

Abstract
The Bromo- and Extra-Terminal (BET) proteins BRD2, BRD3, and BRD4 play important roles in transcriptional regulation, epigenetics, and cancer and are the targets of pan-BET selective bromodomain inhibitor JQ1. However, the lack of intra-BET selectivity limits the scope of current inhibitors as probes for target validation and could lead to unwanted side effects or toxicity in a therapeutic setting. We designed Proteolysis Targeted Chimeras (PROTACs) that tether JQ1 to a ligand for the E3 ubiquitin ligase VHL, aimed at triggering the intracellular destruction of BET proteins. Compound MZ1 potently and rapidly induces reversible, long-lasting, and unexpectedly selective removal of BRD4 over BRD2 and BRD3. The activity of MZ1 is dependent on binding to VHL but is achieved at a sufficiently low concentration not to induce stabilization of HIF-1α. Gene expression profiles of selected cancer-related genes responsive to JQ1 reveal distinct and more limited transcriptional responses induced by MZ1, consistent with selective suppression of BRD4. Our discovery opens up new opportunities to elucidate the cellular phenotypes and therapeutic implications associated with selective targeting of BRD4.
The Bromo- and Extra-terminal (BET) family of proteins, including the ubiquitously expressed BRD2, BRD3, and BRD4 and the testis-specific BRDT, recruit transcriptional regulatory complexes to acetylated chromatin thereby controlling specific networks of genes involved in cellular proliferation and cell cycle progression.1 Deregulation of BET protein activity, in particular BRD4, has been strongly linked to cancer and inflammatory diseases, making BET proteins attractive drug targets.2 For example, RNAi screens have identified BRD4 as a therapeutic target in acute myeloid leukemia,3 ovarian carcinoma,4 and siRNA knock down of BRD4, but not of BRD2 or BRD3, induced upregulation of apolipoprotein A1 (ApoA1), which protects from atherosclerosis progression and other inflammatory processes.5 The silencing of BRD4 furthermore identified BRD4 as a target to treat chronic obstructive pulmonary disease (COPD).6 These results underscore the potential of targeting BRD4 as a therapeutic strategy and motivate further research in validating BRD4 as a drug target.
Crucial to the function of BET proteins are two highly homologous bromodomains that are present in their amino-terminal regions and direct recruitment to nucleosomes by binding to specific acetylated lysines (KAc) within histone tails.7 Small molecule BET inhibitors, including the triazolodiazepine-based JQ18 and I-BET7629 (Figure 1a) among others,10−13 bind to the KAc-binding pocket of the bromodomains and disrupt interaction with histones, thereby displacing BET proteins and their associated transcriptional regulatory complexes from chromatin. BET inhibitors are highly potent (Kd ∼100 nM), cell-penetrant, and active in vivo against a range of solid, hematological, and other tumors, which has prompted compounds entering phase I clinical trials for cancer.14−16 However, BET inhibitors show no selectivity for individual BET family members, thereby limiting their scope as chemical probes for validating the roles of individual BET targets in physiology and disease. To this end, chemical genetic strategies have been recently developed to engineer orthogonal selective BET bromodomain-ligand pairs.17 While this approach has the advantage of enabling disruption at will of a single or more bromodomains, it requires a mutation to be introduced into the target protein. Therapeutically, the effects of BET inhibitors on different transcriptional pathways have raised concerns about the safety and tolerability of BET inhibitors in humans. Crucially, none of the inhibitors described to date is selective for binding BRD4 bromodomains over those of its paralogs BRD2 and BRD3.
Figure 1.

Design, synthesis, and biophysical and biological evaluation of BET bromodomain PROTACs. (a) Chemical structures of BET-bromodomain inhibitors JQ1 and I-BET762 and binders of von Hippel-Lindau protein VHL-1 and VHL-2. (b) Scheme of the synthesis of PROTAC compounds MZ1–3 and cisMZ1; for detailed synthetic procedures see the Supporting Information. (c) Isothermal titration calorimetry data for titration of MZ1 into the individual members of the BET-bromodomain subfamily. Titrations were performed at 30 °C with a protein concentration of 15 μM and ligand concentration of 150 μM (entry 1–6). Titration of MZ1 and cisMZ1 into VBC at 25 °C with identical concentrations (entry 9, 12) and reverse titration of VBC protein (150 μM) into MZ3 (15 μM) at 25 °C (entry 10) were conducted. For ΔS and ΔG values, see the Supporting Information. (d) HeLa cells were treated with either siRNA targeting individual BET proteins or negative control siRNA 24 h prior to treatment with the compounds MZ1–3, cisMZ1, and JQ1 or vehicle control (0.01% DMSO) for an additional 24 h. Abundance of individual BET protein was analyzed by Western blotting using corresponding specific antibodies accordingly after SDS-PAGE. i, data from ref (8); ii, data from ref (26).
