

Abstract
The focus of the study was to implement a new workflow for circulating tumor cells (CTCs) characterization that would allow the analysis of CTCs on a cytomorphological and molecular level in patients with diagnosed gynecological cancer. Our findings may be useful in future cancer patient management. The study introduces a size-based enrichment (MetaCell®) method for the separation of viable CTCs, followed by CTCs culturing in vitro and gene expression characterization. It is based on the observation of CTCs and DTCs (Disseminated Tumor Cells) in several case studies of ovarian, endometrial and cervical cancer by means of cytomorphology and gene expression profiling. The viability of the enriched CTCs was estimated using vital and lethal fluorescence nuclear staining. This type of staining may be predictive for the success rate of subsequent CTC growth in vitro. To identify CTCs in the enriched CTC fraction, cytomorphological evaluations based on vital fluorescence staining were followed by gene expression analysis of tumor-associated (TA) genes. Cytokeratin expression (KRT7, KRT19) was analyzed in combination with MUC1, MUC16, CD24, CD44 and ALDH1. Gene expression analysis has shown that short-term in vitro culture enhanced the differentiation process of the captured CTCs growing on a membrane. On the other hand, redundant white blood cells captured on the membrane were eliminated during a short-term culture. The most frequently elevated genes in ovarian cancer (serous type) are EPCAM, KRT19 and MUC1. It has been demonstrated that CTC presence revealed by cytomorphological evaluation may be usefully complemented by TA-gene expression analysis, to increase the sensitivity of the analysis.
Keywords: CTCs, gynecological cancers, ovarian cancer, cervical cancer, endometrial cancer, cultivation, in vitro
Introduction
Circulating tumor cells (CTCs) are viable solid-tumor cells that have disseminated from the primary tumor and are carried through the circulatory system [1]. CTCs are relatively rare, and can be found at frequencies of approximately one tumor cell per 107 blood elements [2]. The presence of CTCs in cancer patients is associated with a poor prognosis [3-5]. There is, however, limited data on the prognostic potential and prevalence in patients with gynecological cancers.
Identification of CTCs by isolating and separating them from the peripheral blood of patients with solid tumors may help to personalize cancer treatment. In the near future CTCs will be a tool aiding the overall survival and prognosis of cancer patients by improving clinical management. Using currently available methods, the identification of CTCs in peripheral blood is quite challenging. The majority of methods are targeted at the isolation and enumeration of CTCs from previously stabilized peripheral blood [6]. The study of fixed CTCs provides little data about their functional capability, METASTA- TIC potential and treatment-induced changes. The in vitro culturing of CTCs can therefore allow their metastatic potential and the drug sensitivity of tumor cells to be investigated.
The focus of our study was to implement a new CTC-characterization workflow (Figure 1) that should allow the analysis of CTCs on a cytomorphological and molecular level. The study outcome has the potential for use in cancer patient management. Our study introduces a size-based enrichment method for the separation of viable CTCs followed by culturing in vitro and gene expression characterization. The approach is demonstrated on CTCs in patients with gynecological cancers, specifically ovarian, cervical and endometrial cancers.
Figure 1.
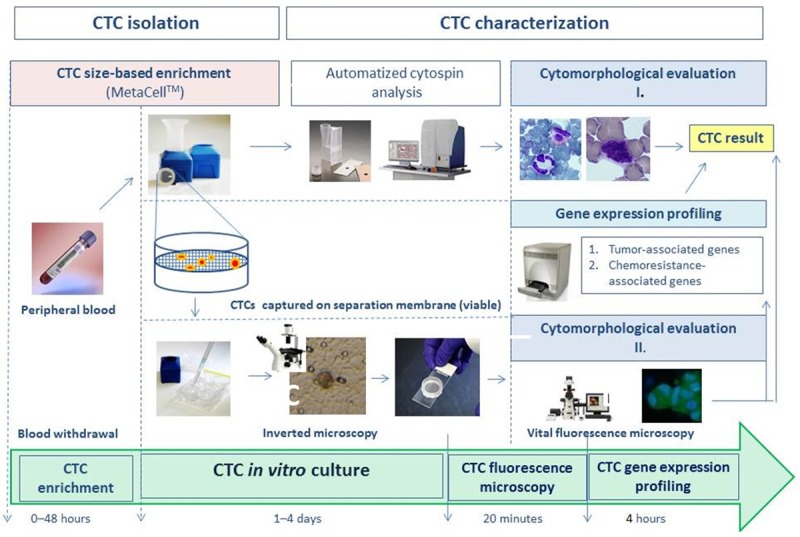
CTC isolation and characterization workflow.
Materials and methods
Patients
Patients diagnosed with gynecological tumors (of the three types mentioned above) were involved in the study: one patient with ovarian cancer, one patient with endometrial cancer and one patient with cervical cancer. Approximately 8 ml of venous blood was drawn from the antecubital veins of each patient and placed into S-Monovette tubes (Sarstedt AG & Co., Numbrecht, Germany) containing 1.6 mg EDTA/ml blood as an anticoagulant. The samples were processed at room temperature using an isolation procedure completed within 24 hours of the blood draw.
