungal product, inhibits angiogenesis by blocking Raf-1 activation. Cancer Sci. 2008;99:1853–1858. doi: 10.1111/j.1349-7006.2008.00890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu X, Zhu J, Mi M, Chen W, Pan Q, Wei M. Anti-angiogenic genistein inhibits VEGF-induced endothelial cell activation by decreasing PTK activity and MAPK activation. Med Oncol. 2012;29:349–357. doi: 10.1007/s12032-010-9770-2. [DOI] [PubMed] [Google Scholar]
- 29.Berra E, Milanini J, Richard DE, Le Gall M, Viñals F, Gothié E, Roux D, Pagès G, Pouysségur J. Signaling angiogenesis via p42/p44 MAP kinase and hypoxia. Biochem Pharmacol. 2000;60:1171–1178. doi: 10.1016/s0006-2952(00)00423-8. [DOI] [PubMed] [Google Scholar]
- 30.Gupta K, Kshirsagar S, Li W, Gui L, Ramakrishnan S, Gupta P, Law PY, Hebbel RP. VEGF prevents apoptosis of human microvascular endothelial cells via opposing effects on MAPK/ERK and SAPK/JNK signaling. Exp Cell Res. 1999;247:495–504. doi: 10.1006/excr.1998.4359. [DOI] [PubMed] [Google Scholar]
- 31.Alavi A, Hood JD, Frausto R, Stupack DG, Cheresh DA. Role of Raf in vascular protection from distinct apoptotic stimuli. Science. 2003;301:94–96. doi: 10.1126/science.1082015. [DOI] [PubMed] [Google Scholar]
- 32.Pałgan K, Bartuzi Z. Angiogenesis in bronchial asthma. Int J Immunopathol Pharmacol. 2015:Epub ahead of print. doi: 10.1177/0394632015580907. [DOI] [PubMed] [Google Scholar]
- 33.Amarapurkar AD, Amarapurkar DN, Vibhav S, Patel ND. Angiogenesis in chronic liver disease. Ann Hepatol. 2007;6:170–173. [PubMed] [Google Scholar]
- 34.Kukla M, Gabriel A, Sabat D, Liszka Ł, Wilk M, Petelenz M, Musialik J, Dzindziora-Frelich I. Association between liver steatosis and angiogenesis in chronic hepatitis C. Pol J Pathol. 2010;61:154–160. [PubMed] [Google Scholar]
- 35.Sanz-Cameno P, Trapero-Marugán M, Chaparro M, Jones EA, Moreno-Otero R. Angiogenesis: from chronic liver inflammation to hepatocellular carcinoma. J Oncol. 2010;2010:272170. doi: 10.1155/2010/272170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gabriel A, Kukla M, Wilk M, Liszka Ł, Petelenz M, Musialik J. Angiogenesis in chronic hepatitis C is associated with inflammatory activity grade and fibrosis stage. Pathol Res Pract. 2009;205:758–764. doi: 10.1016/j.prp.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 37.Shah VH, Bruix J. Antiangiogenic therapy: not just for cancer anymore? Hepatology. 2009;49:1066–1068. doi: 10.1002/hep.22872. [DOI] [PubMed] [Google Scholar]
- 38.Elpek GÖ. Angiogenesis and liver fibrosis. World J Hepatol. 2015;7:377–391. doi: 10.4254/wjh.v7.i3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thaker PH, Sood AK. Neuroendocrine influences on cancer biology. Semin Cancer Biol. 2008;18:164–170. doi: 10.1016/j.semcancer.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Powe DG, Voss MJ, Zänker KS, Habashy HO, Green AR, Ellis IO, Entschladen F. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget. 2010;1:628–638. doi: 10.18632/oncotarget.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liao X, Che X, Zhao W, Zhang D, Bi T, Wang G. The β-adrenoceptor antagonist, propranolol, induces human gastric cancer cell apoptosis and cell cycle arrest via inhibiting nuclear factor κB signaling. Oncol Rep. 2010;24:1669–1676. doi: 10.3892/or_00001032. [DOI] [PubMed] [Google Scholar]
- 42.Al-Wadei HA, Al-Wadei MH, Schuller HM. Prevention of pancreatic cancer by the beta-blocker propranolol. Anticancer Drugs. 2009;20:477–482. doi: 10.1097/CAD.0b013e32832bd1e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pasquier E, Street J, Pouchy C, Carre M, Gifford AJ, Murray J, Norris MD, Trahair T, Andre N, Kavallaris M. β-blockers increase response to chemotherapy via direct antitumour and anti-angiogenic mechanisms in neuroblastoma. Br J Cancer. 2013;108:2485–2494. doi: 10.1038/bjc.2013.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu CP, Jan CR. Effect of carvedilol on Ca2+ movement and cytotoxicity in human MG63 osteosarcoma cells. Basic Clin Pharmacol Toxicol. 2004;95:59–65. doi: 10.1111/j.1742-7843.2004.950203.x. [DOI] [PubMed] [Google Scholar]
- 45.Erguven M, Yazihan N, Aktas E, Sabanci A, Li CJ, Oktem G, Bilir A. Carvedilol in glioma treatment alone and with imatinib in vitro. Int J Oncol. 2010;36:857–866. doi: 10.3892/ijo_00000563. [DOI] [PubMed] [Google Scholar]
- 46.Cheng JS, Huang CC, Chou CT, Jan CR. Mechanisms of carvedilol-induced [Ca2+] i rises and death in human hepatoma cells. Naunyn Schmiedebergs Arch Pharmacol. 2007;376:185–194. doi: 10.1007/s00210-007-0191-5. [DOI] [PubMed] [Google Scholar]
- 47.Sherman M. Hepatocellular carcinoma: epidemiology, surveillance, and diagnosis. Semin Liver Dis. 2010;30:3–16. doi: 10.1055/s-0030-1247128. [DOI] [PubMed] [Google Scholar]
- 48.Tejeda-Maldonado J, García-Juárez I, Aguirre-Valadez J, González-Aguirre A, Vilatobá-Chapa M, Armengol-Alonso A, Escobar-Penagos F, Torre A, Sánchez-Ávila JF, Carrillo-Pérez DL. Diagnosis and treatment of hepatocellular carcinoma: An update. World J Hepatol. 2015;7:362–376. doi: 10.4254/wjh.v7.i3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamdy N, El-Demerdash E. New therapeutic aspect for carvedilol: antifibrotic effects of carvedilol in chronic carbon tetrachloride-induced liver damage. Toxicol Appl Pharmacol. 2012;261:292–299. doi: 10.1016/j.taap.2012.04.012. [DOI] [PubMed] [Google Scholar]