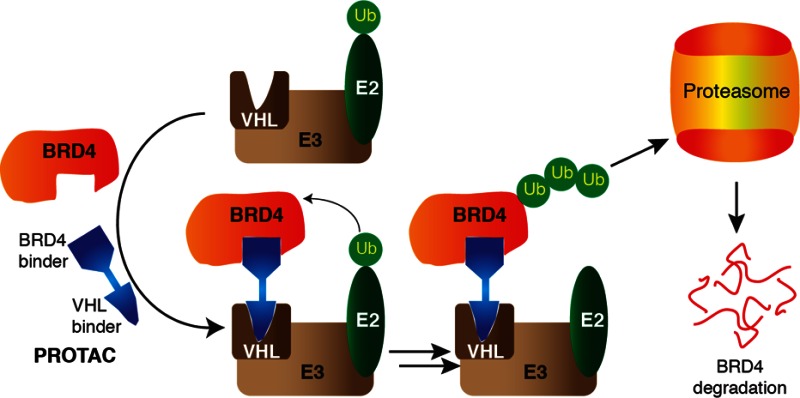
Small molecule chemical probes or inhibitors acting at the post-translational level hold several advantages for target validation over genetic techniques such as dominant-negative mutants or knockouts and RNAi knockdowns, including affording spatial and temporal control in a reversible fashion. A general limitation associated with conventional occupancy-driven target inhibition is that it often demands full target engagement, requiring sustained high concentration of a potent small molecule inhibitor over a prolonged time. This in turn enhances off-target effects and can lead to unwanted side effects or toxicity in a therapeutic setting. To provide an alternative small molecule approach that could address these issues, we hypothesized that it would be possible to design a molecule that can remove BET proteins entirely from the cell as opposed to just inhibit them, yielding new tools for studying BET bromodomain proteins and validating them as drug targets. In order to achieve intracellular BET-protein degradation, we applied a small molecule PROTAC (Proteolysis Targeting Chimera) approach.18,19 A PROTAC is a heterobifunctional compound that contains two ligands connected by a linker unit. One ligand binds an E3 ubiquitin ligase protein, while the other ligand binds to the target protein of interest, thereby bringing the ligase and the target in close proximity. This in turn triggers the polyubiquitination and subsequent proteasome-dependent degradation of the target. Proof-of-concept examples have been described where PROTACs were used to degrade the estrogen20- and androgen-receptor,21 methionine aminopeptidase-2,22 as well as the aryl hydrocarbon receptor.23 However, all first-generation PROTACs included a peptidic moiety as the E3 ligase ligand. For example, a hydroxyproline-containing heptapeptide sequence ALA-Hyp-YIP from the transcription factor Hypoxia-Inducible Factor 1 alpha subunit (HIF-1α) has been widely used,24 as this represents the minimal epitope for HIF-1α binding to the ubiquitously expressed E3 ligase von Hippel-Lindau protein (VHL).25 The high peptidic nature of the first-generation PROTACs resulted in poor physicochemical properties such as low intracellular stability and poor cell permeability, which limited their applicability as chemical probes and their potential therapeutic development. To overcome these limitations here, we develop a nonpeptidic PROTAC approach that exploits our recently discovered and optimized drug-like VHL ligands26 and show that it can be applied to target BET bromodomains and potently induce effective and selective degradation of BRD4.
We began by designing a series of PROTACs that would link together specific VHL ligands and BET bromodomain ligands. Recent work has established compounds VHL-1 and VHL-2 as strong binders with Kd values below 300 nM to VHL (Figure 1a).26 Inspection of the protein–ligand crystal structures show that the methyl group of the terminal acetyl groups in compounds VHL-1 and VHL-2 is solvent exposed, and we therefore reasoned that it could provide a suitable connecting point for a linker (Figure S1). The BET inhibitor JQ18 was chosen as the bromodomains-recruiting scaffold, and its t-butyl ester group was selected as a connecting point for a linker because it is solvent-exposed and not involved in key interaction with the BET bromodomains as revealed by cocrystal structures (Figure S1). Linkers with different lengths comprised of polyethylene glycol chains with either three or four ethylene glycol units were chosen to connect JQ1 with the VHL ligand. To achieve the desired ligands, a generally applicable two-step synthetic strategy was devised. First, the linker bearing a carboxylic acid at one end and an azide group at the other end was connected with the terminal free amine of the VHL ligand by a HATU-mediated amide bond formation. In the second step, reduction of the azide group to an amine and subsequent amide bond formation with the carboxylic acid of the ester-hydrolyzed JQ1 analogue afforded the desired PROTAC compounds MZ1, MZ2, MZ3, and cisMZ1 (Figure 1b).