The ethics committees of all participating universities and hospitals approved the study as compliant with the Declaration of Helsinki. All patients also provided written consent.
CTC enrichment and culture
A simplified size-based separation method for viable CTC enrichment from unclotted peripheral blood (PB) has recently been introduced (MetaCell®, MetaCell s.r.o., Ostrava, Czech Republic) [7,8]. This process is based on the filtration of PB through a porous polycarbonate membrane (with pores of 8 μm diameter). The minimum and maximum volume of the filtered PB may be adjusted up to 50 ml. An 8 ml PB sample from patients suffering from gynecological cancer was transferred into the filtration tube. Gradual transfer of the blood in several steps is preferred to prevent clogging of the membrane filter. The PB flow is driven naturally by blood viscosity and supported by capillary action of the absorbent material touching the membrane filter. Afterwards the membrane filter, which is kept in a plastic ring, is transferred into a 6-well cultivation plate; 4 ml RPMI media is added to the filter top and CTCs are cultured on the membrane in vitro, under standard cancer-cell culture conditions (37°C, 5% atmospheric CO2) and observed by inverted microscope. The CTCs grow in an FBS-enriched RPMI medium (10%) for a minimum of 3 days on the separating membrane. The grown cells can be analyzed by means of histochemistry (May-Grünwald staining - MGG) and immunohistochemistry using specific antibodies to determine the cell origin (pancytokeratin 1-FITC conjugated antibody (Sigma), cytokeratin7 antibody (Dako)) and unspecific, DAPI staining). A novelty that we introduced was a cytomorphological analysis method based on vital fluorescent staining, which enables us to use stained cells for further downstream analysis (e.g. gene expression profiling). The cells captured on the membrane are stained with unspecific nuclear stain (NucBlueTM, Life Technologies) and unspecific green cytoplasmic stain (CellTrackerTM, Life Technologies).
Alternatively, the CTC-enriched fraction can be transferred from the membrane directly onto a microscopic slide with a cytospin centrifuge. The slide is then left to dry for 24 hours and automated cytomorphological analysis can then be performed (Cellavision DM96, Sysmex). The workflow is shown schematically in Figure 1.
Cytomorphological analysis
The cells, either fixed or viable, and stained on the membranes, were examined using microscopy in three steps: 1. screening at x20 magnification to locate cells; 2. observation at x40 magnification for detailed cytomorphological analysis; 3. observation at x100 magnification for a subcellular-level analysis.
Isolated cells and/or clusters of cells of interest were selected, photographed digitally, and the images were then examined by an experienced researcher and/or pathologist. CTCs were defined as cells with the following characteristics: (A) with a nuclear size ≥10 μm; (B) irregular nuclear contour; (C) presence of visible cytoplasm; (D) prominent nucleoli; (E) high nuclear-cytoplasmatic ratio (NC-ratio, which changes anyway with the in vitro culture time) (F) high deformability/plasticity (growth through the membrane to the bottom of the well and setting up of new colonies); and (G) proliferation.
Gene expression analysis (GEA)
To confirm the origin of the cells on the separation membrane, CTC-gene expression analysis can be performed. Gene expression analysis (GEA) allows up to 20 tumor-associated (TA) markers in RNA from different cell fractions to be tested within a single quantitative polymerase chain reaction (qPCR) run. Differential diagnostic markers (TA genes) for qPCR testing are chosen depending on the patient’s clinical status and primary tumor histology. The following genes were included in the TA/qPCR tests: (ACTB, CD45, CD68, EPCAM, MUC1, MUC16, mammaglobin, HER2, ER, PR, CD24, CD44, ALDH1).
The purpose of the GEA analysis is to compare gene expression of the TA markers in the CTC-enriched fractions to that in the blood as a whole.
In detail, RNA is isolated from the expllanation for PBW ( white blood cell fraction) and the CTC-enriched fraction on the membrane. The RNA can be isolated from the CTC fraction immediately after the separation process (these are so-called “virgin” CTCs) or/and from the CTC fraction grown for a minimum of three days on the separation membrane in vitro (the so-called “membrane fraction”). Some of the cells grown on the membrane in vitro may overgrow the membrane and set up new cell colonies on the culture-well bottom. These cells are analyzed as the “bottom fraction”.
Finally, the CTC-gene expression analysis allows identification of the relative amount of TA markers in the whole blood and in CTC-enriched fractions. One possible approach to report the collected gene expression data (Cycle of quantification [Cq] values) is to compare the relative differences (Δ Cq = CqTA-gene - CqControl gene) in CTC fraction and white blood cell fraction (PBW) (Δ = Δ CqCTC fraction -Δ CqPBW fraction). In detail, TA-gene expression (CqTA-gene) is normalized to control gene (CqControl gene) in CTC fraction (Δ CqCTC fraction = CqTA-gene - CqControl gene). Similarly, difference in PWB fraction (Δ CqPBW fraction = CqTA-gene - CqControl gene ) is calculated.
