

Abstract
Calcium release channel on the sarcoplasmic reticulum of cardiomyocytes (ryanodine receptor type 2, RyR2) plays a critical role in the regulation of calcium and was identified as a crucial factor for development of chronic anthracycline cardiomyopathy. Its early stages are less well described although these determine the later development. Hence, we tested the effect of repeated, short-term anthracycline (daunorubicin) administration on cardiac performance, cardiomyocyte function and accompanied changes in calcium regulating proteins expression. Ten-twelve weeks old male Wistar rats were administered with 6 doses of daunorubicin (DAU, 3 mg/kg, i.p., every 48 h), controls (CON) received vehicle. Left ventricular function (left ventricular pressure, LVP; rate of pressure development, +dP/dt and decline, -dP/dt) was measured using left ventricular catheterization under tribromethanol anaesthesia (15 ml/kg b.w.). Cell shortening was measured in enzymatically isolated cardiomyocytes. The expressions of RyR2 and associated intracellular calcium regulating proteins, cytoskeletal proteins (alpha-actinin, alpha-tubul in) as well as oxidative stress regulating enzymes (gp91phox, MnSOD) were detected in ventricular tissue samples using immunoblotting. mRNA expressions of cardiac damage markers (Nppa and Nppb, atrial and brain natriuretic peptides; Myh6, Myh7 and Myh7b, myosin heavy chain alpha and beta) were detected using RT-PCR. Thiobarbituric acid reactive substances concentration was measured to estimate oxidative stress. DAU rats exhibited significantly depressed left ventricular features (LVP by 14%, +dP/dt by 36% and -dP/dt by 30%; for all P<0.05), in line with concomitant increase in Nppa and Nppb gene expressions (3.23- and 2.18-fold, for both P<0.05), and a 4.34-fold increase in Myh7 (P<0.05). Controversially, we observed increased cell shortening of isolated cardiac cells by 31% (p<0.05). DAU administration was associated with a twofold upregulation of RyR2 (P<0.05), but not of other examined Ca2+ regulating proteins remained. In addition, we observed a significant reduction in alpha-tubulin (by 46% when compared to CON P<0.05). Indicators of oxidative injury were unaffected. In conclusion, unbalanced RyR2 overexpression plays a particular role in early development of daunorubicin cardiomyopathy characterized by discrepant in situ versus in vitro cardiac performance.
Keywords: Calcium, ryanodine receptor 2 (RyR2), oxidative stress, daunorubicin cardiomyopathy
Introduction
The therapeutic usage of anthracyclines is limited because of their well-known serious cardiotoxicity leading to cardiomyopathy and consequent cardiac failure. Because cardiac tissue has limited capacity to regenerate, it is very sensitive to toxic attacks of anthracyclines [1,2]. As treatment of consequent dysfunction is only transiently or moderately effective [2-4], it is of crucial importance to determine the responsible toxic mechanism to find an effective and targeted prevention of this specific drug-induced cardiac damage.
Intriguingly, a huge number of mechanisms possibly responsible for anthracycline cardiotoxicity have been introduced [1]. The oxidative stress-based hypothesis involving intramyocardial iron-promoted production of reactive oxygen species (ROS) was believed over decades as the most plausible [5]. The key point in the free radical hypothesis is that anthracyclines and their iron chelates undergo redox cycling, resulting in generation of free radicals and reactive oxygen species. This is supported by successful clinical use of iron-chelator dexrazoxane what remains actually the only clinically well documented protective agent against anthracycline cardiotoxicity [6]. Recently, this concept has been challenged. Several lines of evidence suggest that mechanisms other than the traditionally emphasized “ROS and iron” hypothesis are playing a crucial role in this pathology [7].
In general, abnormal calcium cycling is directly associated with the development of cardiac failure [8-10]. Pharmacologically, the detrimental role of calcium overload in anthracycline cardiotoxicity is extremely interesting as anthracyclines and their metabolites perform a direct pharmacological action on cardiomyocytes as they interact with sarcoplasmic reticulum (SR) proteins and influence intracellular calcium levels [11,12]. Indeed, since decades ago, alterations in calcium cycling have been observed in the presence of anthracyclines what was linked to mitochondrial dysfunction, depletion of high energy phosphates, increased cardiac stiffness, impaired contractility and cell death [13-16]. Interestingly, a direct bimodal effect of anthracyclines on cardiac Ca2+ release channel (ryanodine receptor 2; RyR2) was documented [11,12,17]. The influence of anthracyclines on calcium-regulating proteins expression as well as their action might be the reason also for controversial effects of anthracyclines on cardiac preparations exhibiting both depressed or increased performance [18-20].
SR Ca2+ release is controlled by a tetrameric protein complex that is localized at the junctional SR and consists of the RyR, TRN, junctin (JUN), and CSQ. Functional interactions among TRN, JUN, and CSQ modulate the function of RyR2 [21,22]. According to this model, the binding of CSQ to TRN and JUN inhibits the RyR2 activity at low luminal Ca2+ concentrations. When luminal Ca2+ increases, this inhibition is gradually relieved as the Ca2+ binding sites on CSQ become occupied by Ca2+, attenuating interactions among CSQ and TRN and/or JUN. This results in an increased open probability of the RyR2 [23]. Additionally, an important regulators of calcium cycling are FK506-binding protein (FKBP12), serving as a stabilizer of RyR2 [24], and cardiac sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2a), with its inhibitor phospholamban (PLB), responsible for Ca2+ uptake into SR [25].
