

The current methods of drug development are costly, slow, inefficient, and result in more failures than success. For rare diseases, drug development is even more challenging because of the heterogeneity of the clinical manifestations, limited availability of patient cohort, and the limited marketability of the drugs even when successful. Moreover, while the current pipeline is primarily focused on a few well-known rare diseases, there are thousands of rare genetic diseases. Compounding this problem, the number of patients ascertained with rare genetic diseases will certainly increase in the future with the advent of high-throughput DNA sequencing. Therefore, innovative approaches to drug development are clearly needed to expedite the efficient clinical testing of potentially effective products for these rare conditions.
In contrast to the roughly 7000 rare diseases, there are a relatively limited number of disease mechanisms/pathways, some of which are shared in common across multiple diseases. Therefore, drugs which target molecular pathways that are common to multiple diseases can in principle be used to treat more than one disease, even if the clinical manifestations of the diseases are very different.
The current approach to clinical trials is to test one drug on one disease. As such, patients in a trial are grouped on the basis of having a disease as traditionally defined by a clinical phenotype. However, for a drug that targets a molecular mechanism that is common to multiple diseases, it would be more efficient to group patients by molecular etiology, rather than traditional disease classification based on clinical manifestations, for the purpose of a clinical trial. Doing so would likely make clinical trials more efficient, and facilitate testing of rational therapies in as many patients as possible that might benefit from them.
Here we focus on two examples of pathologic mechanisms spanning multiple diseases to illustrate this concept, and discuss some of the practical implications of this new approach.
Nonsense Mutations and Abnormal Protein Folding: Examples of Grouping Patients by Molecular Etiology
Nonsense mutations resulting in premature termination codons (PTC) are a common molecular mechanism underlying multiple genetic diseases. It has been estimated that roughly 11% of disease-causing mutations are nonsense mutations1. Following upon early studies in bacteria showing that aminoglycoside antibiotics can allow some read-through of termination codons, there has been an effort to develop read-through drugs for the treatment of disease resulting from PTCs. The best known of these compounds is PTC-124 (Ataluren) 2. This particular drug has been the subject of controversy, based in part of issues related to the assay used to develop it originally 3, 4, although it has shown efficacy in some clinical studies, particularly in CF 5. It should be stressed that while ataluren is in clinical trials, it is not approved for the treatment of any disease. Importantly, however, other PTC read-through compounds that act in a similar manner, and which may turn out to be more effective than ataluren, are in preclinical development 6,7,8. For this paper, our focus is on PTC read-through compounds as a class of drug that acts on a mechanism common to multiple diseases, and not on PTC-124 specifically.
The actual amount of functional protein produced by a given read-through drug will depend upon many factors, including the sequence of PTC and surrounding sequence 9,10,11, the identity of the amino acid incorporated in place of the PTC, the ability of the drug to stabilize the mutant mRNA6, and the extent to which the mRNA is subject to nonsense-mediated decay (NMD)12. It should be noted here that a small molecule inhibitor of NMD has been developed, and was recently shown to enhance the activity of PTC read-through drugs 13.
Another pathologic mechanism that is common to multiple diseases is abnormal protein folding 14. In genetic diseases, abnormal protein folding typically results from mutations that alter the amino acid sequence of proteins. Abnormalities in protein folding can result in either recessive (loss of function) diseases, or dominant (gain of function diseases). In recessive diseases, misfolded proteins can be retained in the ER, in which case they do not reach sites in the cell where they are normally active, resulting in disease. A well-known example of this mechanism is in cystic fibrosis caused by the delF508 mutation 15. In addition, there are multiple examples of loss of function in lysosomal storage diseases (LSDs) 16. In other cases, misfolded proteins can form toxic aggregates17.
Cells have a variety of mechanisms to maintain the correct folding of proteins, which are collectively referred to as proteastasis network. In principle, manipulation of the proteastatis network is an attractive mechanism for the treatment of multiple protein folding diseases 18. There are several published examples in which a single drug has been shown to be able to significantly improve misfolded proteins in cells from more than one disease, demonstrating in principle that a drug could be effective in multiple diseases 19,20,21. As noted in Nature Biotechnology, a small biotech company has been awarded a patent by the European patent office for Hsp70 to treat LSDs as a group, rather than to treat an individual disease (http://www.nature.com/nbt/journal/v31/n3/full/nbt0313-189.html), thereby demonstrating the plausibility of this grouping approach.