To assess whether PROTAC molecules retained their binding to the target proteins VHL and BET bromodomains in a similar fashion as the parental ligands, isothermal titration calorimetry (ITC) experiments were performed (Figure 1; all ITC titrations are shown in the Supporting Information). MZ1, as a representative of all PROTAC molecules that share the same JQ1 moiety for binding bromodomains, was titrated into individual first and second bromodomains of BRD2, BRD3, and BRD4 (Figure 1c, entries 1−6). The measured binding affinities (Kd of 115–382 nM) and ΔH (−6.1 to −10.0 kcal/mol) compared well with those reported for unmodified JQ18 (literature values for BRD4 bromodomains shown in Figure 1c, entries 7, 8), suggesting that JQ1 binding mode is conserved within the context of our PROTACs. Similarly, as binding to the VHL protein is crucial for the recruitment of target proteins to the E3 ligase, the binding of MZ1 and MZ3 to the VHL-ElonginB-ElonginC complex (VBC) was also quantified using ITC (Figure 1c, entries 9, 10). The measured affinities (Kd of 150 and 310 nM for MZ1 and MZ3, respectively) and ΔH (−6.9 and −4.9 kcal/mol, respectively) compared very closely to those of the parental unmodified ligands VHL-1 (Kd = 185 nM, ΔH = −5.5 kcal/mol, entry 11) and VHL-2 (Kd = 290 nM, ΔH = −5.3 kcal/mol).26 As the stereochemistry of the hydroxyl group of the central hydroxyproline moiety is crucial for ligand binding to VHL, compound cisMZ1 was synthesized that is structurally identical to MZ1 except for a reversed stereocenter at the C-4 position bearing the hydroxyl group. As expected, cisMZ1 did not exhibit any measurable binding affinity for VHL in the ITC experiment (Figure 1c, entry 12) and thus was elected as a negative control compound in cellular assays.
To demonstrate that PROTACs are able to induce degradation of BET proteins, HeLa cells transfected with control siRNA were treated with 1 μM of compounds MZ1–3 alongside negative controls JQ1 and cisMZ1 for 24 h (Figure 1d). In parallel, HeLa cells with BRD2, BRD3, and BRD4 individually and separately silenced by transfection with the respective siRNA were treated with vehicle DMSO to compare the protein depletion effect of RNAi knockdown and PROTACs. BET protein abundance was evaluated by SDS-PAGE followed by Western blot using corresponding specific antibodies to probe for BRD2, BRD3 or BRD4, respectively. All three PROTAC compounds demonstrated complete removal of BRD4 with no detectable protein observed after 24 h of treatment. In contrast, removal of BRD2 and BRD3 was not complete after 24 h. MZ1 exhibited the highest efficacy among the three compounds. MZ2, which is structurally analogous to MZ1 except for a longer linker containing four PEG units, showed a weaker removal effect compared to MZ1. MZ3, containing an additional phenylalanine moiety between the linker and the VHL ligand, showed to be the least effective at removing BRD2 and BRD3. Together, the data demonstrate potent and effective degradation of BET proteins and suggested a preferential degradation effect on BRD4 over BRD2 and BRD3. The latter observation was unexpected given the parental compound JQ1 is a pan-BET inhibitor and our PROTACs bind with similar affinities to BET bromodomains. Nevertheless, the attractive opportunity to achieve single target selectivity prompted us to conduct further characterization.
To assess the compound dose- and time-dependent intracellular activities, HeLa cells were first treated with various concentrations of MZ1, MZ2, and MZ3 (Figure 2a). All three compounds showed concentration dependent BET removal activity with higher activity at higher concentrations. As in the initial experiment, MZ1 proved the most active compound, with more than 90% of all BET proteins being removed at compound concentration down to 1 μM. Remarkably, preferential removal of BRD4 over BRD2 and BRD3 was confirmed with all three compounds. Such preference is more prominent with treatment at lower concentrations, e.g., 0.1–0.5 μM. To study the activities of our PROTACs over time, HeLa cells were treated with 1 μM or 0.1 μM MZ1, and cellular BET protein levels were monitored in a time course experiment (Figure 2b for representative data with MZ1, see Figure S2 for additional data with other compounds and concentrations). Progressive removal of BET proteins over time was observed in all experimental setups, and BRD4 consistently exhibited the strongest and fastest reduction in protein level. Reassuringly, no BET protein degradation was observed in the presence of either DMSO or JQ1 (Figure S3) or cisMZ1 (Figure 3a). To verify whether the observation of preferential removal for BRD4 by our PROTACs can be observed in another cell line, the same study was carried out in U2OS osteosarcoma cells, and the same activity profile was observed (Figure S4). To visualize the BET protein degradation process, U2OS cells were transfected with a plasmid coding for a green fluorescent protein (GFP) tagged BRD4 protein, allowing fluorescence readout of BRD4 within the cell nuclei. Cells were induced to express GFP-BRD4 for 24 h and then were treated with either 5 μM MZ1 or 5 μM cisMZ1, and the fluorescence was observed over time. In the presence of the active compound MZ1, a complete depletion of the fluorescence signal was observed after just 3 h, whereas cisMZ1 caused no change in the fluorescence signal over the course of the experiment (Figure 2c, Figure S5 and Supporting Information videos a and b). These data confirmed that BRD4 is removed from the cell nuclei in a time dependent manner due to the presence of MZ1. Taken together, time and dose–response activity profiles revealed rapid and effective PROTAC-induced preferential degradation of BRD4 over BRD2 and BRD3.
