

Abstract
Hypertrophic cardiomyopathy (HCM) is the most common monogenic heart disease with a frequency as high as 1 in 200. In many cases, HCM is caused by mutations in genes encoding the different components of the sarcomere apparatus. HCM is characterized by unexplained left ventricular hypertrophy (LVH), myofibrillar disarray, and myocardial fibrosis. The phenotypic expression is quite variable. While the majority of patients with HCM are asymptomatic, serious consequences are experienced in a subset of affected individuals who present initially with sudden cardiac death (SCD) or progress to refractory heart failure (HF). The HCMR study is a National Heart Lung and Blood Institute (NHLBI)-sponsored 2750 patient, 41 site, international registry and natural history study designed to address limitations in extant evidence to improve prognostication in HCM (NCT01915615). In addition to collection of standard demographic, clinical, and echocardiographic variables, patients will undergo state-of-the-art cardiac magnetic resonance (CMR) for assessment of left ventricular (LV) mass and volumes as well as replacement scarring and interstitial fibrosis. In addition, genetic and biomarker analysis will be performed. HCMR has the potential to change the paradigm of risk stratification in HCM, using novel markers to identify those at higher risk.
Keywords: Hypertrophic cardiomyopathy, magnetic resonance imaging, biomarkers, genetics, outcomes, fibrosis
Background
Hypertrophic cardiomyopathy (HCM) is the most common monogenic heart disease with a frequency as high as 1 in 200(1;2). Sarcomere gene mutations are an important cause of disease. HCM is characterized by unexplained left ventricular hypertrophy (LVH), myofibrillar disarray, and myocardial fibrosis(3;4). Phenotypic expression is highly variable. While the majority of patients with HCM are asymptomatic, the prognosis is poor in a subset of affected individuals who present initially with sudden cardiac death (SCD) or rapidly progress to heart failure (HF).
Current clinical methods to assess risk of these adverse events and to target therapy are somewhat limited. The currently accepted risk predictors for SCD as indication for primary prevention with implantable cardioverter defibrillators (ICD’s) include: (1) family history of HCM-related SCD, (2) unexplained recent syncope, (3) massive left ventricular hypertrophy (LVH) (thickness ≥30 mm), (4) multiple bursts of nonsustained ventricular tachycardia on ambulatory electrocardiography and (5) hypotensive or attenuated blood pressure response to exercise(1). Howevermost patients with HCMR will not suffer SCD, and events still occur in patients with no risk factors, currently considered low risk, or with only 1 risk factor, (5). ICD placement for primary prevention based on these 5 risk factors was associated with appropriate therapy rates of 17% over 5 years and rates of inappropriate shocks and complications of 27% and 20%, respectively(5). Thus, improvement is needed over and above current clinical risk stratification for SCD in order to reduce morbidity, mortality, and lifetime costs.
Cardiovascular magnetic resonance (CMR) has emerged as a powerful tool for the diagnosis of HCM, and has been recognized for its potential utility for improved risk stratification (6). As myocardial fibrosis may underlie the arrhythmogenic substrate as well as promote development of HF, recent studies have focused on late gadolinium enhancement (LGE) by CMR, a marker of fibrosis, as an independent risk factor for adverse outcomes. A meta-analysis suggested that the presence of LGE (in up to 2/3 of cases) is associated with cardiac death (odds ratio (OR) 2.9, p<0.05), HF (OR 5.7, p<0.01), and a trend for SCD (OR 2.4, p=0.09)(7). A retrospective study of 1293 patients demonstrated that LGE>15% of LV mass was associated with a 2-fold increase in SCD risk (8), suggesting that the amount of LGE rather than just its presence may be an important risk marker. LGE identifies replacement and interstitial fibrosis(9), but post-mortem analysis indicates that diffuse fibrosis is also present in other regions not identified by LGE. Recent advances in T1 mapping with CMR (10) have enabled validation of diffuse interstitial fibrosis measurements in HCM (11) and demonstration of increases, even in genotype (+), phenotype (−) patients(12). Despite these advances, recent guidelines do not recommend the use of CMR for risk stratification(13;14).
For over two decades, it has been recognized that the majority of HCM cases are inherited as an autosomal dominant trait, caused by mutations in genes encoding cardiac sarcomeric proteins(4;15). The most commonly affected genes include those encoding the β myosin heavy chain (MYH7), cardiac myosin binding protein C (MYBPC3), troponin T, troponin I, myosin regulatory light chain, myosin essential light chain, α tropomyosin, and cardiac α actin. However, within sarcomeric HCM, the relationship between genetic mutation, disease phenotype and clinical outcomes remains poorly understood. In addition, the majority of patients with a clinical diagnosis of HCM appear not to have sarcomere mutations, particularly if there is no incidence of familial disease. This group is likely to be of mixed etiology, and early data suggest a more favorable natural history(16)..