In experimental chronic anthracycline cardiomyopathies, the development of delayed cardiac failure was linked to downregulation of RyR2 and SERCA2a [14,16,26,27]. However, early changes are less well described albeit they may substantially determine the later development. As anthracyclines and their metabolites modify RyR2 and SERCA2a activity by binding to the proteins and so influencing Ca2+ cycling [11,12], we hypothesized that modulation of expressions of calcium regulating proteins occurs already at early stages, i.e. in early anthracycline cardiomyopathy and thereby influences its development. Hence, we tested the effect of repeated, short-term anthracycline (daunorubicin) administration on cardiac performance, cardiomyocytes function and accompanied changes in calcium regulating proteins expression. Additionally, we evaluated the protein or mRNA expression of other potentially influential contributors in early anthracycline-induced cardiomyopathy such as ROS-generating enzymes, cardiac myosin heavy chain isoforms, components of renin-angiotensin system and markers of cardiac damage.
Material and methods
Experimental animals and drug application
In our study we used male Wistar rats (10-12 weeks, Dobra Voda, Slovak Republic), divided randomly into two experimental groups: control (CON) and daunorubicin (DAU). In DAU group, six doses of daunorubicin (Daunoblastina inj, Pharmacia Italia, Italy) were injected intraperitoneally (3 mg/kg every 48 hours) [18]. Controls received vehicle (0.9% saline solution). Rats were kept under standard conditions and received food and water ad libitum. All experiments were performed 48 h after administration of the last dose.
All animal care and experiments were approved by the State Veterinary and Food Administration of the Slovak Republic and by the Ethics Committee of the Faculty of Pharmacy, Comenius University in Bratislava and conducted according to the EU adopted Directive 2010/63/EU of the European Parliament and of the Council on the protection of animals used for experimental and other scientific purposes and to the Slovak law regulating animal experiments.
Left ventricular catheterization
A computerized ECG system was used to measure and analyse standard 12-lead ECG. Electrocardiograms were recorded under tribromoethanol (15 ml/kg, i.p.) anaesthesia. Duration of QT, as a measure of cardiac repolarisation, was determined and corrected to rat cardiac cycle length as QTc (in milliseconds) = QT/(RR/150)1/2 [28].
Subsequently, left ventricular function (left ventricular pressure, LVP; heart rate, HR; rate of pressure development, +dP/dt; rate of pressure decrease, -dP/dt) was measured in situ via catheterization of the left ventricle (Spel Advanced HaemoSys; Experimetria Ltd., Budapest, Hungary) as described [18]. The functional response of the left ventricle to beta-adrenergic stimulation was measured upon infusion of increasing doses of isoproterenol (5, 15, 30 and 60 ng/min) into the vena jugularis.
Isolation of left ventricular cardiac myocytes
Ventricular myocytes were isolated enzymatically using a slight modification of protocols published before [18,25]. Shortly, animals were anesthetized by thiopental 45 mg/kg and pre-treated with heparin (1.5 IU). After bilateral thoracotomy, the hearts were quickly excised and cannulated through the aorta fixed in the Langendorff apparatus. Isolated hearts were perfused for 5 min at 2.5 ml/min with Ca2+ free perfusion buffer (37°C) followed by a perfusion with the same buffer supplemented by 0.1 mmol/L ethylene glycol tetraacetic acid (EGTA) for 5 min and finally with enzyme solution containing 0.9 mg/mL collagenase (type II; GIBCOTM Invitrogen Corp., Thermo Scientific, Waltham, MA, USA) in low CaCl2 (50 mmol/L). After enzymatic digestion, the left ventricle was separated, gently mechanically agitated and filtered through a nylon mesh (200 µm pore diameter). Filtrate was centrifugated at 42 g for 60 s, supernatant removed and the pellets of myocytes resuspended in enzyme-free isolation solution containing 0.75 mmol/L of CaCl2 (see Solutions for composition). The isolated left ventricular cardiac myocytes were stored at RT and all experiments were performed within the next 6 h. For measurement, only rod-shaped cells with sharp edges and clear sarcomere striping, without spontaneous contractions and with good reaction to electrical stimulation were used.