Figure 2.

PROTACs induce concentration- and time-dependent selective degradation of BRD4. (a) HeLa cells treated for 24 h with different concentrations of MZ1 (panel I), MZ2 (panel II), and MZ3 (panel III). The bands observed in the BRD4 short isoform lane at a high concentration of each compound are correlated to nonspecific binding. (b) Time dependent treatment over 36 h of HeLa cells with 1 μM (panel I) and 100 nM (panel II) of MZ1. (c) U2OS cells transfected with GFP-BRD4 were treated with either 5 μM of MZ1 or cisMZ1 over a time course of 4 h. BRD4 degradation over time was followed by live fluorescence imaging.
Figure 3.

Mechanistic studies on PROTAC biological activity. (a) Time dependent treatment over 36 h of HeLa cells with 1 μM inactive compound cisMZ1. (b) HeLa cells treated with JQ1 or MZ1 at 1 μM in the absence or presence of the proteasome inhibitor MG132. (c) Time dependent treatment over 36 h of HeLa cells with 1 μM MZ1 observing the levels of the von Hippel-Lindau (VHL) protein. (d) HeLa cells treated with 100 μM CoCl2 as a hypoxia control or 0.1, 1, and 10 μM MZ1. (e) BRD4 protein levels were observed (panel I) with single treatment of MZ1 at t = 0 for 4 h and then exchange of media, (panel II) single treatment with MZ1 at t = 0 but no exchange of media, and (panel III) single treatment with 0.01% DMSO for 4 h and then exchange of media.
To gain mechanistic insights, the VHL and proteasome dependency of PROTAC-mediated protein degradation was first examined. cisMZ1 was unable to induce degradation of any of the BET proteins over time (Figure 3a), demonstrating that PROTAC efficacy is dependent on productive recruitment of VHL. Next, the reliance of the PROTAC-induced protein degradation on proteasome activity was assessed using proteasome inhibitor MG132. Treatment with MG132 completely abrogated MZ1-induced degradation of all BET proteins (compare lanes 3 and 6 in Figure 3b), establishing the expected proteasome-dependence of the approach. Interestingly, MG132 treatment in the absence of PROTAC showed no significant accumulation in BET protein levels, either alone or in combination with JQ1 (compare lanes 1 and 2 with 4 and 5 in Figure 3b, respectively), suggesting that basal proteasome activity level against BET proteins is negligible under those conditions and only becomes significant as a result of PROTAC treatment.
To further evaluate the biological activity of our compounds, we asked whether PROTAC treatment had any effect on the levels of its target E3 ligase (VHL) and on the level of HIF-1α, the natural substrate of VHL. VHL levels in the presence of MZ1 (1 μM) remained unaffected over the course of up to 36 h, thus indicating that the amount of E3 ligase is not influenced by MZ1 binding (Figure 3c). On the other hand, as the VHL ligand portions of our PROTACs occupy the same binding site on VHL that is used to recruit HIF-1α, PROTACs could block HIF-1α binding to VHL to an extent that it may lead to potential stabilization of HIF-1α within cells. For the approach herein described, this effect would not be desirable as up-regulation of HIF-1α transcriptional activity would confound the effects resulting from degradation of BET proteins and would be expected to result in induction of the hypoxic response, potentially giving rise to unwanted side effects. To assess whether any HIF-1α stabilization could be observed, HeLa cells were treated with MZ1 and with cobalt(II) chloride as a hypoxia mimicking positive control. Reassuringly, we could not observe any evidence of HIF-1α stabilization even at concentrations of MZ1 up to 10 μM, while clear HIF-1α stabilization is observed in the presence of CoCl2 (Figure 3d).
A number of non-BET potential off-targets of JQ1 have been recently reported, among which proteins DDB1 and RAD23B (hHR23b) were validated by proteome labeling and Western blotting.27 To assess whether MZ1 causes degradation of these off-targets, protein levels were examined in HeLa cells treated with MZ1 at 1 μM and 100 nM over a time course of 36 h, and no degradation was observed (Figure S6). Next, to determine whether the removal of BET proteins by PROTAC treatment is reversible, and to establish how long it would take for cells to reverse the effect, we treated HeLa cells for 4 h with 1 μM of MZ1, removed the compound from the media, and then monitored protein levels over a period of 48 h. The washed cells showed detectable recovery of intracellular BRD4 only by 20 h after washout, while in the absence of the wash step, no protein could be detected even after 48 h (Figure 3e, see Figure S7 for the same experiment monitoring time-dependent levels of BRD2 and BRD3). Taken together, these results demonstrate that PROTAC-induced protein degradation is strictly dependent on binding to VHL, on proteasome activity, and does not interfere with the normal endogenous levels of both VHL and HIF-1α. Furthermore, the degradation effect is not only rapid but also sustained and long lasting even upon removal of the compound.