Abnormal protein folding and PTC mutations are used here as two examples to illustrate the concept and value of grouping by molecular etiology. For the purpose of example, we will assume that we have two different drugs; a PTC read-through compound that has been shown to be effective in stimulating PTC read-through at more than one PTC mutation, and a protein folding drug that reduces the abnormal folding of more than one misfolded protein. We will assume that both drugs have acceptable toxicology profiles, and are ready for clinical trials. The question we are concerned with here is the most efficient way to test such compounds in patients with different clinical diseases who might benefit from them.
For simplicity, we consider three diseases, cystic fibrosis, Gaucher, and Tay-Sachs. In all three diseases, a subset of patients have nonsense mutations, and another subset have missense mutations which result in abnormal protein folding 22,23,24. According to the current approach based on traditional clinical definition of disease, 6 trials would be needed to test the one read-through drug and the one folding drug (Fig. 1a). However, since the drugs under study are targeted to the molecular defects common to patients with different clinical diseases, the appropriate patient population should be all those with the relevant mutations. Therefore, if we group patients by molecular etiology, rather than by classical disease, only two trials would be necessary, one for the read-through drug and one for the protein folding drug ( Fig. 1b).
Figure 1.
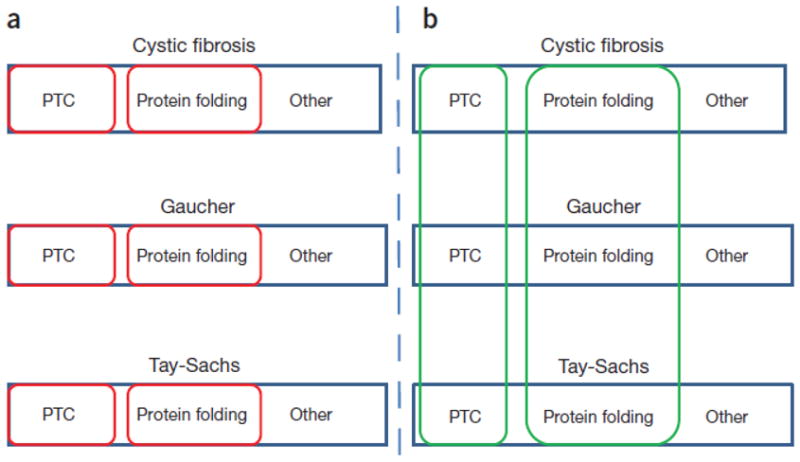
Benefits of grouping by molecular etiology. Shown are schematic representations of three different diseases, cystic fibrosis, Tay-Sachs, and Gaucher disease. In each disease, multiple patients have PTC mutations, protein folding mutations, or other mutations. According to the current approach (panel A), testing a PTC read-through drug and a protein folding drug would involve 6 different trials (red boxes). In contrast, if patients are grouped on the basis of molecular etiology (panel B), only two trials would be needed (green boxes). For further discussion, see text.
While simple in principle, the idea of grouping across disease by molecular etiology raises a number of issues from clinical trial design standpoint. First, a collaborative effort would be necessary, involving clinicians with expertise in each of the traditional diseases being studied in the trial. This expertise is essential, since clinical trial end points would have to be defined for all of the diseases studies, as would be the case in a standard clinical trial. Such end-points would be based on the known clinical features and progression of the diseases, as well as appropriate biomarkers if available. For rare diseases, the lack of accurate knowledge about disease progression is often a limiting factor, emphasizing the need for adequate natural history studies.
For a trial of a drug based upon molecular etiology, the main difference is that in addition to standard inclusion criterion, it would also be necessary to demonstrate that the drug actually corrects the molecular abnormality in the cells of the patient. In the case of a misfolded protein, this would mean demonstrating that the drug significantly reduces the amount of misfolded protein in a cell culture model. For nonsense mutation disease, it would seem necessary to show that a patient has at least one nonsense mutation, and that the drug to be tested significantly increases read-through of the PTC to produce full-length protein in the patients’ cells. In those diseases where genotype-phenotype data provides information on the amount of full-length protein necessary to restore function, this should also be considered. Testing drugs could be done in vitro, using patient-derived cells7. Optimally, iPSCs differentiated into the affected cell type might be used for such studies. An efficient strategy might be to distribute compound to different laboratories which are working with patient-derived cells for in vitro testing.
In terms of the practical reality of conducting a clinical trial, a reasonable setting might be an academic medical center, a rare disease center of excellence, or an existing consortium in which multiple investigators are studying different rare genetic diseases. Such organizational structures would facilitate interaction between the multiple investigators involved, standardization of data collection and analysis, and adverse event reporting.