Measurement of relative cell shortening of isolated cardiac myocytes
Measurements of the ventricular myocyte function were performed using a videomicroscopy. The cell suspension was placed in custom-made perfusion chamber mounted on a microscope. This chamber allowed simultaneous perfusion with perfusion solution (see Solutions for composition) and electrical field-stimulation (0.5, 1, 2 or 3 Hz, pulse amplitude 10V, pulse width 5ms). The cells were then superfused with perfusion solution and electrically paced by different frequencies. Myocyte shortening was visualized on a PC monitor. The image was processed in software Cell Length Monitor 1.0 (International Laser Centre, Bratislava, Slovak Republic), where the longitudinal line for detecting the changes of the cell edges position during electrical stimulation was used for measurement of the cell shortening. The maximum contraction was normalized to resting cell length and expressed as percentage of shortening. Cell shortening was measured at stimulation frequency 0.5 Hz before and after acute isoproterenol application (10-7 mol/L ISO in perfusion solution) and at stepwise increasing stimulation frequency (0.5 Hz, 1 Hz, 2 Hz, and 3 Hz). Moreover, the time to 50% decay (T50) and time to 90% decay (T90) of the cell shortening (defined as the time from peak level to 50% or to 90% of the peak amplitude, respectively) at stimulation frequency 0.5 Hz was calculated.
Solutions
The basis for solutions used for isolation and storage of myocytes was Krebs-Henseleit solution contained (in mmol/L): 130 NaCl, 5.4 KCl, 1.4 MgCl2·6H2O, 1.2 NaH2PO4·H2O, 0.4 NaH2PO4, 10.0 creatine, 20.0 taurine, 10.0 glucose, 10.0 4-(2-hydroxyethyl)-1-piperazineethenesulfonic acid (HEPES), titrated to pH 7.3 with NaOH.
Basic external solution perfused on single cardiomyocytes during recording contained (mmol/L): 130 NaCl, 5.4 KCl, 1.4 MgCl2·6H2O, 0.4 NaH2PO4, 10.0 glucose, 10.0 HEPES and 1.8 CaCl2 adjusted to pH 7.3 with NaOH.
All chemicals were purchased from Sigma-Aldrich (Munich, Germany). Thiopental and heparin were obtained from Lachema (Brno, Czech Republic).
SDS PAGE and Western blotting
For Western blotting analysis, left ventricles of rats after catheterization were further used. Tissue samples from left ventricles were flash-frozen in liquid nitrogen and homogenized in a buffer containing 10 mM Tris-HCl (pH 7.4), 0.125 M sucrose, 1 mM EDTA-Na, 10% sodium dodecylsulphate and 1 mM phenylmethylsulphonyl fluoride, as described previously [25]. For immunoblot analysis, 50 µg (for detection of sarco-endoplasmic reticulum Ca2+ ATP-ase, SERCA2a; endothelial NO-synthase, eNOS; caveolin-1 and -3, cav-1, cav-3; heat shock protein, hsp90; glycosylated subunit of the NADPH oxidase, gp91phox; α-tubulin; α-actinin; β-actin; manganese superoxide dismutase, MnSOD), 100µg (for detection of calsequestrin, CSQ; triadin, TRD; phospholamban, PLB; FK506 binding protein, FKBP12) 150 µg (for detection of inducible NO-synthase, iNOS) or 250 µg (for detection of RyR2) of homogenate protein were separated on SDS-PAGE. After transferring of proteins to polyvinylidene fluoride (PVDF) transfer membrane (Immobilon-P, Millipore Corp., Billerica, MA, USA), the blots were incubated with different antibodies. We used various antibodies raised against the following proteins: eNOS, iNOS, cav-1, cav-3, hsp90, gp91phox, MnSOD (BD Transduction Laboratories, Franklin Lakes, NJ, USA), α-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), α-actinin (Sigma-Aldrich, Saint Louis, MO, USA). Specific monoclonal antibodies against RyR2, SERCA2a, PLB, TRD, FKBP12 and CSQ were used as described [25]. β-actin (Sigma-Aldrich, Saint Louis, MO, USA) was used as a housekeeping protein for all investigating proteins except proteins of SR (TRD, PLB, FKBP12, RyR2, SERCA2a) where the CSQ took over his role. The amount of bound protein was detected by using chemiluminescent detection (ECL Plus, Amersham, Buckinghamshire, UK) or by 125I-labeled protein A and quantified using Storm 860 (Molecular Dynamics, Sunnyvale, CA). Quantification was performed using threedimensional densitometry in Optiquant (Packard Instruments) or ImageQuant® (Molecular Dynamics, Sunnyvale, USA), respectively. To allow comparison of the protein levels between groups, these arbitrary density levels were then normalized to the average control level.
Isolation of RNA and real-time PCR
Separate groups of rats were used. As described [25], total RNA was isolated from rat cardiac left ventricle using Tri-Reagent (Ambion, Grand Island, NY, USA). Isolated RNA was verified to be intact by using agarose gel electrophoresis. Reverse transcription to cDNA was performed using High Capacity cRNA RT Kit with RNAse inhibitor (Applied Biosystems, Grand Island, NY, USA), 1 µg of total RNA were reverse-transcribed to cDNA. mRNA levels of selected genes (see Table 1) were evaluated using real-time PCR (ABI Prism 7300, Applied Biosystems, USA) with gene-specific primers with SYBR green detection (Power SYBR Green PCR Master Mix, Applied Biosystems, Foster City, California, USA). Relative quantification of mRNA expression in RT-PCR experiments was performed by using the 2-ΔΔCt (Ct-threshold cycle) method. Results were normalized to the expression of endogenous reference genes (Actb, B2m) and calibrated to the control group. All primers (Table 1) were verified to yield a single PCR product with correct molecular weight and the absence of signal was verified when reverse transcription was omitted.