BET inhibitors such as JQ1 influence the expression of an assortment of genes.28 Selective targeting of individual BET family members would be predicted to elicit distinct and more limited transcriptional responses, because the genome occupancy patterns of BET proteins are not identical.29 To evaluate the functional consequences of removing BET proteins using PROTACs, and in particular of inducing selective degradation of BRD4 over BRD2 and BRD3, we next monitored the mRNA expression profiles of a selection of cancer-related genes which respond to JQ1 treatment and BET protein inhibition: MYC, P21, AREG, FAS, TYRO3, and FGFR1. The dependence of MYC and P21 expression on BRD4 activity is well characterized. MYC stimulates cell cycle progression and is constantly expressed upon misregulation in cancer, thus leading to continuous overexpression of downstream MYC-dependent genes.30 In bone associated tumors31 as well as leukemia and lymphoma cell lines,32 JQ1 treatment or silencing of BRD4 resulted in downregulation of MYC. MYC represses transcription of the cell cycle CDK inhibitor P21, a tumor suppressor.33 Downregulation of MYC and consequent derepression of P21 promotes cell cycle arrest. In contrast to the well characterized BRD4 dependency of MYC and P21, FAS, which encodes a proapoptotic protein belonging to the tumor necrosis factor receptor family,34 is downregulated by depletion of BRD2,35 while for the growth factors AREG and FGFR1 as well as the protein tyrosine kinase TYRO3 little is known about any BET protein specific regulation. However, these four genes are known to strongly respond to treatment with JQ136 and therefore were included as a representative set of genes to compare between the pan-BET inhibitory effect caused by JQ1 and a selective BRD4 degradation caused by MZ1. Treatment with MZ1 at 100 nM for 24 h was chosen as this provided an optimal condition and the lowest effective concentration for achieving selective degradation of BRD4 over BRD2 and BRD3 and at the same time minimizing potential interference due to BET bromodomain inhibition (Figure 2a panel I and Figure 2b panel II). In addition, treatments with negative control compound VHL-1′ (Figure S8) lacking the JQ1 moiety, as well as with JQ1 itself, were also conducted to provide comparisons. Treatment of MZ1 resulted in down regulation of MYC, similar to JQ1, after 12 h (Figure S9), although MYC levels recovered after 24 h. Treatment with MZ1 and JQ1 resulted in similar upregulation of P21 and AREG both after 12 h (Figure S9) and 24 h (Figure 4a). Interestingly, in contrast to JQ1, which resulted in significant changes on FAS, TYRO3, and FGFR1, MZ1 showed more subtle and less significant effects on these genes relative to VHL-1′ (Figure 4a and Figure S9). We hypothesize that such differences observed in gene modulation may be the result of preferential degradation of BRD4 over the other two BET proteins caused by MZ1. To test this hypothesis, we suppressed individual BRD2, BRD3, or BRD4 genes using siRNA to mimic the protein removal effect (Figure S10) and analyzed the gene expression level of the target genes of interest (Figure 4b). While MYC, P21, and AREG levels were confirmed to be affected by suppression of BRD4, we found that FAS was downregulated upon suppression of BRD2 only but not BRD4 (Figure 4b), while FGFR1 is upregulated upon suppression of either BRD3 or BRD4. These results are consistent with preferential degradation of BRD4 over BRD2 and BRD3 by MZ1 and point to a more BRD4-selective pharmacological profile of MZ1 compared with pan-selective inhibitor JQ1.
Figure 4.

Selective degradation of BRD4 leads to a differential response between JQ1 and MZ1 on selected genes. mRNA expression profiles of MYC, P21, AREG, FAS, FGFR1, and TYRO3 upon treatment with PROTAC MZ1 and JQ1 were compared. (a) HeLa cells were treated with 100 nM MZ1, VHL-1′, or JQ1 or 0.01% DMSO vehicle control (Veh.) for 24 h. (b) To mimic the protein removal effect, HeLa cells were transfected with siRNA targeting individual BRD2, BRD3, or BRD4 or negative control siRNA and were harvested after 48 h. Quantitative PCR was performed to analyze relative gene expression level of treated HeLa cells using target specific primers. Gene expression levels relative to GAPDH were normalized to control treatment. The data shown represent the mean ± SEM (n = 3 technical replicates) of one experiment. Statistical significance compared to the control was determined with two-tailed t tests: *P < 0.05, **P < 0.01, ***P < 0.001, and n.s. = not significant.