If the relative amount of tumor-associated gene is increased in CTC fraction if compared to the PBW, the results table is marked with an X (see Table 1). If TA genes are highly expressed in the CTC fraction, a subsequent analysis of chemoresistance-associated (CHA) genes is performed. The following genes are included in the qPCR-testing: (ERCC1, MDR1, MRP1, MRP2, MRP4, MRP5, MRP10). Molecular analysis allows identification of which type of chemotherapy could be used as an effective treatment. In future, the GEA analysis should make it possible to develop and implement a personalized cancer therapy.
Table 1.
Gene expression analysis report
| Genes tested by quantitative qPCR (PBW vs. CTC-enriched fraction) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| T | ↑ | T | ↑ | T | ↑ | ||||||
| 1 | ACTB | X | 14 | CHGA | 27 | SCGB | X | ||||
| 2 | CD45 | X | 15 | MLANA | 28 | CD24 | X | X | |||
| 3 | CD68 | X | 16 | S100B | 29 | CD44 | X | ||||
| 4 | EPCAM | X | X | 17 | VIM | 30 | ALDH | X | |||
| 5 | MUC1 | X | 18 | VEGF | 31 | MRP1 | |||||
| 6 | TTF1 | 19 | WT1 | 32 | MRP2 | ||||||
| 7 | KRT5 | 20 | CD56 | 33 | MRP4 | ||||||
| 8 | KRT6 | 21 | CD57 | 34 | MRP5 | X | |||||
| 9 | KRT7 | X | X | 22 | SYP | 35 | MRP7 | X | |||
| 10 | KRT18 | 23 | HER2 | X | 36 | MDR1 | |||||
| 11 | KRT19 | X | X | 24 | ER | X | 37 | ERCC1 | |||
| 12 | KRT20 | 25 | PR | X | |||||||
| 13 | EGFR | 26 | MUC16 | X | X | ||||||
T: genes tested by qPCR, ↑: genes with elevated amount of RNA (relative RNA amount).
The cells on the membrane are lysed in RLT-buffer with beta-mercapto-ethanol (Qiagen). RNA is then isolated using the RNeasy Mini Kit (Qiagen). The RNA from the whole blood is isolated with a modified procedure, and the quality/concentration of RNA is measured with NanoDrop (ThermoScientific). As there are only a few hundred cells on the membrane, the median concentration of RNA is quite low (5-10 ng/μl). For cDNA production we used the High Capacity cDNA Reverse Transcription Kit (Life Technologies). For gene expression analysis we employed Taqman chemistry including Taqman MGB probes for all the above-mentioned genes (Life Technologies). (A list of Taqman probes employed is given in Table 2).
Table 2.
The list of Taqman probes
| Gene | Taqman probe specification |
|---|---|
| ACTB | Hs01060665_g1 |
| CD45 | Hs04189704_m1 |
| CD68 | Hs02836816_g1 |
| KRT7 | Hs00559840_m1 |
| KRT19 | Hs01051611_gH |
| MUC1 | Hs00159357_m1 |
| MUC16 | Hs01065189_m1 |
| EPCAM | Hs00158980_m1 |
| HER2 | Hs01001580_m1 |
| ER | Hs00174860_m1 |
| PR | Hs01556702_m1 |
| SCGB (Mammaglobin) | Hs00935948_m1 |
| CD24 | Hs03044178_g1 |
| CD44 | Hs01075861_m1 |
| ALDH1 | Hs00946916_m1 |
| ABCC1 (MRP1) | Hs01561502_m1 |
| ABCC2 (MRP2) | Hs00166123_m1 |
| ABCC4 (MRP4) | Hs00988717_m1 |
| ABCC5 (MRP5) | Hs00981087_m1 |
| ABCC10 (MRP7) | Hs00375701_m1 |
| MDR1 | Hs00184500_m1 |
| ERCC1 | Hs01012158_m1 |
Statistical analysis of the gene expression data was performed using multivariate techniques: hierarchical clustering and principal component analysis (PCA) using the GenEx Pro v.6 Software (MultiD, Sweden). The cut-off value was set as 40 Cq. Missing data were obtained by interpolation. Off-scale data (above the cut-off value of 30 Cq) were replaced by maximal Cq value of the tested gene plus 0.5 (when maximal Cq was below the cut-off value). The relative RNA-quantity data between the PBW and CTC fractions in patient cohorts were compared by non-parametric Man-Whitney test (not applicable if reporting on individuals).
Results
We report successful implementation and optimalization of the workflow for CTC characterization. The study is based on the description of CTCs and DTCs in several case studies of ovarian, endometrial and cervical cancer by means of cytomorphology and gene expression profiling data.