Table 1.
Primer sequences for quantitative RT-PCR
| GenBank official symbol mRNA accession number | Primer sequences (5’→3’) | PCR product length (nt) |
|---|---|---|
| Actb | forward: CCGCGAGTACAACCTTCTTG | 81 |
| NM_031144.2 | reverse: GCAGCGATATCGTCATCCA | |
| B2m | forward: ATGGAGCTCTGAATCATCTGG | 105 |
| NM_012512.1 | reverse: AGAAGATGGTGTGCTCATTGC | |
| Myh6 | forward: GCCCTTTGACATCCGCACAGAGT | 152 |
| NM_017239.2 | reverse: TCTGCTGCATCACCTGGTCCTCC | |
| Myh7 | forward: GCGGACATTGCCGAGTCCCAG | 133 |
| NM_017240.1 | reverse: GCTCCAGGTCTCAGGGCTTCACA | |
| Myh7b | forward: CCCGATTCTCAACACCAACACCTCT | 150 |
| NM_001107794.2 | reverse: CATCAGGCACCCAGACCCGT | |
| Ryr2 | forward: ACTGCTGGGCTACGGCTAC | 99 |
| NM_001191043.1 & NM_032078.2 | reverse: CTGAAGATGCGGAACCTCTC | |
| Cacna1c | forward: GTTGCCCTGGGTGTATTTTG | 109 |
| NM_012517.2 | reverse: GGCTTTCTCCCTCTCTTTGG | |
| Atp2a2 | forward: CCCGAAACTACCTGGAGCCTGCA | 83 |
| NM_001110139.2 | reverse: ATGCACGCACCCGAACACCC | |
| Trpc3 | forward: GCGTCGCTGAGTCGGGTCAAA | 130 |
| NM_021771.2 | reverse: CGATGGTCTGCTCCCGAAGGC | |
| Trpc6 | forward: TCGTGGCGCATCCGAACTGC | 285 |
| NM_053559.1 | reverse: GGAGGAGCTTGGTGCCTTCAAATCT | |
| Acta1 | forward: TCACTTCCTACCCTCGGCACCC | 142 |
| NM_019212.2 | reverse: GGGGCATCATCCCCGGCAAAG | |
| Nppb | forward: GACCGGATCGGCGCAGTCAGT | 78 |
| NM_031545.1 | reverse: GGAGTCTGCAGCCAGGAGGTCT | |
| Nppa | forward: GGGGGTAGGATTGACAGGAT | 104 |
| NM_012612.2 | reverse: GGATCTTTTGCGATCTGCTC | |
| Ctf1 | forward: GGAAGTCTGGAAGACCACCA | 137 |
| NM_017129.1 | reverse: TGCTGCACATATTCCTCCAG | |
| Kcnq1 | forward: CTGGTCTGCCTCATCTTCAG | 110 |
| NM_032073.1 | reverse: TCTGTCCCAAAGAACACCAC | |
| Kcnh2 | forward: GACCTGCTTACTGCCCTCTAC | 124 |
| NM_053949.1 | reverse: GACGTGCATACAGGTTCAGAG | |
| Gja1 | forward: CCTTTGACTTCAGCCTCCAA | 86 |
| NM_012567.2 | reverse: CATGTCTGGGCACCTCTCTT | |
| Scn5a | forward: TGTATGTCCTCAGCCCCTTC | 112 |
| NM_013125.2 | reverse: ATGAACACGCAGTTGGTCAG | |
| Ace | forward: tcctgctagacatggagacga | 142 |
| NM_012544.1 | reverse: cagctcttccacacccaaag | |
| Ace2 | forward: cgctgtcaccagacaagaa | 129 |
| NM_001012006.1 | reverse: cgtccaatcctggttcaag | |
| Agtr1a | forward: cacacaaccctcccagaaag | 147 |
| NM_030985.4 | reverse: ttggggcagtcatcttgg | |
| Mas1 | forward: cagagctgggtttacctgga | 132 |
| NM_012757.2 | reverse: atggctttctcctcagcaaa | |
| Thrb | forward: TCAGAGCGTCTCAAGCGCCCA | 108 |
| NM_012672.3 | reverse: TTGTCCCCGCACACTACGCAG | |
| Thra1 | forward: CGTCCTTGCTCAGCTCGGTCCA | 120 |
| NM_001017960.1 | reverse: AGAGCTAGGGGCAAGCTGGCT | |
| Thra2 | forward: AACATTCCGCACTTCTGGCCCA | 166 |
| NM_031134.2 | reverse: GGACCTGCGGACCCTGAACAAC |
Abbreviations: Actb (actin, beta); B2m (beta-2 microglobulin); Myh6 (myosin, heavy chain 6, cardiac muscle, alpha); Myh7 (myosin, heavy chain 7, cardiac muscle, beta); Myh7b (myosin, heavy chain 7B, cardiac muscle, beta); Ryr2 (ryanodine receptor 2, cardiac); Cacna1c (calcium channel, voltage-dependent, L type, alpha 1C subunit); Atp2a2 (ATPase, Ca++ transporting, cardiac muscle, slow twitch 2); Trpc3 (transient receptor potential cation channel, subfamily C, member 3); Trpc6 (transient receptor potential cation channel, subfamily C, member 6); Acta1 (actin, alpha 1, skeletal muscle); Nppb (natriuretic peptide B); Nppa (natriuretic peptide A); Ctf1 (cardiotrophin 1); Kcnq1 (potassium channel, voltage-gated KQT-like subfamily Q, member 1); Kcnh2 (potassium channel, voltage gated eag related subfamily H, member 2); Gja1 (gap junction protein, alpha 1); Scn5a (sodium channel, voltage-gated, type V, alpha subunit); Ace (angiotensin I converting enzyme); Ace2 (angiotensin I converting enzyme 2); Agtr1a (angiotensin II receptor, type 1a); Mas1 (MAS1 proto-oncogene, G protein-coupled receptor); Thrb (thyroid hormone receptor beta); Thra1 (thyroid hormone receptor alpha, transcript variant TRalpha1); Thra2 (thyroid hormone receptor alpha, transcript variant TRalpha2).