Since myocardial fibrosis is a key feature of HCM, collagen metabolism and turnover may have an impact on myocardial hemodynamics and remodeling, and ultimately, prognosis. Sarcomere-mutation carriers with overt HCM, and importantly, mutation carriers without LVH have higher serum levels of C-terminal propeptide of type 1 procollagen (P1CP), a marker of collagen synthesis(17), relative to controls. This pro-fibrotic state may represent an important risk marker. Moreover, sarcomere mutations may render the cardiomyocyte more susceptible to injury, including through ischemia(18). Thus, measures of myocardial injury and wall stress may be important markers of risk for SCD and HF.
HCMR was designed as the first prospective multinational registry to identify novel prognostic markers in HCM including CMR markers of fibrosis, genetic markers, and biomarkers. In this way, risk stratification including decisions on ICD implantation and/or HF prevention or treatment may be substantially improved and potential targets for disease-modifying therapeutic intervention using existing or novel drug therapy may be demonstrated..
Study Design
Overview
A prospective cohort of 2750 HCM patients will undergo a CMR and blood draw for genetics and biomarker evaluation and be followed for up to 5 years with hopes to identify novel prognostic factors of cardiovascular outcome.
Executive Committee and Core Laboratories
The co-principal investigators of the study are Christopher M. Kramer MD, University of Virginia Health System and Stefan Neubauer MD, University of Oxford. The data coordinating center at Christiana Center for Outcomes Research is led by William S. Weintraub MD. ICON Medical Imaging is the contract research organization administering the study. Other members of the executive committee and core laboratories are listed on Figure 1. The study is being monitored by an independent Observational Data and Safety Monitoring Board (OSMB) appointed by the National Heart, Lung and Blood Institute. The OSMB reviews all aspects of study conduct, data collection and analyses and makes recommendations to the investigators and the Institute.
Figure 1.
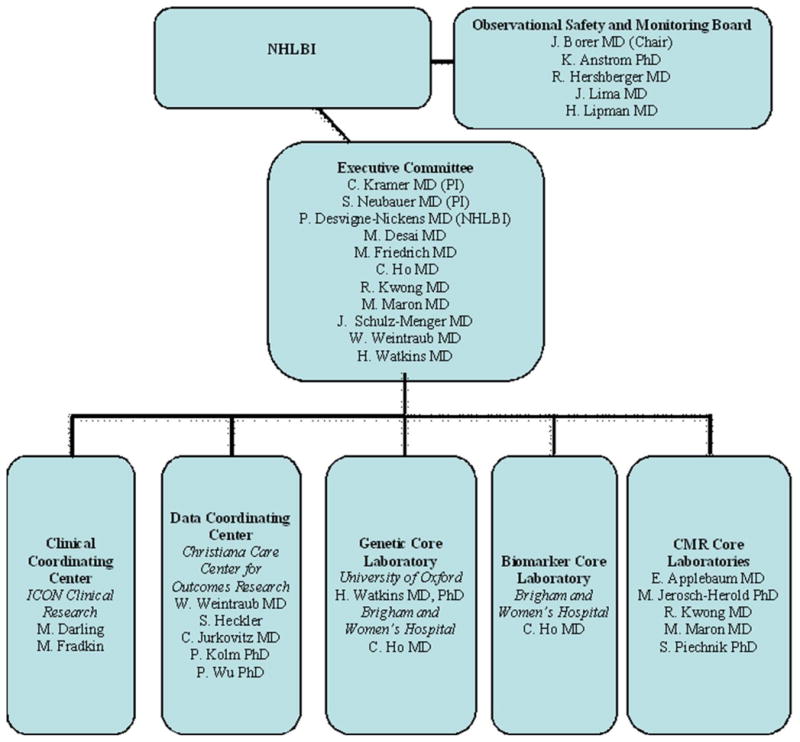
Diagram of the organizational structure of the HCMR study.
Inclusion/Exclusion Criteria
Patients included are ages 18–65 with an established diagnosis of HCM defined as unexplained LVH (wall thickness >15cm) without cavity dilatation or a predisposing cause (hypertension, aortic stenosis, etc.)(14). Other causes of infiltrative/hypertrophic cardiomyopathies such as amyloidosis, sarcoidosis, Fabry disease, Danon disease, or Noonan’s syndrome will be excluded. Patients older than 65 are excluded as they have lower event rates related to HCM and more hypertensive heart disease, which can mimic the phenotypic presentation of LVH with or without LV outflow obstruction, and competing mortality risks, in particular from coronary artery disease and cancer.