After blood filtration, the membrane kept in a plastic ring was transferred to a 6-well plate and the CTCs were observed by inverted microscope (Figure 2). The CTCs were cultured in vitro for a minimum of 3 days. The cells on the membrane were co-incubated with unspecific fluorescent vital stains and the morphology was evaluated after 3 days of culturing and digital images were captured (Figures 3, 4 and 5). Thanks to their vital staining the cells can be used immediately after microscopy for mRNA isolation and gene expression profiling. As shown in Figure 6 it is possible to follow the procedure of filtration by cytospin-slide preparation. All the captured cells were washed from the separation membrane, transferred using a cytospin to a slide, and analyzed automatically with a Cellavision DM 96 analyzer (Sysmex). In practical terms, this means that the machine takes high quality photographs and the analytical specialist’s only task is to evaluate the cell morphology on the digitized images (Figure 6).
Figure 2.
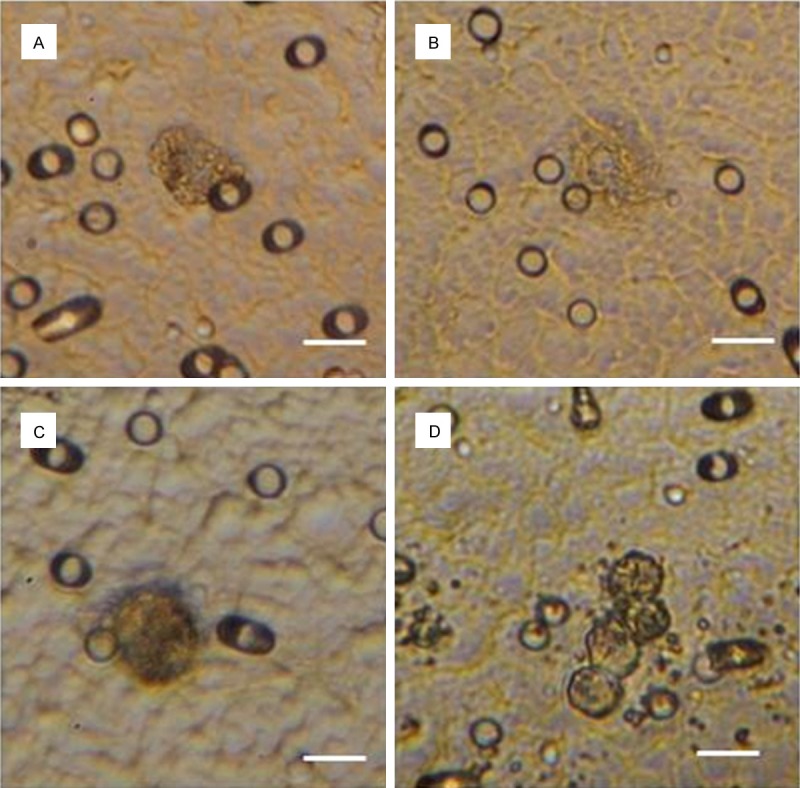
CTCs isolated from patients with: (A) ovarian cancer; (B) endometrial cancer; (C, D) cervical cancer. Scale bar 10 µm.
Figure 3.
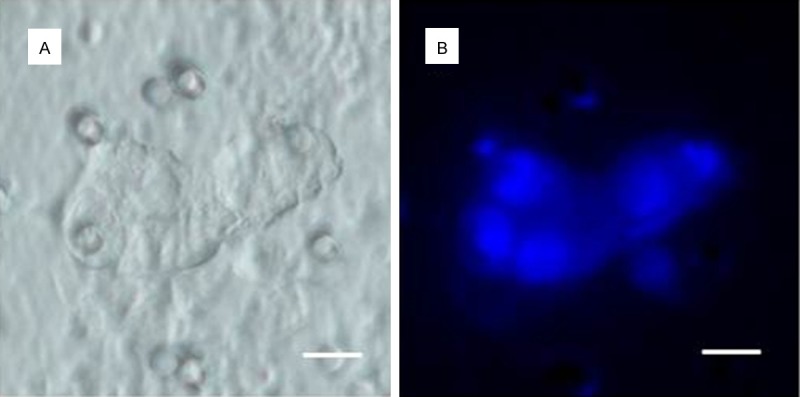
Comparison of: (A) inverted microscope and (B) fluorescence microscope images of DTCs. DTCs were enriched out of the ascitic fluid of an ovarian cancer patient. For visualization of the nucleus a vital nuclear stain (NucBlueTM) is used. Scale bar 10 µm.
Figure 4.