Estimation of lipid peroxidation
The concentration of thiobarbituric acid reactive substances (TBARSs) was used as a biomarker to measure the level of oxidative stress in left ventricular tissue [29]. Shortly, left ventricular samples were homogenized in 10 mM potassium phosphate buffer, pH 7.4. Thereafter, 0.5% solution of thiobarbituric acid in 20% trichloroacetic acid was added to homogenates. These mixtures were incubated for 30 min at 90°C. Then, samples were cooled and after centrifugation (at 11000 RPM for 10 min) measured using spectrophotometer Multiscan RC (Labsystems, Vantaa, Finland) at 530 nm. The measured TBARs levels were finally expressed as nmol/mg per total protein.
Data analysis
All variables are reported as mean and standard error of the mean (SEM). Data were compared by Mann-Whitney U test. Values P<0.05 were considered significant. The data were handled by GraphPad Prism 4 for Windows (GraphPad Software, Inc., version 4.00, San Diego, California, USA).
Results
Wasting syndrome and impaired left ventricular function
As expected, cumulative dose of daunorubicine (18 mg/kg) used in our study caused significant cachexia manifested as significant loss in body weight as well as reduced weights of internal organs, including the heart (see Table 2). In situ, left ventricular catheterisation unveiled a depressed cardiac function with significantly reduced LVP (by 13%), +dP/dt (by 42%) and -dP/dt (by 31%) in DAU group when compared to controls (Table 2). This dysfunction persisted also under beta-adrenergic stimulation (Figure 1).
Table 2.
Basal gravimetric, cardiac and myocyte parameters of control (CON) and daunorubicin (DAU) experimental group
| CON | DAU | |
|---|---|---|
| Gravimetry (n) | 13 | 14 |
| Body weight (g) | 277±9 | 204±4* |
| Heart (mg) | 857±20 | 600±14* |
| Heart/body weight (mg/g) | 3.11±0.08 | 2.95±0.06 |
| Lung (mg) | 1202±54 | 976±36* |
| Liver (g) | 10.5±0.4 | 6.4±0.3* |
| Spleen (mg) | 636±19 | 315±24* |
| Kidney (mg) | 937±25 | 650±16* |
| 12-lead electrocardiography | 7 | 8 |
| RR (ms) | 152±4 | 182±6* |
| QT (ms) | 76±2 | 93±2* |
| QTc (ms) | 75±1 | 84±2* |
| Left ventricular catheterization (n) | 7 | 7 |
| Heart rate (b/min) | 410±9 | 396±14 |
| Left ventricular pressure (mmHg) | 132±5 | 114±6* |
| +dP/dt (mmHg/s) | 4976±503 | 2893±435* |
| -dP/dt (mmHg/s) | -4144±304 | -2868±297* |
| Isolated cells (animals/cells) | 5/40 | 5/45 |
| Cell lenght (μm) | 117.1±2.9 | 120.7±2.5 |
| Cell width (μm) | 24.8±1.0 | 27.3±1.2 |
| Cell shortening (%) | 5.1±0.4 | 6.7±0.5* |
| Time to 50% decay (ms) | 140±6 | 117±7* |
| Time to 90% decay (ms) | 406±20 | 333±19* |
Data are shown as mean ± SEM. n = number of animals;
P<0.05 vs. CON.
Figure 1.