Additional exclusion criteria are 1) prior septal myectomy or alcohol septal ablation, 2) prior myocardial infarction or known CAD 3) incessant ventricular arrhythmias, 4) inability to lie flat, 5) contraindication to CMR including pacemakers, defibrillators, intraocular metal, certain types of intracranial aneurysm clips, severe claustrophobia, and Stage IV/V chronic kidney disease, 6) diabetes mellitus with end organ damage, 7) pregnancy, or 8) inability to provide informed consent.
Patient Enrollment
2750 patients with an established or new diagnosis of HCM will be enrolled at a total of 41 sites in the U.S., Canada, and Europe (Tables 1 and 2) from April 2014 through June 2016. The sites were chosen as experienced centers of excellence with focused care of HCM patients as well as state-of-the-art CMR capabilities. Emphasis will be placed on recruiting across the risk spectrum (as judged by classical risk factors), including high-risk patients referred for ICD implantation as well as recruiting high percentages of minorities and women. After signing consent, patients will have blood drawn for genetic and biomarker analysis and undergo CMR. Data regarding baseline demographics and clinical variables will be gathered from clinical records including results of Holter monitoring and stress testing, as available. Clinical echocardiographic data will be recorded including LV outflow tract gradient and Doppler variables.
Table 1.
Listing of North American sites for the HCMR study.
| North American Sites | City, State |
|---|---|
| University of Virginia Health System | Charlottesville, VA |
| Brigham and Women’s Hospital | Boston, MA |
| Cleveland Clinic | Cleveland, OH |
| Duke University Hospital | Durham, NC |
| Houston Methodist Hospital | Houston, TX |
| Johns Hopkins Hospital | Baltimore, MD |
| Montreal Heart Institute | Montreal, Canada |
| Mayo Clinic | Rochester, MN |
| Morristown Medical Center | Morristown, NJ |
| New York Presbyterian/Weill Cornell | New York, NY |
| Northwestern University | Chicago, IL |
| Oregon Health Sciences University | Portland, OR |
| St. Luke’s Hospital/Mt. Sinai | New York, NY |
| Stanford University Hospital | Palo Alto, CA |
| Toronto General Hospital | Toronto, Canada |
| Tufts University | Boston, MA |
| University of Alberta | Calgary, Canada |
| University of Michigan | Ann Arbor, MI |
| University of Pennsylvania | Philadelphia, PA |
| Yale University | New Haven, CT |
Table 2.
Listing of European sites.
| European Sites | City, Country |
|---|---|
| University of Oxford | Oxford, UK |
| London Chest Hospital | London, UK |
| Glenfield Hospital | Leicester, UK |
| Royal Infirmary of Edinburgh | Edinburgh, UK |
| University of Aberdeen | Aberdeen, UK |
| St. George’s Hospital | London, UK |
| University of Leeds | Leeds, UK |
| University Hospital Birmingham | Birmingham, UK |
| University of Glasgow | Glasgow, UK |
| Royal Brompton Hospital | London, UK |
| King’s College, St. Thomas’ Hospital | London, UK |
| University Hospital, Southampton | Southampton, UK |
| Sapienza University of Rome | Rome, Italy |
| University of Bologna | Bologna, Italy |
| University Vita-Salute San Raffaele | Milan, Italy |
| Azienda Ospedaliero-University Careggi | Florence, Italy |
| Charite | Berlin, Germany |
| Universitats Klinikum Heidelberg | Heidelberg, Germany |
| Robert-Bosch-Krankenhaus | Stuttgart, Germany |
| VU University Medical Center | Amsterdam, NL |
| Erasmus MC | Rotterdam, NL |
CMR methods
All patients will undergo the entire CMR protocol to assure the correct assessment of LV volumes, mass, hypertrophy distribution, LGE (regional, patchy fibrosis), and measurement of diffuse fibrosis using native and post-contrast T1 mapping at specific time points. CMR will be performed at 1.5 or 3 Tesla on MR systems from the 3 primary vendors (General Electric, Philips Medical Systems, Siemens Healthcare) using multi-channel channel phased-array chest coils and electrocardiographic (ECG) gating. An example of the direct CMR outputs is shown in Figure 2. Table 3 lists the hypothesized roles of the respective CMR measures and derived information in HCM.