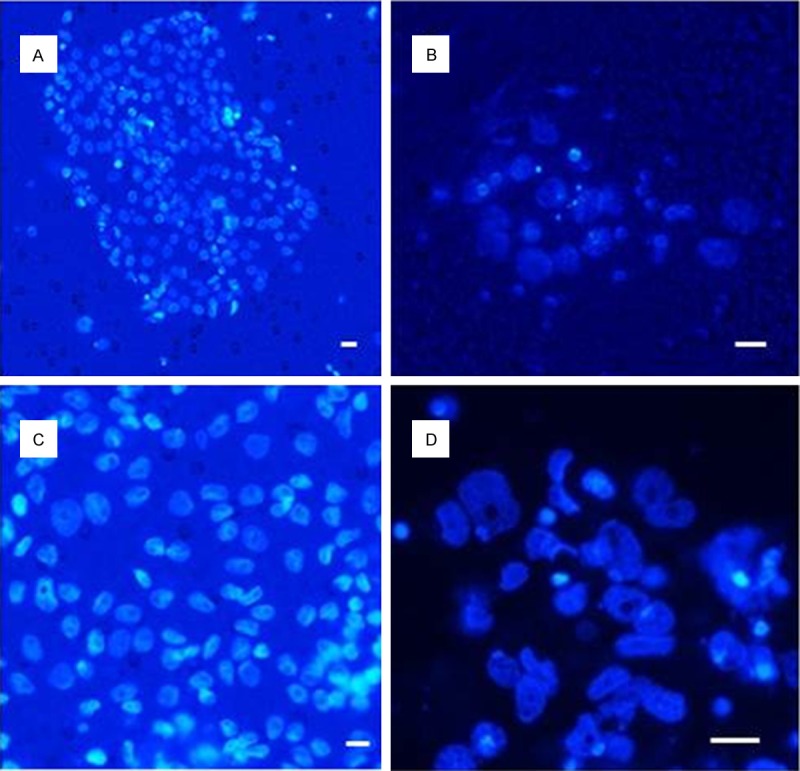
(A, C, D) DTCs and (B) CTCs from an ovarian cancer patient after 10 days of in vitro culture (NucBlueTM - nuclear staining). Scale bar 10 µm.
Figure 5.
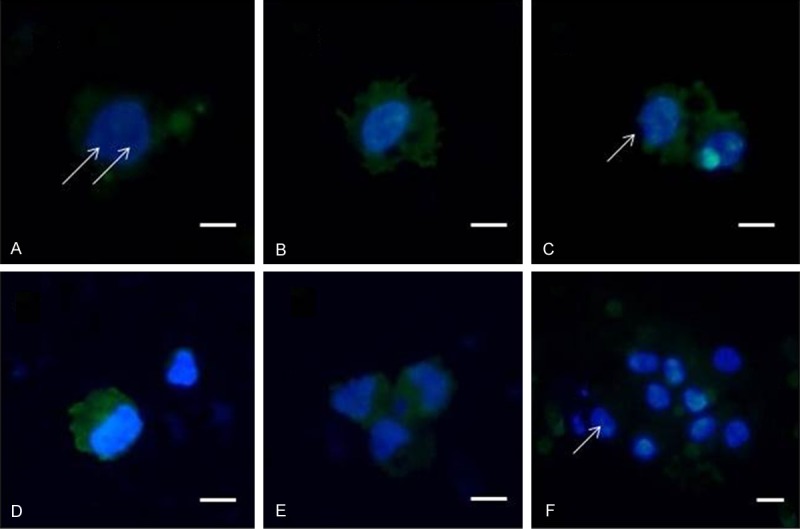
CTCs from an ovarian cancer patient, stained for subcellular cytomorphology evaluation with vital nuclear stain (NucBlueTM) and unspecific green cytoplasmic stain (CelltrackerTM). Prominent nucleoli are arrowed. Scale bar 10 µm.
Figure 6.
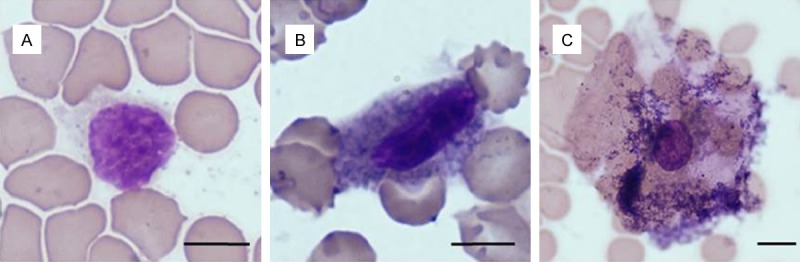
CTCs from an ovarian cancer patient analyzed automatically by Cellavision DM 96 analyzer after May-Grünwald staining. Note the different cytomorphology of the captured cancer cells. Scale bar 5 µm.
Similarly, DTCs from the peritoneum can be analyzed. We report a successful in vitro culture of DTCs, but the success of in vitro growth is dependent on the viability of the cells. The viability of the captured cells can be estimated using vital (NucBlueTM) and lethal (NucGreenTM Dead) nuclear staining (Figure 7). The green nuclei can be found in dead cells, the cells with blue nuclei may grow further. This type of staining may be predictive of the success of subsequent in vitro growth. The DTC cultures may then be used for additional chemotherapy testing (Figure 8).
Figure 7.