An illustrative example of a consortium approach to grouping by molecular etiology, albeit in the context of cancer, is the recent clinical trial of crizotinib in pediatric cancer25 (ClinicalTrials.gov, NCT00939770). The study was carried out by a preexisting consortium, the Children’s Oncology Group (http://www.childrensoncologygroup.org). The trial included several anatomically and clinically distinct types of pediatric cancers (anaplastic large-cell lymphoma, inflammatory myofibroblastic tumour, non-small-cell lung cancer, and neuroblastoma). The patients selected for the trial were grouped on the basis of activating mutations in the ALK (analplastic lymphoma kinase) gene as the underlying molecular etiology. Crizotinib is an ALK inhibitor, which had shown efficacy in individual trials of the different cancers with activating ALK mutations (for references see25 ). Notably, since different types of cancers were included in this trial, multiple clinical end-points were defined and utilized, establishing a precedent for studying multiple rare diseases in a single trial as we proposed above.
Some implications of grouping by molecular etiology for clinical trials
The idea of grouping patients by underlying molecular etiology rather than the traditional concept of disease based clinical phenotype may at first seem quite different from the current clinical practice. However, the molecular classification of disease is already gaining acceptance, (see http://www.nap.edu/catalog.php?record_id=13284), and in our view likely to become more common in the future. For many genetic diseases, the current nomenclature of disease is based on historical considerations, and is often confusing. In many cases, the same name may refer to a collection of genetically distinct conditions (e.g. Usher syndromes, Charcot-Marie-Tooth disease). Diseases can also be named after biochemical abnormalities, but as illustrated in Fig. 1, the same biochemical defect can result from very different underlying molecular defects. In contrast, a molecular defect caused by a mutation in a specific gene, such as a PTC or misfolded protein, is unambiguous and objectively measureable, and therefore a better foundation upon which to group disease patients for drug development and clinical trials.
The major advantage of this strategy is that it represents an efficient way to determine in which patients a drug may be successful. The amount of normally folded protein produced by a protein folding drug will likely vary depending upon other aspects of the proteastasis network, as well as the amount of other proteins being synthesized in the cell. Similarly, as discussed above, the actual amount of functional protein produced by a given read-through drug will depend upon many factors. Indeed, a particular read-through compound might have no effect for some PTC mutations 6,9. However, the process we are proposing here includes validation of the effectiveness of the read-through drug in the patient cells as an enrollment criterion. Therefore, if the PTC read-though drug is not effective for an individual patients’ PTC mutation, perhaps because of an unfavorable surrounding sequence, then the patient would not be enrolled in the actual trial of that drug. The overall goal of our proposal is to outline the most efficient process to identify those patients for whom a PTC read-through drug or a protein folding drug could have clinical benefit, and test the drug specifically in those patients.
A separate and more important question is the amount of full length protein needed to produce a clinical benefit. A priori, it seems likely that this will depend on many factors, such as the function(s) of the protein, its inherent stability, and the affinity of the protein for its cellular targets. However, the most important determinant of whether either type of drug is of clinical benefit may be the pathophysiology of the disease. Duchenne muscular dystrophy is caused by mutations in the gene encoding dystrophin, a large structural protein that plays an important role in muscle function. Available information indicates that Ataluren treatment does result in some clinical benefit for patients with Duchenne muscular dystrophy although not to the extent of the originally proposed clinical end-point 26. Similarly, recent results using splice switching oligonucleotides in DMD have also not met expectations 27. In contrast, for other genetic diseases such as lysosomal storage disorders or DNA repair disorders 28,29, 6 the mutations affect enzymes that prevent the accumulation of a toxic metabolic byproduct. In these conditions, disease progression can take years, as the level of the toxic material accumulates. In such diseases, a small increase in active enzyme that significantly decreases the rate of toxin accumulation could in turn substantially slow the rate of disease progression. Indeed, a recent study demonstrated that PTC read-though drugs, in concert with a NMD inhibitor, were able to reduce the levels of toxic glycosaminoglycans to near wild-type levels in a mouse model of mucopolysaccaridosis resulting from a PTC mutation 13.