Figure 2.
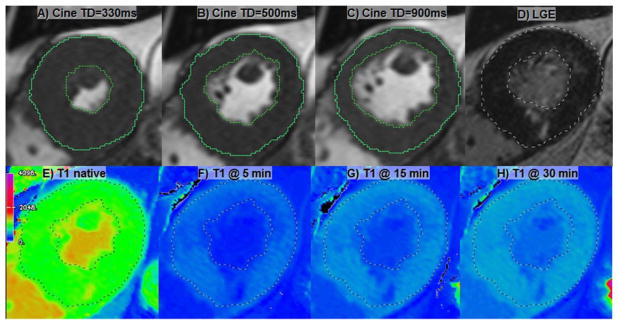
An illustration of the basic CMR outputs in HCMR study. A–C) Selected mid short-axial cine frames with cardiac delay times corresponding to A) systolic, B) mid-diastolic and C) end-diastolic frames. D) LGE image. E) Native T1 map as displayed on Siemens platform. Currently General Electric and Philips ShMOLLI acquisitions require off-line processing. F–H) Postcontrast T1 maps at specified time points.
Table 3.
CMR endpoints for HCMR
| CMR measure | Derived Information | Hypothesized Roles in HCM |
|---|---|---|
| CMR cine | LV+RV mass; regional wall-thickness; pattern of hypertrophy | Marker of disease severity; relation to genotype; marker of both SCD and HF |
| CMR cine | LV and RV volumes (end-diastolic and end-systolic | Marker of progression to HF |
| CMR cine | Systolic function (ejection fraction, regional wall thickening) | Marker of progression to HF |
| Late gad enhancement | Presence and localization of scar; Pattern of scar (patchy, confluent, etc.); | Potential marker of SCD and HF |
| T1 mapping pre and post-Gd | Extracellular matrix expansion | Marker of diffuse interstitial fibrosis; Potential marker of SCD and progression to HF |
After rapid localization of the heart, short axis cine steady state free precession imaging (SSFP) will be performed covering the whole heart in 8mm thick slices. Baseline T1 mapping will be performed in 3 short axis slices representing 16 of the 17 AHA segments. The Shortened Modified Look-Locker Inversion recovery technique (ShMOLLI) will be used as the recommended standard(19). Gadolinium contrast will then be infused intravenously at a dose of 0.15 mmol/kg. Long-axis function by SSFP cine imaging will then be obtained. Post-contrast T1 mapping acquisitions will be performed in the same 3 slices as pre-contrast at 5, 14, and 29 minutes post-contrast. (Figure 2E–H). LGE imaging will be acquired in the same long axis and short-axis stack locations beginning at minute 17 post-contrast with a 2D breath-hold, segmented inversion-recovery sequence (inversion time (TI) optimized by the Look-Locker sequence (TI scout) to null normal myocardium). Total imaging time will be approximately 60 minutes.
CMR Image Analysis
Analysis of CMR data will be coordinated by the CMR Core Lab led by Raymond Kwong, MD, MPH and Michael Jerosch-Herold, PhD. Cine and extracellular volume measurements will be performed at the Brigham. LGE image analysis will be performed at the PERFUSE CMR Core Laboratory/Beth Israel Deaconess Medical Center overseen by Evan Appelbaum MD. Native T1 mapping analysis will be led by Stefan Piechnik, PhD, at the University of Oxford. Images will be stored centrally at the CMR Core Lab and remote access will be granted to the other core laboratories.
Commercially available software (QMassMR, Medis Inc., Leiden, NL) will be used for analysis of all CMR images (cine, T1 maps, and LGE). LV mass, volumes, wall thickness and thickening will be measured according to SCMR standards(20). For LGE images, 3 distinct quantification methods will be performed to determine the most reproducible. Quantification will be performed according to SCMR standards(20). The location and pattern of LGE will be classified: Focal (1 or 2 LV segments), multi-focal (≥2 areas of noncontiguous LGE each occupying 1 or 2 segments), diffuse (LGE involving ≥3 contiguous segments) and extensive (contiguous LGE occupying ≥75% of the LV on one or more short-axis images).
T1 quantification will be initially performed on a segmental basis, resulting in multiple T1 measurements (one pre- and 3 post-contrast) calculated as averages of ShMOLLI T1 maps. Gadolinium partition coefficient λ will be calculated segmentally and globally by linear least squares regression of R1 (=1/T1) relaxivity changes in myocardium, against R1 changes in the blood pool, and converted to extracellular volume (ECV) using the patient’s hematocrit (Hct) (10). T1 calibration will be performed as part of the study. Each participating center is given a dedicated calibration phantom containing 9 compartments and will scan the phantoms using reference protocols in order to track T1 measurement stability.