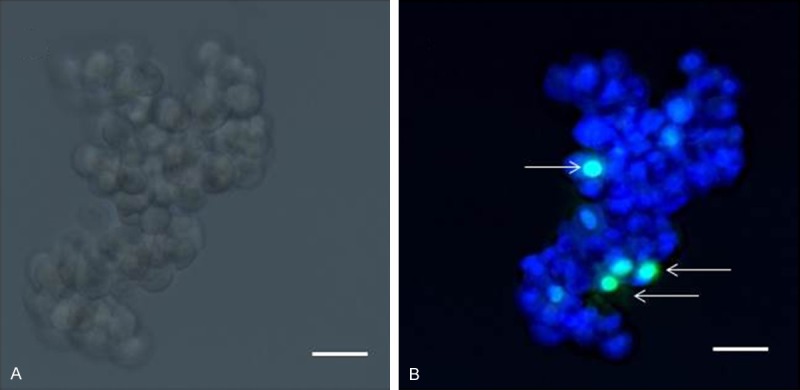
DTCs from an ovarian cancer patient (ascites) - (A) Inverted microscope image; (B) Fluorescence microscope image. Viable cells are stained with NucBlueTM, dead cells (arrowed) with NucGreenTM. Scale bar 10 µm.
Figure 8.
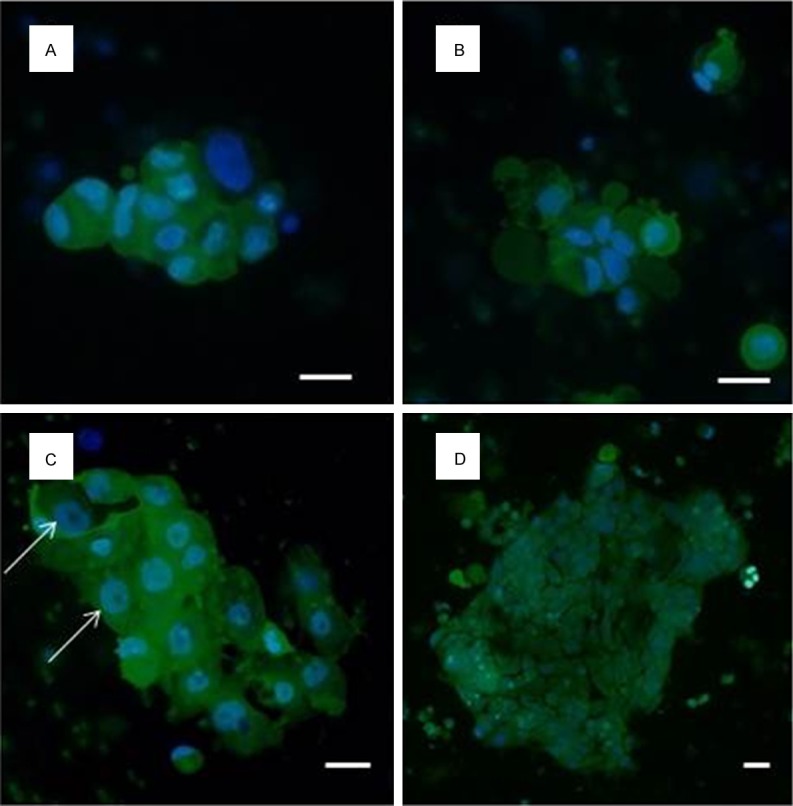
In vitro cultured DTCs isolated from ascitic fluid from an ovarian cancer patient stained with vital nuclear stain (NucBlueTM) and unspecific green cytoplasmic stain (CelltrackerTM) to allow subcellular cytomorphology evaluation. Prominent nucleoli are arrowed. Scale bar 10 µm.
To identify CTCs in the enriched CTC fraction, an RNA relative quantity of TA genes was evaluated. Following the generally adopted procedure, we looked for cells expressing EpCAM. Since CTCs undergo epithelial mesenchymal transition (EMT), cytokeratin expression (KRT7, KRT19) was analyzed in combination with MUC1 [9,10]. Additionally, MUC16 was found a promising marker of ovarian tissue [11]. CD24, CD44 and ALDH1 were tested for the stemness of the captured cells. This could not be achieved without reporting expression of CD45, CD68 and a control gene (ACTB). It is quite challenging to find a relevant control gene for gene expression analysis of CTCs as they change their phenotype relatively quickly. Similarly, it is quite challenging to find a general pattern characterizing gene expression in CTCs for all the included patients. It has been shown that the approach to analyze gene expression based on comparison within one individual could report more relevant information on CTC character.
We expected that the ratios of control gene and TA genes would change in enriched fractions. Ultimately, it was shown that in vitro culture enhanced the differentiation process of the captured CTCs and on the other hand white blood cells died during a short-term culture. The detection of CTCs by cytomorphological evaluation could be further enhanced with verification by complementary GEA of TA-gene expression in CTC-enriched fractions. The most frequently elevated genes in ovarian cancer (serous type) are EPCAM, KRT19, MUC1.
An example of GEA data shown by cluster analysis is shown in Figure 9.
Figure 9.