Another issue that can influence drug efficacy is penetration into different tissues and cell types. Here again, testing drugs based on shared molecular etiology in multiple rare diseases affecting different tissues and cell types could provide valuable information on this point, which may benefit subsequent drug development efforts. For all of these reasons, as well as the fact that for many rare diseases pathophysiology is simply not well understood, it seems very difficult to predict a priori diseases in which a given PTC read-through drug or protein folding drug will be clinically effective. Therefore, testing these types of drugs in as many patients as possible is important, not only to maximize the number of individuals that may experience clinical benefit, but also to learn more about the relationship between pathophysiology and drug response.
One important long-term implication of grouping by molecular etiology on drug development should be mentioned. At present, drug development is focused on one disease at a time. However, as the concept of grouping by molecular etiology gains acceptance, this will in turn encourage drug development efforts on mechanisms that are shared across diseases. A recent example of this approach is the discovery that delta-tocopherol can reduce pathological phenotypes in patient fibroblasts from multiple lysosomal storage diseases30
Other Possible Examples of Grouping by Molecular Etiology
Drugs that promote read-though of PTCs and prevent misfolding were chosen to illustrate the concept of grouping by molecular etiology because of the numbers of diseases caused by such mutations. However, the basic principle could be extended to other types of mutations, such as those that affect RNA splicing. Two groups have identified small molecules that affect splicing of the IKAP gene in familial dysautonomia31,32, and one of these compounds, kinetin, affects the splicing of a subset of other genes as well33. While there are several different types of splicing mutations34, further research into the molecular targets of kinetin 35 and other drugs that affect RNA splicing 36 can identify protein targets that carry out specific steps of the splicing reaction. Such information could be used to identify and group patients for trials of drugs that target specific types of splicing mutations.
Several genetic diseases of the nervous system are caused by transcriptional deregulation, which can result from epigenetic gene silencing (e.g. Friedreich’s ataxia), or mutations in transcriptional regulators (e.g. Rubinstein-Taybi syndrome). Grouping these as transcriptional regulation disorders could facilitate the testing of drugs such as histone deacetylase inhibitors that are promising therapeutic candidates in several of these diseases 37.
Grouping by molecular etiology could also be applied for drugs that act on pathogenic proteins. For example, several neurodegenerative diseases show prominent tau pathology (tau hyperphosphorylation and aggregation resulting in neurofibrillary tangles) and are collectively referred to as tauopathies. There are at least 85 known phosphorylation sites in tau and strategies to modulate the various kinases for tau phosphorylation is an active area of therapy development 38. Other –opathies, such as ubiquitinopathies39, are potential candidates for drug development involving grouping by molecular etiology. Finally, several degradation pathways converge at the level of the lysosome, including endocytosis, phagocytosis and autophagy. Lysosomal dysfunction is a common hallmark associated with many neurodegenerative conditions, such as Alzheimer’s and Parkinson’s disease as well as Lafora and Pompe disease, thus therapies aimed at restoring lysosomal function may be beneficial for a host of disorders 40.
Grouping by Common Etiology as an Additional Strategy for Accelerate Drug Testing, not a Replacement for the Standard Approach
The standard approach to therapeutics development in genetic diseases is based upon targeting the biochemical defect resulting from the mutation. There is no doubt that this approach has been very successful in some genetic diseases (e.g. cystinosis41 NAGS deficiency 42). Drug repurposing represents another very attractive strategy for rare diseases that is being actively pursued43. It is important to stress that the grouping by molecular etiology is not intended to replace either of these approaches. Instead, the grouping approach we propose will likely be most useful for those diseases where the biochemical pathway targeted therapeutics are not available either because the biochemical defect does not result in a “druggable” target, or simply due to a lack of knowledge of pathophysiology, which is a common problem in rare diseases. For individuals with such diseases, grouping by molecular etiology can provide an accelerated route to clinical trials of therapeutics targeted to specific types of mutations.
Summary
Although there are thousands of Mendelian disorders, they can be classified into a smaller number of groups based on the molecular etiology resulting from the mutation. The good news is that novel therapies are being developed that are potentially applicable to many patients with different diseases. Testing these drugs in patients who might potentially benefit from them, in a timely and efficient manner, is a major practical hurdle. Grouping patients by molecular etiology as a single group for therapeutics testing has multiple advantages. For the biotech industry, larger groups of patients increase the financial incentive for drug development, and may also spur competition for this patient population. Last but not least, for patients with rare diseases, grouping by molecular etiology should facilitate access to clinical trials of drugs which offer rational therapeutic potential, regardless of how rare their particular disease is, or how little we understand about the pathophysiology.