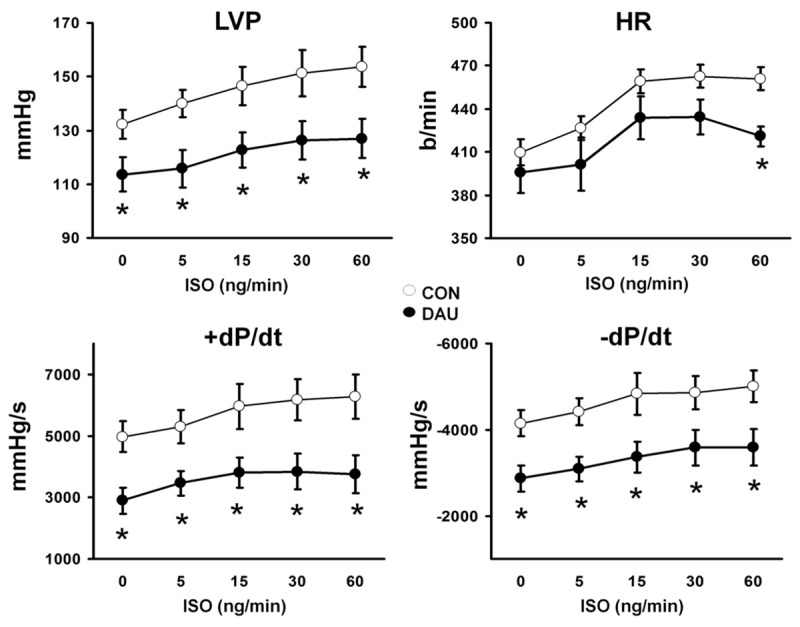
Left ventricular catheterisation performed in closed-chest anaesthetized rats from control (CON, n = 7, white circles) and daunorubicin (DAU, n = 7, black circles). Increasing doses of isoproterenol (ISO) were injected into the left jugular vein, determined in ng/min. LVP-left ventricular pressure, HR-heart rate, +dP/dt -rate of pressure development , -dP/dt -rate of pressure decline, data are shown as mean ± SEM, *P<0.05 vs CON.
Enhanced cell shortening in single cardiac myocytes
We examined whether the impaired cardiac function could be due to deterioration in the function of isolated ventricular cardiomyocytes. Interestingly, these experiments revealed a significant augmentation in the relative cell shortening at the stimulation frequency of 0.5 Hz by 33% in DAU group (Table 2, Figure 2A) with a significant reduction in the T50 by 16% as well as in the T90 by 18%. Similarly, we observed an enhanced cell shortening in isolated cardiac myocytes in the DAU group compared to CON at all other stimulation frequencies (Figure 2B). The increased cell shortening in DAU cardiomyocytes as compared to CON persisted also under ISO stimulation (Figure 2C). However, in contrast to CON, DAU cardiomyocytes failed to respond significantly to beta-adrenergic stimulation.
Figure 2.
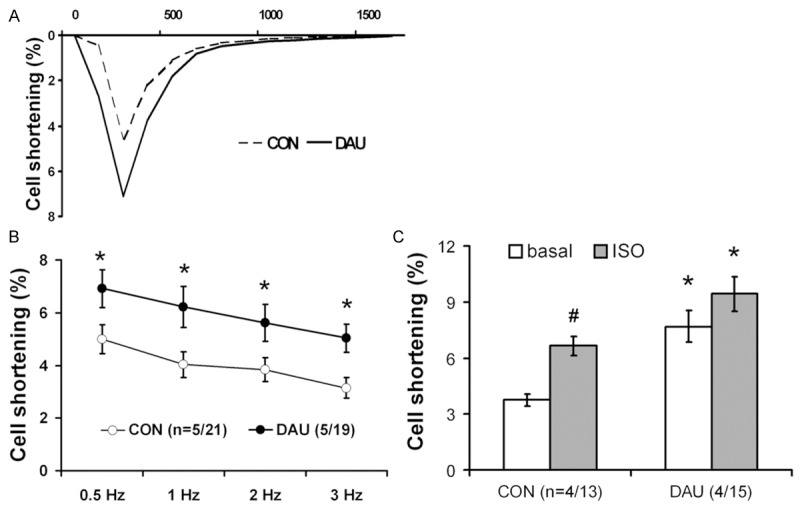
Cell shortening of single ventricular myocytes. A. Representative time curve of cell shortening of single myocyte from control (CON, dashed line) and daunorubicin (DAU, solid line) group. B. Frequency-dependent effects on myocyte cell shortening. Response to different stimulation frequencies is displayed for CON (white circles) and DAU (black circles) myocytes, displayed as average ± SEM. Frequency was increased stepwise every 5 min from 0.5 to 3 Hz *P<0.05 vs CON, in brackets: no. of animals/myocytes. C. Myocyte cell shortening in response to β-adrenergic receptor stimulation in CON and DAU myocytes. The cell shortening was measured at 0.5-Hz stimulation frequency first at basal conditions (superfusion with perfusion solution, white columns, basal) and then in response to isoproterenol (superfusion with perfusion solution supplemented by isoproterenol to final concentration 10-7 mol·l-1, grey columns, ISO). Data are shown as average ± SEM, *P<0.05 vs CON, #P<0.05 vs. basal, in brackets: no. of animals/myocytes.
An upregulation of RyR2 but a stable expression of other SR proteins
As cardiac excitation-contraction cycle is directly dependent on a proper cellular calcium handling, we investigated the protein levels of the main SR proteins involved in Ca2+-induced Ca2+ release (Figure 3). Importantly, we found a significant, 94% increase of RyR2 expression in DAU. However, this was not accompanied by alterations in the expressions of RyR2 accessory proteins (TRD, CSQ and FKBP12) or of other tested Ca2+ regulating proteins (SERCA2a and PLB).