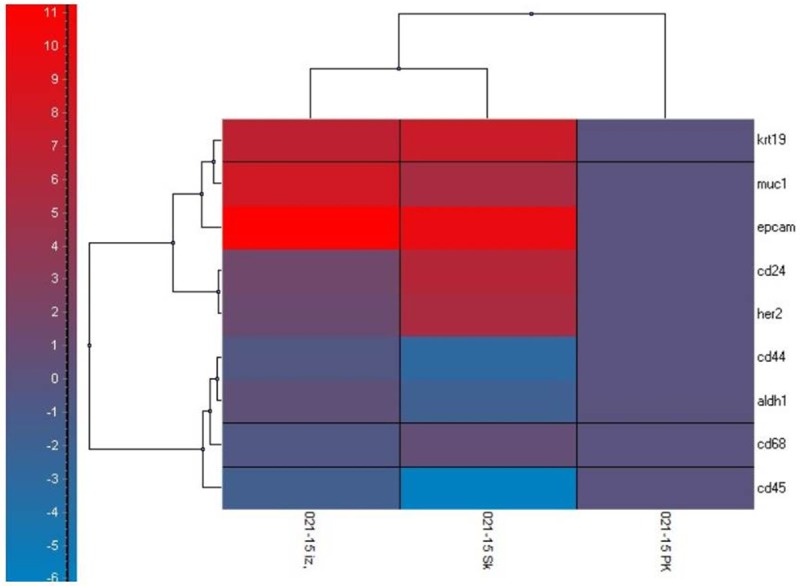
Gene expression profiling data reported by hierarchical clustering shown in relative RNA-quantity values (scale at left). Gene expression level of TA genes in peripheral blood - the white blood cell fraction (PK) is compared to the CTC-enriched fraction (without in vitro culture) (PK izo) and the CTC-enriched fraction after in vitro culture (PK Sk). The following genes were tested: KRT19, MUC1, EpCAM, CD24, CD44, HER2, ALDH1, CD68, CD45, ACTB. Significant changes were seen in gene expression of EPCAM, MUC1 and KRT19 in the CTC-enriched fraction. After in vitro culturing of the CTC-enriched fraction, CD24 and HER2 expression increased, while CD44, ALDH1, CD68, CD45 expression fell. The fall of mRNA levels of these genes could be relevant to the dying of the white blood cells fraction.
Discussion
The early detection and characterization of CTCs is important as it is a general tool aimed at monitoring and preventing the development of overt metastatic disease [12,13].
Although the rate at which tumor cells are released into the bloodstream in cancer patients is unknown, experimental models indicate that millions are continuously dispersed through the body, though very few reach distant organs, and survive in a dormant state [14].
There are many strategies for detecting and isolating CTCs. Various approaches to the enrichment process are under development in laboratories all over the world [15,16]. Most of capture technologies are dependent on the epithelial character of solid tumors, which is demonstrated by EpCAM expression [17]. This technical issue leads to the underdetection of CTCs.
Another specific aspect is viability of the enriched CTCs. The captured CTCs lose their viability during the immunofluorescence detection process so that subsequent culturing of collected cells is disabled.
Quantification of CTCs could be of high value for clinicians as these CTCs generally represent the tumor (metastases) and facilitate real-time monitoring during systemic therapies by sequential peripheral blood sampling [18,19].
In current clinical practice, the decision to employ targeted therapies is based solely on the analysis of the primary tumor although the therapy is directed against metastatic cells.
Furthermore, molecular characterization of CTCs might enable the identification of therapeutic targets and contribute to personalized anti-metastatic therapies. Molecular characterization of CTCs might be essential to identify therapeutic targets and contribute to more ‘tailored’ and personalized anti-metastatic therapies [20].
CTC enrichment combined with an RT-PCR (qPCR) analysis could already be used for the identification of tumor-related markers (EpCAM, MUC1, and ERBB2), EMT-associated transcripts (PI3Ka (phosphatidylinositol 3-kinase alpha), Akt-2, or Twist1), or stem-cell markers such as ALDH1 (aldehyde dehydrogenase 1) [21].
qPCR is highly specific and sensitive and can detect the expression or mutation at a single cell level [22].
The testing of chemoresistance in cancer patients has its own place in treatment. The problem with implementing chemoresistance testing in the past was that it did not reflect the real-time situation but only the character of primary tumors. Testing metastatic tissue would of course be more beneficial but biopsy of metastatic sites is not usually feasible.
As reported in our study, a liquid biopsy (as CTCs are known) itself offers a way to overcome the above-mentioned obstacles. The captured CTCs can mirror the therapy effect - first by enabling numerical CTC analysis, second by qPCR testing of chemoresistance-associated genes. Of course, one could ask: why test, if in some cancer types the only therapy possible is based on just one drug? We believe that even information about inefficacy of drugs could be of value for patient management and could spare the patient many side effects. The main effect of chemoresistance testing in CTCs could provide a real-time message about the effectivity of administered chemotherapy until chemoresistance is developed.
Acknowledgements
This study was supported by Czech Ministry of Health grant IGA NT14441-3/2013.
Disclosure of conflict of interest
None to declare.