Genetic Testing
Blood samples for genomic DNA analysis will be collected from each subject and batch tested with a standardized genetic screening protocol designed to optimize yield (by including all the firmly validated HCM disease genes) and to utilize state-of-the-art next generation sequencing. All DNA extractions and sequencing analyses will be performed at the Oxford Regional Genetics Laboratory using a protocol validated for the UK National Health Service. This will ensure consistency and standardization as well as a very large database for discriminating disease-causing from innocent polymorphisms. The genetics core laboratory will be headed by Hugh Watkins MD, PhD.
The complete coding sequence, and flanking splice-site sequences, will be analyzed of the sarcomeric HCM-genes (MYH7, MYPBC3, TNNT2, TNNI3, MYL2, MYL3, ACTC1, TPM1, CSRP3) as well as genes encoding some clinically related disorders, i.e. “phenocopies” (PRKAG2, GLA, LAMP2, FHL1). Sequencing will be achieved using a strategy of long PCR amplification and/or array capture and a next generation sequencing platform. Interpretation of sequence variants will be by standard, clinically adopted, criteria using existing mutation databases and literature, dbSNP, and standard algorithms to evaluate conservation and predicted functional impact, reinforced where possible with segregation data from the affected family.
Adequate numbers will be available to compare sarcomeric with non-sarcomeric HCM (with the expectation that sarcomeric HCM will have a higher event rate) and to compare the numerically important subgroups of HCM attributable to MYH7, MYBPC, and TNNT2 mutations. Genetic identification of related, but distinct, disorders (such as syndromic cardiomyopathies and storage disorders) will likely only relate to about 5% of participants but clinically important differences are expected in natural history, clinical/imaging phenotypes, and inheritance patterns in these groups compared to sarcomeric HCM.
Serum Biomarkers
Blood samples will be transported on ice, processed within 60 minutes of phlebotomy, and stored at −70°C until batched assay at the endo of the study period. The HCMR Biomarker Core Laboratory at Brigham and Women’s Hospital headed by Carolyn Ho MD will coordinate standardized sample collection, provide long-term sample storage, and perform analyses. Validated, commercially-available immunoassays will be used to assess metrics of collagen metabolism, myocardial injury, and hemodynamic stress. These analyses will include measurement of carboxy-terminal propeptide of procollagen type I (P1CP), matrix metalloproteinase (MMP-1), tissue inhibitor of metalloproteinase (TIMP-1), MMP-1:TIMP-1 ratio, the carboxy-terminal telopeptide of type I collagen (CITP), bone alkaline phosphatase (BAP), and galectin-3 (Gal-3) as well as other markers that may reflect collagen metabolism in HCM. New generation ultrasensitive cardiac troponin I (cTnI) assays will be used to detect reversible myocardial injury(21;22). Amino- terminal propeptide of B-type natriuretic peptide (NT-proBNP) and serum soluble ST2 levels will also be measured to similarly test for surrogate evidence of increased filling pressures or myocardial wall stress. The final selection of assays to be run will be directed by state-of-the-art knowledge available at the end of the study period. Remaining samples will be banked to allow for future biomarker discovery and validation investigations.
Data Management and Statistical Analysis
Patients will be followed for a minimum of 3 years and maximum of 5 years depending on the time of their study entry. Follow-up will be conducted by each individual site and will consist of annual telephone follow-up and interview with acquisition of any hospital records as necessary as well as review of the National Death Index. Two percent dropout per year is expected, yielding a final group of 2500 patients. The primary endpoint of this prospective study is the composite of cardiac death (SCD and HF death), aborted SCD including appropriate ICD firing, and need for heart transplantation. Secondary endpoints will include all-cause mortality, ventricular tachyarrhythmias, septal myectomy or alcohol ablation, hospitalization for heart failure, atrial fibrillation, and stroke. Relevant hospital and physician office records are gathered. An events committee will review all primary data regarding the endpoints including formal adjudication of ICD events.
The data management and statistical analysis are being performed at the Data Coordinating Center (CCC) at Christiana Center for Outcomes Research. Clinical data are entered in to an on-line data management system. Upon entry, data undergo a series of range and quality checks on entry. Data are sent to the DCC by ICON on a monthly basis and a missing data report is generated and reported back to the sites. Blood samples and images are tracked to make sure that they are collected and transferred to the core laboratories and that the blood and image data can be properly identified and integrated with the clinical data.