Figure 3.
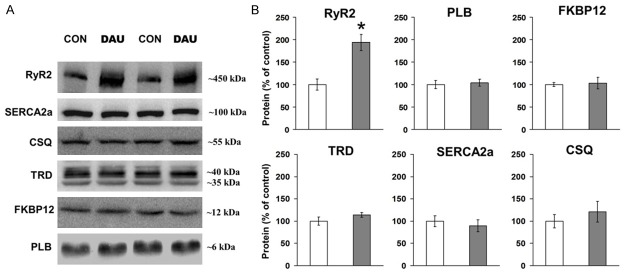
Western blotting analysis of Ca2+ regulating proteins levels. A. Representative immunoblots of total protein level of ryanodine receptor (RyR2), sarco-endoplasmic reticulum ATP-ase 2a (SERCA2a), calsequestrin (CSQ), triadin (TRD), FK506 binding protein (FKBP12) and phospholamban (PLB) from control (CON) and daunorubicin (DAU) group. B. Quantification of sarcoplasmic reticulum proteins. Data from DAU (grey columns) are normalized to CON (white columns) and are displayed as % of CON ± SEM, *P<0.05 vs CON, n = 7-9 per group.
Unaffected indicators of oxidative stress
Because anthracyclines may activate oxidative stress, we determined the protein levels of several proteins involved in its signalling pathways such as ROS regulating enzymes (gp91phox, MnSOD) and also proteins of nitric oxide cascade (eNOS, iNOS, hsp90, cav-3, cav-1). We found no significant alterations in the above mentioned proteins, except for an increased protein levels of cav-1 and cav-3 in DAU (by 22% and 23%, resp., P<0.05 vs. CON; Figure 4A, Table 3). Moreover, TBARS determination, to reveal possible lipid peroxidation, also showed no significant changes between DAU and CON group (Figure 4C).
Figure 4.
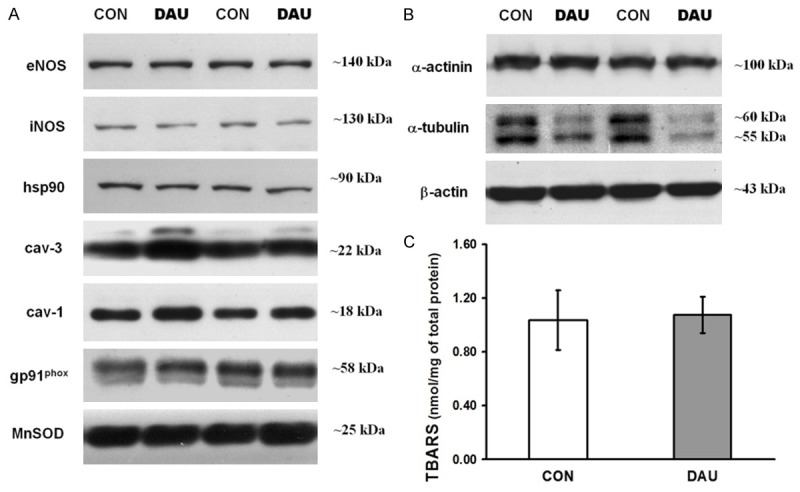
Western blotting analysis of proteins linked to anthracycline-induced oxidative stress and cytoskeletal proteins. A. Representative immunoblots of total protein level of endothelial NO-synthase (eNOS), inducible NO-synthase (iNOS), heat shock protein 90 (hsp90), caveolin 3 and 1 (cav-3, cav-1), glycosylated subunit of the NADPH oxidase (gp91phox) and manganese superoxide dismutase (MnSOD) from control (CON) and daunorubicin (DAU) group. B. Representative immunoblots of total protein level of cytoskeletal proteins α-tubulin, α-actinin and β-actin from CON and DAU. C. The concentration of thiobarbituric acid reactive substances (TBARSs) in ventricular samples. Data are shown as mean ± SEM, n = 6 per group.
Table 3.
Level of protein expression in left ventricular tissue determined by western blot
| CON | DAU | |
|---|---|---|
| NO-signalling proteins | ||
| eNOS | 100±6 | 95±4 |
| iNOS | 100±16 | 84±9 |
| hsp90 | 100±13 | 102±6 |
| cav-1 | 100±8 | 122±5* |
| cav-3 | 100±10 | 123±8* |
| ROS regulating enzymes | ||
| gp91phox | 100±5 | 93±8 |
| MnSOD | 100±9 | 100±15 |
| Cytoskeletal proteins | ||
| α-actinin | 100±5 | 104±10 |
| α-tubulin | 100±9 | 54±8* |
| β-actin | 100±7 | 105±4 |
Mean ± SEM. n = 7-9 per group;
P < 0.05 vs. CON.
Initially, cytoskeletal proteins were used as loading controls as they showed stable expression (α-actinin and β-myosin). Interestingly, we found a significant decrease in protein expression of α-tubulin by 46% in DAU ventricles as compared to CON (P<0.05 vs. CON; Figure 4B).