References
- 1.Eliasova P, Kolostova K, Kobierzycki C, Bobek V. Clinical studies monitoring circulating and disseminated tumor cells in gastrointestinal cancers. Folia Histochem Cytobiol. 2013;51:265–77. doi: 10.5603/FHC.2013.0037. [DOI] [PubMed] [Google Scholar]
- 2.Mostert B, Sleijfer S, Foekens JA, Gratama JW. Circulating tumor cells (CTCs): detection methods and their clinical relevance in breast cancer. Cancer Treat Rev. 2009;35:463–74. doi: 10.1016/j.ctrv.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Liberko M, Kolostova K, Bobek V. Essentials of circulating tumor cells for clinical research and practice. Crit Rev Oncol Hematol. 2013;88:338–56. doi: 10.1016/j.critrevonc.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Mego M, Reuben JM. Prognostic and Predictive Role of Circulating Tumor Cells in Breast Cancer. Curr Breast Cancer Rep. 2014;6:251–259. [Google Scholar]
- 5.Friedlander TW, Premasekharan G, Paris PL. Looking back, to the future of circulating tumor cells. Pharmacol and Ther. 2014;142:271–280. doi: 10.1016/j.pharmthera.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Song Y, Ji L, Yang L. Advances in techniques for detecting circulating tumor cells. Chinese J Clin Oncol. 2012;39:1859–1863. [Google Scholar]
- 7.Kolostova K, Zhang Y, Hoffman RM, Bobek V. In vitro culture and characterization of human lung cancer circulating tumor cells isolated by size exclusion from an orthotopic nude-mouse model expressing fluorescent protein. J Fluoresc. 2014;24:1531–6. doi: 10.1007/s10895-014-1439-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bobek V, Matkowski R, Gürlich R, Grabowski K, Szelachowska J, Lischke R, Schützner J, Harustiak T, Pazdro A, Rzechonek A, Kolostova K. Cultivation of circulating tumor cells in esophageal cancer. Folia Histochem Cytobiol. 2014;52:171–7. doi: 10.5603/FHC.2014.0020. [DOI] [PubMed] [Google Scholar]
- 9.Ksiazkiewicz M, Markiewicz A, Zaczek AJ. Epithelial-mesenchymal transition: A hallmark in metastasis formation linking circulating tumor cells and cancer stem cells. Pathobiology. 2012;79:195–208. doi: 10.1159/000337106. [DOI] [PubMed] [Google Scholar]
- 10.Joosse SA, Gorges TM, Pantel K. Biology, detection, and clinical implications of circulating tumor cells. EMBO Mol Med. 2015;7:1–11. doi: 10.15252/emmm.201303698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, El-Bahrawy MA. Expression profile of mucins in ovarian mucinous tumors: Distinguishing primary ovarian from metastatic tumors. Int J Gynecol Pathol. 2014;33:166–175. doi: 10.1097/PGP.0b013e318288b384. [DOI] [PubMed] [Google Scholar]
- 12.Hong B, Zu Y. Detecting circulating tumor cells: Current challenges and new trends. Theranostics. 2013;3:377–394. doi: 10.7150/thno.5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coumans FAW, Siesling S, Terstappen LWMM. Detection of cancer before distant metastasis. BMC Cancer. 2013;13:283. doi: 10.1186/1471-2407-13-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alunni-Fabbroni M, Sandri MT. Circulating tumour cells in clinical practice: Methods of detection and possible characterization. Methods. 2010;50:289–297. doi: 10.1016/j.ymeth.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 15.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 16.Esmaeilsabzali H, Beischlag TV, Cox ME, Parameswaran AM, Park EJ. Detection and isolation of circulating tumor cells: principles and methods. Biotechnol Adv. 2013;31:1063–84. doi: 10.1016/j.biotechadv.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 17.Qi XW, Jiang J. Advances in detection methods of circulating breast-tumor cells. Chinese Journal of Cancer Prevention and Treatment. 2010;17:1586–1590. [Google Scholar]
- 18.van de Stolpe A, Pantel K, Sleijfer S, Terstappen LW, den Toonder JM. Circulating tumor cell isolation and diagnostics: toward routine clinical use. Cancer Res. 2011;71:5955–60. doi: 10.1158/0008-5472.CAN-11-1254. [DOI] [PubMed] [Google Scholar]
- 19.Cegan M, Kolostova K, Matkowski R, Broul M, Schraml J, Fiutowski M, Bobek V. In vitro culturing of viable circulating tumor cells of urinary bladder cancer. Int J Clin Exp Pathol. 2014;7:7164–7171. [PMC free article] [PubMed] [Google Scholar]
- 20.Mavroudis D. Circulating cancer cells. Ann Oncol. 2010;21:95–100. doi: 10.1093/annonc/mdq378. [DOI] [PubMed] [Google Scholar]
- 21.Lowes LE, Allan AL. Recent advances in the molecular characterization of circulating tumor cells. Cancers. 2014;6:595–624. doi: 10.3390/cancers6010595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ušiaková Z, Mikulova V, Pintérová D, Brychta M, Valchar J, Kubecova M, Tesarova P, Bobek V, Kološtová K. Circulating tumor cells in patients with breast cancer: Monitoring chemotherapy success. In Vivo. 2014;28:605–614. [PubMed] [Google Scholar]