Anticipating a 2% dropout rate, a total of 2,750 HCM patients will be enrolled to obtain 2,500 patients available for analysis. Over a three year period, the primary composite endpoint event rate is estimated at 4–5% or 100–125 events in the patient sample. Because the anticipated event rate is small and the number of potential predictive risk factors is large, commonly used regression methods may not result in valid models. (23;24). Principal components analysis (PCA) will be used to create linear combinations of clinical, imaging, genetic, and biomarker variables to decrease the number of risk factors for inclusion in a prediction model. Approximately 10–15 variables from each of the categories will be selected to include in PCA. These will be selected based on expert clinical judgment. The most important components would be used in a predictive model of the outcome(25). The distribution of continuous variables will be assessed for normality. Appropriate transformations (e.g., logarithm, root) will be made if necessary.
The composite endpoint will be analyzed by multilevel (site included as a random effect) Cox proportional hazards regression. Time-to-event will be calculated from enrollment to endpoint occurrence (first occurrence for non-fatal events). Patients not experiencing the endpoint will be censored at the time of their last known follow-up. Link tests and Schoenfeld residuals will be used to assess the proportional hazards assumption(26). Cox-Snell residuals will be used to assess overall model goodness-of-fit.
Models will be validated by bootstrap methods to estimate bias-corrected calibration and discrimination indices(23). A second strategy will use penalized Cox regression to reduce overfitting when event rates are small (27;28). Models will be used to develop a prediction algorithm that rapidly calculates risk on a hand-held electronic device.
Missing data can potentially bias results of regression models as well as PCA. Missing data will be assessed by age, gender, race, and site and compared via Poisson or negative binomial regression to determine similarity. Statistical methods for imputing missing data may include multiple imputation assuming data are missing at random (MAR), as well as selection or pattern mixture models assuming data are not missing at random (NMAR)(29).
Discussion
HCMR is planned as a natural history study of 2750 patients with clinically diagnosed In addition to baseline collection of demographic data and traditional clinical risk factors, markers from CMR, genotyping, and serum biomarkers will be assessed to understand the relationship between these risk markers and clinical outcome, providing novel insights into disease progression and risk. This is the largest such prospective outcomes study ever performed in this disease. The study is powered to identify risk markers (imaging, serum biomarkers, and genetic beyond standard clinical risk factors) that predict the primary endpoint, which will be cardiac death (including SCD and HF death), aborted SCD (appropriate discharge of an implantable cardioverter-defibrillator), and need for heart transplantation. This study will enable development of a predictive model that will help to identify patients at risk as well as patients for future clinical trials to prevent SCD and HF. In addition, it will identify surrogate endpoints to monitor treatment response in HCM.
The study was funded in July of 2013. The first patient was enrolled in April 2014 and as of April 2015, 35 of 41 sites have been initiated and 457 patients enrolled (77% of projected at this time point).
Limitations
One limitation is that the primary endpoint used is a composite of cardiac death (SCD and HF death), aborted SCD including appropriate ICD firing, and need for heart transplantation. These reflect a mix of different pathophysiologies. However, they were chosen as they are the critical events determining serious morbidity and mortality in HCM patients. We have combined them to ensure adequate power, but will also examine each individually as secondary endpoints. To power the study using any one of these endpoints would be prohibitive in terms of the sample size.
Another limitation is that no validation set of patients is planned currently. HCM patients with previously implanted ICD’s are excluded from entry into the study. These patients are high risk and could yield important data regarding appropriate ICD discharge, which could then be related to imaging, genetic and biomarker data. However, in addition to safety concerns which can be overcome, imaging patients with devices would introduce artifacts that interfere with optimal CMR, especially for T1 mapping and LGE imaging. We will capture data on newly enrolled patients who subsequently receive ICD’s. Another excluded patient group is that with either prior septal myectomy or alcohol septal ablation. However, these subgroups have clearly altered myocardial pathology that changes their natural history for reasons that may be distinct from genetic drivers. In addition, these prior therapies significantly alter CMR findings and biomarker results. Patients who undergo these procedures after enrollment will be included and the procedures documented as secondary endpoints.
Different therapies chosen at different recruiting sites could introduce a bias. However, the sites selected to participate are HCM centers of excellence and as such follow international guideline-based care. To account for potential differences we will use a hierarchical model considering sites as a random effect. Age 65 was chosen as an upper age limit. This may reduce the incidence of HF events although most SCD events occur at a younger age. This cutoff was used to increase the homogeneity of the cohort and reduce potential confounding events in an older population such as CAD. Age will be a variable included in the statistical analysis as discussed above. Including more than one member of a family might introduce a bias. This will be taken into account in the statistical modeling. In addition, only 5 members from any one family are allowed to enroll.