In summary, we report a small molecule PROTAC approach achieving rapid, effective, and prolonged intracellular degradation of BET bromodomain proteins. The PROTAC-induced protein degradation is dependent on binding to VHL, is reversed upon blocking proteasome activity, and does not interfere with the endogenous, physiological levels of VHL and of its natural substrate HIF-1α. All investigated compounds showed preferential degradation of BRD4 over BRD2 and BRD3 at low concentrations. The downstream gene expression pattern resulting from treatment with our potent and selective PROTAC MZ1 is similar to JQ1 inhibition for BRD4-dependent genes MYC, P21, and AREG but not for FAS, FGFR1, and TYRO3. Our results suggest a different pharmacological response resulting from selectively depleting BRD4 with MZ1 compared to inhibiting the whole BET protein subfamily with JQ1. Given that no preference for binding the bromodomains of BRD4 over the highly homologous bromodomains of BRD2 and BRD3 was observed by ITC within the context of the purified proteins, we speculate that the observed selectivity could arise from preferential and more efficient polyubiquitination of lysine residues on the surface of BRD4 compared to those of BRD2 and BRD3. Alternatively or in addition, preferential direct interaction or reduced steric constraints between VHL and BRD4 compared to BRD2/3 may occur as a result of PROTAC binding, triggering a more productive formation of a VHL:PROTAC:BRD4 ternary complex. Elucidation of the molecular basis for the BRD4-selective activity of PROTACs will warrant further mechanistic investigation in the future. Our findings demonstrating effective and selective degradation of BRD4 with a PROTAC approach open up unprecedented opportunities to study the downstream physiological and pathological consequences of BRD4 modulation. It will allow determination of whether more selective pharmacological perturbations of BET protein function will have improved therapeutic efficacy, potentially leading to more efficient and specific new drugs in the future. Finally, potent chemical probes that bind to human bromodomains outside the BET subfamily are beginning to emerge,37 which could be similarly conjugated to a VHL ligand to induce selective intracellular degradation of their respective target bromodomain-containing proteins.
Methods
For detailed descriptions of synthetic and biological methods, see the Supporting Information.
Acknowledgments
This work was supported by awards to A.C. from the UK Biotechnology and Biological Sciences Research Council (BBSRC, grant BB/J001201/2 and David Phillips Fellowship BB/G023123/2) and the European Research Council (ERC, Starting Grant ERC-2012-StG-311460 DrugE3CRLs). Light microscopy is supported by a Wellcome Trust strategic award to the University of Dundee (097945/Z/11/Z). We thank P. Soares and S. Scaffidi for providing VHL ligand precursors, M. Ferguson, L. Guther and S. Damerow for providing access and assistance with the LI-COR Odyssey system, S. Swift and C. Thomson of the Dundee Imaging Facility for assistance with the light microscopy, and K. Airey of the Dundee MRC tissue culture facility for training and assistance. The University of Dundee and the authors have filed a patent application (GB1504314.4) related to the use of BET bromodomain targeting PROTACs to induce degradation of BET proteins.
Supporting Information Available
Additional figures, tables, materials and methods, and .avi videos. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.5b00216.
Author Contributions
A.C. conceived and supervised the project; M.Z., K.-H.C. and A.C. designed the research; M.Z. and K.-H.C. performed the experiments; M.Z., K.-H.C. and A.C. analyzed the data; and M.Z., K.-H.C. and A.C. wrote and edited the manuscript.
The authors declare no competing financial interest.
Around the time the "Just Accepted" version of this paper was published online, two independent reports have come out online describing pan-BET selective PROTAC compounds dBET1 (DOI: 10.1126/science.aab1433) and ARV-825 (DOI: 10.1016/j.chembiol.2015.05.009) that conjugate the same BET bromodomain recruiting ligand JQ1 to a ligand for a different E3 ligase, cereblon.