In closing, we realize that our proposal represents a different way of thinking about rare disease therapeutics development and testing, and that is raises many complex new issues to be worked out. As such, we view this proposal as an effort to begin a dialog about these issues with rare disease clinical researchers, patient advocates, and industry. In the long term, we look forward to the day when no patient is denied access to a rational therapy simply because their disease is too rare to treat.
References
- 1.Mort M, Ivanov D, Cooper DN, Chuzhanova NA. A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat. 2008;29:1037–1047. doi: 10.1002/humu.20763. [DOI] [PubMed] [Google Scholar]
- 2.Peltz SW, Morsy M, Welch EM, Jacobson A. Ataluren as an agent for therapeutic nonsense suppression. Annu Rev Med. 2013;64:407–425. doi: 10.1146/annurev-med-120611-144851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Auld DS, Thorne N, Maguire WF, Inglese J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc Natl Acad Sci U S A. 2009;106:3585–3590. doi: 10.1073/pnas.0813345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McElroy SP, et al. A lack of premature termination codon read-through efficacy of PTC124 (Ataluren) in a diverse array of reporter assays. PLoS Biol. 2013;11:e1001593. doi: 10.1371/journal.pbio.1001593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilschanski M, et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respir J. 2011;38:59–69. doi: 10.1183/09031936.00120910. [DOI] [PubMed] [Google Scholar]
- 6.Kuschal C, Digiovanna JJ, Khan SG, Gatti RA, Kraemer KH. Repair of UV photolesions in xeroderma pigmentosum group C cells induced by translational readthrough of premature termination codons. Proc Natl Acad Sci U S A. 2013;110:19483–19488. doi: 10.1073/pnas.1312088110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du L, et al. Nonaminoglycoside compounds induce readthrough of nonsense mutations. J Exp Med. 2009;206:2285–2297. doi: 10.1084/jem.20081940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nudelman I, et al. Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J Med Chem. 2009;52:2836–2845. doi: 10.1021/jm801640k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dietz HC. New therapeutic approaches to mendelian disorders. N Engl J Med. 2010;363:852–863. doi: 10.1056/NEJMra0907180. [DOI] [PubMed] [Google Scholar]
- 10.Linde L, Kerem B. Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008;24:552–563. doi: 10.1016/j.tig.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 11.Keeling KM, Bedwell DM. Suppression of nonsense mutations as a therapeutic approach to treat genetic diseases. Wiley Interdiscip Rev RNA. 2011;2:837–852. doi: 10.1002/wrna.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuzmiak HA, Maquat LE. Applying nonsense-mediated mRNA decay research to the clinic: progress and challenges. Trends Mol Med. 2006;12:306–316. doi: 10.1016/j.molmed.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 13.Keeling KM, et al. Attenuation of nonsense-mediated mRNA decay enhances in vivo nonsense suppression. PLoS One. 2013;8:e60478. doi: 10.1371/journal.pone.0060478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindquist SL, Kelly JW. Chemical and biological approaches for adapting proteostasis to ameliorate protein misfolding and aggregation diseases: progress and prognosis. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amaral MD, Farinha CM. Rescuing mutant CFTR: a multi-task approach to a better outcome in treating cystic fibrosis. Curr Pharm Des. 2013;19:3497–3508. doi: 10.2174/13816128113199990318. [DOI] [PubMed] [Google Scholar]
- 16.Parenti G. Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics. EMBO Mol Med. 2009;1:268–279. doi: 10.1002/emmm.200900036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bemporad F, Chiti F. Protein misfolded oligomers: experimental approaches, mechanism of formation, and structure-toxicity relationships. Chem Biol. 2012;19:315–327. doi: 10.1016/j.chembiol.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 19.Wang F, Song W, Brancati G, Segatori L. Inhibition of endoplasmic reticulum-associated degradation rescues native folding in loss of function protein misfolding diseases. J Biol Chem. 2011;286:43454–43464. doi: 10.1074/jbc.M111.274332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang C, et al. Celastrol increases glucocerebrosidase activity in Gaucher disease by modulating molecular chaperones. Proc Natl Acad Sci U S A. 2014;111:249–254. doi: 10.1073/pnas.1321341111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mu TW, Fowler DM, Kelly JW. Partial restoration of mutant enzyme homeostasis in three distinct lysosomal storage disease cell lines by altering calcium homeostasis. PLoS Biol. 2008;6:e26. doi: 10.1371/journal.pbio.0060026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quintana-Gallego E, Delgado-Pecellin I, Calero Acuna C. CFTR Protein Repair Therapy in Cystic Fibrosis. Arch Bronconeumol. 2013 doi: 10.1016/j.arbres.2013.07.013. [DOI] [PubMed] [Google Scholar]