Conclusion
HCMR has the potential to change the paradigm of risk stratification in HCM using novel imaging, genetic, and biomarkers. This may lead to improved patient care and identification of optimal candidates for new therapies for this disease.
Acknowledgments
Funding sources: National Heart Lung Blood Institute U01HL117006-01A1, National Institute of General Medical Sciences U54-GM10494, and Oxford NIHR Biomedical Research Centre
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maron BJ. Contemporary insights and strategies for risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Circulation. 2010;121:445–456. doi: 10.1161/CIRCULATIONAHA.109.878579. [DOI] [PubMed] [Google Scholar]
- 2.Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65:1249–1254. doi: 10.1016/j.jacc.2015.01.019. [DOI] [PubMed] [Google Scholar]
- 3.Maron BJ. Hypertrophic Cardiomyopathy. JAMA. 2002;287(10):1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 4.Watkins H, Ashrafian H, Redwood C. Inherited Cardiomyopathies. N Engl J Med. 2011;364(17):1643–1656. doi: 10.1056/NEJMra0902923. [DOI] [PubMed] [Google Scholar]
- 5.Maron BJ, Spirito P, Shen WK, Haas TS, Formisano F, Link MS, et al. Implantable Cardioverter-Defibrillators and Prevention of Sudden Cardiac Death in Hypertrophic Cardiomyopathy. JAMA. 2007;298(4):405–412. doi: 10.1001/jama.298.4.405. [DOI] [PubMed] [Google Scholar]
- 6.Maron M. Clinical Utility of Cardiovascular Magnetic Resonance in Hypertrophic Cardiomyopathy. J Cardiovasc Magn Reson. 2012;14(1):13. doi: 10.1186/1532-429X-14-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green JJ, Berger J, Kramer CM, Salerno M. Prognostic value of cardiac magnetic resonance late gadolinium enhancement in clinical outcomes for hypertrophic cardiomyopathy. JACC Cardiovasc Imaging. 2012;5:370–377. doi: 10.1016/j.jcmg.2011.11.021. [DOI] [PubMed] [Google Scholar]
- 8.Chan RH, Maron BJ, Olivotto I, Pencina MJ, Assenza GE, Haas T, et al. Prognostic Value of Quantitative Contrast-Enhanced Cardiovascular Magnetic Resonance for the Evaluation of Sudden Death Risk in Patients With Hypertrophic Cardiomyopathy. Circulation. 2014;130(6):484–495. doi: 10.1161/CIRCULATIONAHA.113.007094. [DOI] [PubMed] [Google Scholar]
- 9.Moravsky G, Ofek E, Rakowski H, Butany J, Williams L, Ralph-Edwards A, et al. Myocardial Fibrosis in Hypertrophic Cardiomyopathy: Accurate Reflection of Histopathological Findings by CMR. JACC: Cardiovasc Imaging. 2013;6(5):587–596. doi: 10.1016/j.jcmg.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 10.Jerosch-Herold M, Sheridan DC, Kushner JD, Nauman D, Burgess D, Dutton D, et al. Cardiac magnetic resonance imaging of myocardial contrast uptake and blood flow in patients affected with idiopathic or familial dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. 2008;295(3):H1234–H1242. doi: 10.1152/ajpheart.00429.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flett AS, Hayward MP, Ashworth MT, Hansen MS, Taylor AM, Elliott PM, et al. Equilibrium Contrast Cardiovascular Magnetic Resonance for the Measurement of Diffuse Myocardial Fibrosis. Circulation. 2010;122(2):138–144. doi: 10.1161/CIRCULATIONAHA.109.930636. [DOI] [PubMed] [Google Scholar]
- 12.Ho CY, Abbasi SA, Neilan TG, Shah RV, Chen Y, Heydari B, et al. T1 Measurements Identify Extracellular Volume Expansion in Hypertrophic Cardiomyopathy Sarcomere Mutation Carriers With and Without Left Ventricular Hypertrophy. Circ Cardiovasc Imaging. 2013;6(3):415–422. doi: 10.1161/CIRCIMAGING.112.000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur Heart J. 2014;35(39):2733–2779. doi: 10.1093/eurheartj/ehu284. [DOI] [PubMed] [Google Scholar]
- 14.Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Executive Summary. J Am Coll Cardiol. 2013;58(25):2703–2738. doi: 10.1016/j.jacc.2011.10.825. [DOI] [PubMed] [Google Scholar]