Supplementary Material
References
- Gallenkamp D.; Gelato K. A.; Haendler B.; Weinmann H. (2014) Bromodomains and their pharmacological inhibitors. ChemMedChem. 9, 438–464. [DOI] [PubMed] [Google Scholar]
- Belkina A. C.; Denis G. V. (2012) BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 12, 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J.; Shi J.; Wang E.; Rappaport A. R.; Herrmann H.; Sison E. A.; Magoon D.; Qi J.; Blatt K.; Wunderlich M.; Taylor M. J.; Johns C.; Chicas A.; Mulloy J. C.; Kogan S. C.; Brown P.; Valent P.; Bradner J. E.; Lowe S. W.; Vakoc C. R. (2011) RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratta M. G.; Schinzel A. C.; Zwang Y.; Bandopadhayay P.; Bowman-Colin C.; Kutt J.; Curtis J.; Piao H.; Wong L. C.; Kung A. L.; Beroukhim R.; Bradner J. E.; Drapkin R.; Hahn W. C.; Liu J. F.; Livingston D. M. (2015) An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc. Natl. Acad. Sci. U. S. A. 112, 232–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C.-W.; Coste H.; White J. H.; Mirguet O.; Wilde J.; Gosmini R. L.; Delves C.; Magny S. M.; Woodward R.; Hughes S. A.; Boursier E. V.; Flynn H.; Bouillot A. M.; Bamborough P.; Brusq J.-M. G.; Gellibert F. J.; Jones E. J.; Riou A. M.; Homes P.; Martin S. L.; Uings I. J.; Toum J.; Clément C. A.; Boullay A.-B.; Grimley R. L.; Blandel F. M.; Prinjha R. K.; Lee K.; Kirilovsky J.; Nicodeme E. (2011) Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J. Med. Chem. 54, 3827–3838. [DOI] [PubMed] [Google Scholar]
- Khan Y. M.; Kirkham P.; Barnes P. J.; Adcock I. M. (2014) Brd4 is essential for IL-1β-induced inflammation in human airway epithelial cells. PLoS One 9, e95051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Knapp S. (2012) The bromodomain interaction module. FEBS Lett. 586, 2692–2704. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. (2010) Selective inhibition of BET bromodomains. Nature 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E.; Jeffrey K. L.; Schaefer U.; Beinke S.; Dewell S.; Chung C.-W.; Chandwani R.; Marazzi I.; Wilson P.; Coste H.; White J.; Kirilovsky J.; Rice C. M.; Lora J. M.; Prinjha R. K.; Lee K.; Tarakhovsky A. (2010) Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boi M.; Gaudio E.; Bonetti P.; Kwee I.; Bernasconi E.; Tarantelli C.; Rinaldi A.; Testoni M.; Cascione L.; Ponzoni M.; Mensah A. A.; Stathis A.; Stussi G.; Riveiro M. E.; Herait P.; Inghirami G.; Cvitkovic E.; Zucca E.; Bertoni F. (2015) The BET Bromodomain inhibitor OTX015 affects pathogenetic pathways in pre-clinical B-cell tumor models and synergizes with targeted drugs. Clin. Cancer Res. 21, 1628–1638. [DOI] [PubMed] [Google Scholar]
- Gosmini R.; Nguyen V. L.; Toum J.; Simon C.; Brusq J.-M. G.; Krysa G.; Mirguet O.; Riou-Eymard A. M.; Boursier E. V.; Trottet L.; Bamborough P.; Clark H.; Chung C.-W.; Cutler L.; Demont E. H.; Kaur R.; Lewis A. J.; Schilling M. B.; Soden P. E.; Taylor S.; Walker A. L.; Walker M. D.; Prinjha R. K.; Nicodeme E. (2014) The discovery of I-BET726 (GSK1324726A), a potent tetrahydroquinoline ApoA1 up-regulator and selective BET bromodomain inhibitor. J. Med. Chem. 57, 8111–8131. [DOI] [PubMed] [Google Scholar]
- Mirguet O.; Lamotte Y.; Donche F.; Toum J.; Gellibert F.; Bouillot A.; Gosmini R.; Nguyen V. L.; Delannée D.; Seal J.; Blandel F.; Boullay A.-B.; Boursier E.; Martin S.; Brusq J.-M.; Krysa G.; Riou A.; Tellier R.; Costaz A.; Huet P.; Dudit Y.; Trottet L.; Kirilovsky J.; Nicodeme E. (2012) From ApoA1 upregulation to BET family bromodomain inhibition: Discovery of I-BET151. Bioorg. Med. Chem. Lett. 22, 2963–2967. [DOI] [PubMed] [Google Scholar]
- McLure K. G.; Gesner E. M.; Tsujikawa L.; Kharenko O. A.; Attwell S.; Campeau E.; Wasiak S.; Stein A.; White A.; Fontano E.; Suto R. K.; Wong N. C. W.; Wagner G. S.; Hansen H. C.; Young P. R. (2013) RVX-208, an inducer of ApoA-I in humans, is a BET bromodomain antagonist. PLoS One 8, e83190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z.; Gong Y.; Ma Y.; Lu K.; Lu X.; Pierce L. A.; Thompson R. C.; Müller S.; Knapp S.; Wang J. (2013) Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin. Cancer Res. 19, 1748–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinjha R. K.; Witherington J.; Lee K. (2012) Place your BETs: The therapeutic potential of bromodomains. Trends Pharmacol. Sci. 33, 146–153. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Knapp S. (2014) Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discovery 13, 337–356. [DOI] [PubMed] [Google Scholar]