- 23.Myerowitz R. Tay-Sachs disease-causing mutations and neutral polymorphisms in the Hex A gene. Hum Mutat. 1997;9:195–208. doi: 10.1002/(SICI)1098-1004(1997)9:3<195::AID-HUMU1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 24.Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA) Hum Mutat. 2008;29:567–583. doi: 10.1002/humu.20676. [DOI] [PubMed] [Google Scholar]
- 25.Mosse YP, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14:472–480. doi: 10.1016/S1470-2045(13)70095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pichavant C, et al. Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol Ther. 2011;19:830–840. doi: 10.1038/mt.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffman EP, Connor EM. Orphan drug development in muscular dystrophy: update on two large clinical trials of dystrophin rescue therapies. Discov Med. 2013;16:233–239. [PubMed] [Google Scholar]
- 28.Nakamura K, Hattori K, Endo F. Newborn screening for lysosomal storage disorders. Am J Med Genet C Semin Med Genet. 2011;157:63–71. doi: 10.1002/ajmg.c.30291. [DOI] [PubMed] [Google Scholar]
- 29.Kulkarni A, Wilson DM., 3rd The involvement of DNA-damage and -repair defects in neurological dysfunction. Am J Hum Genet. 2008;82:539–566. doi: 10.1016/j.ajhg.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu M, et al. delta-Tocopherol reduces lipid accumulation in Niemann-Pick type C1 and Wolman cholesterol storage disorders. J Biol Chem. 2012;287:39349–39360. doi: 10.1074/jbc.M112.357707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson SL, Rubin BY. Tocotrienols reverse IKAP and monoamine oxidase deficiencies in familial dysautonomia. Biochem Biophys Res Commun. 2005;336:150–156. doi: 10.1016/j.bbrc.2005.08.054. [DOI] [PubMed] [Google Scholar]
- 32.Axelrod FB, et al. Kinetin improves IKBKAP mRNA splicing in patients with familial dysautonomia. Pediatr Res. 2011 doi: 10.1203/PDR.0b013e31822e1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hims MM, et al. Therapeutic potential and mechanism of kinetin as a treatment for the human splicing disease familial dysautonomia. J Mol Med (Berl) 2007;85:149–161. doi: 10.1007/s00109-006-0137-2. [DOI] [PubMed] [Google Scholar]
- 34.Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 35.Boone N, et al. Genome-wide analysis of familial dysautonomia and kinetin target genes with patient olfactory ecto-mesenchymal stem cells. Hum Mutat. 2012;33:530–540. doi: 10.1002/humu.22010. [DOI] [PubMed] [Google Scholar]
- 36.Rymond B. Targeting the spliceosome. Nat Chem Biol. 2007;3:533–535. doi: 10.1038/nchembio0907-533. [DOI] [PubMed] [Google Scholar]
- 37.Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- 38.Medina M, Garrido JJ, Wandosell FG. Modulation of GSK-3 as a Therapeutic Strategy on Tau Pathologies. Front Mol Neurosci. 2011;4:24. doi: 10.3389/fnmol.2011.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cullen V, et al. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann Neurol. 2011;69:940–953. doi: 10.1002/ana.22400. [DOI] [PubMed] [Google Scholar]
- 40.Amelio I, Melino G, Knight RA. Cell death pathology: cross-talk with autophagy and its clinical implications. Biochem Biophys Res Commun. 2011;414:277–281. doi: 10.1016/j.bbrc.2011.09.080. [DOI] [PubMed] [Google Scholar]
- 41.Kleta R, Gahl WA. Pharmacological treatment of nephropathic cystinosis with cysteamine. Expert Opin Pharmacother. 2004;5:2255–2262. doi: 10.1517/14656566.5.11.2255. [DOI] [PubMed] [Google Scholar]
- 42.Tuchman M, et al. N-carbamylglutamate markedly enhances ureagenesis in N-acetylglutamate deficiency and propionic acidemia as measured by isotopic incorporation and blood biomarkers. Pediatr Res. 2008;64:213–217. doi: 10.1203/PDR.0b013e318179454b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang R, et al. The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci Transl Med. 2011;3:80ps16. doi: 10.1126/scitranslmed.3001862. [DOI] [PMC free article] [PubMed] [Google Scholar]