- 15.Kaski JP, Syrris P, Esteban MTT, Jenkins S, Pantazis A, Deanfield JE, et al. Prevalence of Sarcomere Protein Gene Mutations in Preadolescent Children With Hypertrophic Cardiomyopathy. Circ Cardiovasc Genet. 2009;2(5):436–441. doi: 10.1161/CIRCGENETICS.108.821314. [DOI] [PubMed] [Google Scholar]
- 16.Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, et al. Myofilament Protein Gene Mutation Screening and Outcome of Patients With Hypertrophic Cardiomyopathy. Mayo Clin Proc. 2008;83(6):630–638. doi: 10.4065/83.6.630. [DOI] [PubMed] [Google Scholar]
- 17.Ho CY, Lopez B, Coelho-Filho OR, Lakdawala NK, Cirino AL, Jarolim P, et al. Myocardial Fibrosis as an Early Manifestation of Hypertrophic Cardiomyopathy. N Engl J Med. 2010;363(6):552–563. doi: 10.1056/NEJMoa1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olivotto I, Girolami F, Sciagr+á R, Ackerman MJ, Sotgia B, Bos JM, et al. Microvascular Function Is Selectively Impaired in Patients With Hypertrophic Cardiomyopathy and Sarcomere Myofilament Gene Mutations. J Am Coll Cardiol. 2011;58(8):839–848. doi: 10.1016/j.jacc.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 19.Piechnik S, Ferreira V, Dall’Armellina E, Cochlin L, Greiser A, Neubauer S, et al. Shortened Modified Look-Locker Inversion recovery (ShMOLLI) for clinical myocardial T1-mapping at 1.5 and 3 T within a 9 heartbeat breathhold. J Cardiovasc Magn Reson. 2010;12(1):69. doi: 10.1186/1532-429X-12-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schulz-Menger J, Bluemke DA, Bremerich J, Flamm SD, Fogel MA, Friedrich MG, et al. Standardized image interpretation and post processing in cardiovascular magnetic resonance: Society for Cardiovascular Magnetic Resonance (SCMR) Board of Trustees Task Force on Standardized Post Processing. J Cardiovasc Magn Reson. 2013;15(1):35. doi: 10.1186/1532-429X-15-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Todd J, Freese B, Lu A, Held D, Morey J, Livingston R, et al. Ultrasensitive Flow-based Immunoassays Using Single-Molecule Counting. Clin Chem. 2007;53(11):1990–1995. doi: 10.1373/clinchem.2007.091181. [DOI] [PubMed] [Google Scholar]
- 22.Sabatine MS, Morrow DA, de Lemos JA, Jarolim P, Braunwald E. Detection of acute changes in circulating troponin in the setting of transient stress test-induced myocardial ischaemia using an ultrasensitive assay: results from TIMI 35. Eur Heart J. 2009;30:162–169. doi: 10.1093/eurheartj/ehn504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrell F., Jr . Regression Modeling Strategies With Applications to Linear Models, Logistic Regression and Survival Analysis. New York: Springer; 2001. [Google Scholar]
- 24.Peduzzi P, Concato J, Feinstein AR, Holford TR. Importance of events per independent variable in proportional hazards regression analysis II. Accuracy and precision of regression estimates. J Clin Epidemiol. 1995;48(12):1503–1510. doi: 10.1016/0895-4356(95)00048-8. [DOI] [PubMed] [Google Scholar]
- 25.Witten DM, Tibshirani R. Testing significance of features by lassoed principal components. Ann Appl Stat. 2008;3:986–1012. doi: 10.1214/08-AOAS182SUPP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schoenfeld D. Partial residuals for the proportional hazards regression model. Biometrica. 1982;69:239–241. [Google Scholar]
- 27.Goeman JJ. L1 penalized estimation in the Cox Proportional Hazards Model. Biom J. 2010;52(1):70–84. doi: 10.1002/bimj.200900028. [DOI] [PubMed] [Google Scholar]
- 28.Ambler G, Seaman S, Omar RZ. An evaluation of penalised survival methods for developing prognostic models with rare events. Statist Med. 2012;31(11–12):1150–1161. doi: 10.1002/sim.4371. [DOI] [PubMed] [Google Scholar]
- 29.Molenberghs G, Kenward MG. Missing Data in Clinical Studiees. West Sussex, UK: John Wiley & Sons; 2007. [Google Scholar]