- Baud M. G. J.; Lin-Shiao E.; Cardote T.; Tallant C.; Pschibul A.; Chan K.-H.; Zengerle M.; Garcia J. R.; Kwan T. T. L.; Ferguson F. M.; Ciulli A. (2014) A bump-and-hole approach to engineer controlled selectivity of BET bromodomain chemical probes. Science 346, 638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina K.; Crews C. M. (2010) Chemical inducers of targeted protein degradation. J. Biol. Chem. 285, 11057–11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corson T. W.; Aberle N.; Crews C. M. (2008) Design and applications of bifunctional small molecules: Why two heads are better than one. ACS Chem. Biol. 3, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyrus K.; Wehenkel M.; Choi E. Y.; Swanson H.; Kim K. B. (2010) Two-headed PROTAC: An effective new tool for targeted protein degradation. ChemBioChem. 11, 1531–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K. M.; Kim K. B.; Verma R.; Ransick A.; Stein B.; Crews C. M.; Deshaies R. J. (2003) Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol. Cell. Proteomics 2, 1350–1358. [DOI] [PubMed] [Google Scholar]
- Sakamoto K. M.; Kim K. B.; Kumagai A.; Mercurio F.; Crews C. M.; Deshaies R. J. (2001) Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A. 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.; Puppala D.; Choi E. Y.; Swanson H.; Kim K. B. (2007) Targeted degradation of the aryl hydrocarbon receptor by the PROTAC approach: A useful chemical genetic tool. ChemBioChem. 8, 2058–2062. [DOI] [PubMed] [Google Scholar]
- Schneekloth J. S.; Fonseca F. N.; Koldobskiy M.; Mandal A.; Deshaies R.; Sakamoto K.; Crews C. M. (2004) Chemical genetic control of protein levels: Selective in vivo targeted degradation. J. Am. Chem. Soc. 126, 3748–3754. [DOI] [PubMed] [Google Scholar]
- Hon W.-C.; Wilson M. I.; Harlos K.; Claridge T. D. W.; Schofield C. J.; Pugh C. W.; Maxwell P. H.; Ratcliffe P. J.; Stuart D. I.; Jones E. Y. (2002) Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 417, 975–978. [DOI] [PubMed] [Google Scholar]
- Galdeano C.; Gadd M. S.; Soares P.; Scaffidi S.; Van Molle I.; Birced I.; Hewitt S.; Dias D. M.; Ciulli A. (2014) Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel-Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem. 57, 8657–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Wang D.; Li L.; Pan S.; Na Z.; Tan C. Y. J.; Yao S. Q. (2014) “Minimalist” cyclopropene-containing photo-cross-linkers suitable for live-cell imaging and affinity-based protein labeling. J. Am. Chem. Soc. 136, 9990–9998. [DOI] [PubMed] [Google Scholar]
- Shi J.; Vakoc C. R. (2014) The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 54, 728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders L.; Guenther M. G.; Qi J.; Fan Z. P.; Marineau J. J.; Rahl P. B.; Lovén J.; Sigova A. A.; Smith W. B.; Lee T. I.; Bradner J. E.; Young R. A. (2014) Genome-wide localization of small molecules. Nat. Biotechnol. 32, 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretones G.; Delgado M. D.; León J. (2014) Myc and cell cycle control. Biochim. Biophys. Acta 1849, 506–516. [DOI] [PubMed] [Google Scholar]
- Lamoureux F.; Baud’huin M.; Rodriguez Calleja L.; Jacques C.; Berreur M.; Rédini F.; Lecanda F.; Bradner J. E.; Heymann D.; Ory B. (2014) Selective inhibition of BET bromodomain epigenetic signalling interferes with the bone-associated tumour vicious cycle. Nat. Commun. 5, 3511. [DOI] [PubMed] [Google Scholar]
- Mertz J. A.; Conery A. R.; Bryant B. M.; Sandy P.; Balasubramanian S.; Mele D. A.; Bergeron L.; Sims R. J. (2011) Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. U. S. A. 108, 16669–16674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane J.; Le H.-V.; Massagué J. (2002) Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 419, 729–734. [DOI] [PubMed] [Google Scholar]
- Strasser A.; Jost P. J.; Nagata S. (2009) The many roles of FAS receptor signaling in the immune system. Immunity 30, 180–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnilicová J.; Hozeifi S.; Stejskalová E.; Dušková E.; Poser I.; Humpolíčková J.; Hof M.; Staněk D. (2013) The C-terminal domain of Brd2 is important for chromatin interaction and regulation of transcription and alternative splicing. Mol. Biol. Cell 24, 3557–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore J. E.; Issa G. C.; Lemieux M. E.; Rahl P. B.; Shi J.; Jacobs H. M.; Kastritis E.; Gilpatrick T.; Paranal R. M.; Qi J.; Chesi M.; Schinzel A. C.; McKeown M. R.; Heffernan T. P.; Vakoc C. R.; Bergsagel P. L.; Ghobrial I. M.; Richardson P. G.; Young R. A.; Hahn W. C.; Anderson K. C.; Kung A. L.; Bradner J. E.; Mitsiades C. S. (2011) BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M.; Measures A. M.; Wilson B. G.; Cortopassi W. A.; Alexander R.; Höss M.; Hewings D. S.; Rooney T. P. C.; Paton R. S.; Conway S. J. (2015) Small molecule inhibitors of bromodomain–acetyl-lysine interactions. ACS Chem. Biol. 10, 22–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.