Higher mRNA expressions of RyR2, Myh7 and heart failure markers
We performed real-time PCR to determine mRNA expression of several gene groups including genes for calcium signalling, markers related to heart failure, genes related to heart conduction system, components of renin-angiotensin system and thyroid hormone receptors (Table 3). In concert with increased protein level, mRNA levels of Ryr2 was also significantly higher in DAU group in compare to CON (by 1.29 fold, P<0.05) with no significant differences in mRNA expression of other genes of Ca2+ signalling (Cacna1c, Serca2a, Trpc3, and Trpc6). Gene expression analysis also revealed an increase in mRNAs of all observed heart failure markers (Acta1, Nppb, Nppa, Ctf1) in DAU group as compared to CON (P<0.05). Similarly, mRNA levels of myosin heavy chain beta (Myh7, Myh7b) were in DAU significantly upregulated against controls (4.34 fold and 1.57 fold, resp., P<0.05). In the DAU group, genes regulating cardiac electrogenesis were also affected. We found an enhanced expression of Scn5a and Kcnq1 (Table 4).
Table 4.
mRNA levels of selected genes using RT-PCR normalised to control group
| Gene group | mRNA product of PCR | CON | DAU (fold of CON) |
|---|---|---|---|
| Cardiac myosin heavy chain isoforms | Myh6 | 1.00±0.07 | 0.81±0.13 |
| Myh7 | 1.00±0.11 | 4.34±1.02* | |
| Myh7b | 1.00±0.10 | 1.57±0.15* | |
| Calcium signaling | Ryr2 | 1.00±0.09 | 1.29±0.08* |
| Cacna1c | 1.00±0.09 | 1.15±0.07 | |
| Serca2a | 1.00±0.08 | 1.14±0.07 | |
| Trpc3 | 1.00±0.05 | 1.12±0.12 | |
| Trpc6 | 1.00±0.09 | 1.08±0.11 | |
| Markers related to cardiac failure | Acta1 | 1.00±0.23 | 1.46±0.17* |
| Nppb | 1.00±0.14 | 2.18±0.42* | |
| Nppa | 1.00±0.16 | 3.23±0.86* | |
| Ctf1 | 1.00±0.11 | 1.65±0.25* | |
| Conduction system of the heart | Kcnq1 | 1.00±0.07 | 1.34±0.16* |
| Kcnh2 | 1.00±0.10 | 1.17±0.16 | |
| Gja1 | 1.00±0.06 | 1.26±0.13 | |
| Scn5a | 1.00±0.03 | 1.30±0.08* | |
| Renin-Angiotensin system | Ace | 1.00±0.07 | 1.15±0.21 |
| Ace2 | 1.00±0.12 | 0.84±0.09 | |
| Agtr1 | 1.00±0.11 | 1.31±0.16 | |
| Mas | 1.00±0.23 | 0.97±0.43 | |
| Thyroid hormone receptors | Thrb | 1.00±0.07 | 1.26±0.18 |
| Thra1 | 1.00±0.06 | 1.11±0.12 | |
| Thra2 | 1.00±0.06 | 1.04±0.13 |
For abbreviations see Table 1. Mean ± SEM. n = 7-9 per group;
P < 0.05 vs. CON.
Discussion
Anthracyclines have undoubtedly a key role in the treatment of many neoplastic diseases, but cardiotoxic effects limit their chemotherapeutic use. Although the mechanism of anthracycline-induced toxicity is in all likelihood complex and consists of several factors [1], direct modulation of cellular Ca2+ cycling [11,12] emphasizes the importance of this component for early development of anthracyclines-induced damage. Indeed, several papers describe involvement of reduced RyR2 expression in chronic cardiac dysfunction) [26,27,30]. Some of them suggested the influence of iron-dependent oxidative stress in the RyR2 downregulation [27] that might be a consequence of reciprocal interaction between generation of reactive oxygen species and RyR2 opening [15]. As anthracyclines directly and indirectly modulate RyR2 function, we hypothesized that the cardiac tissue would respond to repeated stimuli by immediately changing the channel expression. Indeed, the main finding of our study is the twofold increase in cardiac RyR2 protein expression accompanied by an exaggerated isolated cardiomyocytes function, concomitantly to in situ cardiac dysfunction, in the model of early daunorubicin cardiomyopathy. Interestingly, the upregulation of RyR2 was not accompanied by alterations in other Ca2+ regulating proteins (and was also not accompanied by significant alterations in biomarkers of oxidative stress). This indicates that RyR2 is a key player in the early development of anthracycline car diomyopathy.
Binding of anthra cyclines and/or their metabolites to SR proteins (RyR2, SERCA2a and CSQ) is a well known phenomenon [11,12,31]. In particular, their binding to RyR2 favours channel opening, leading to increased cytosolic Ca2+ [32]. In this study we showed that this pharmacological feature causes alteration in expression of Ca2+ release channel and, consequently, it is directly involved in early development of cardiomyopathy. As showed, concomitantly to alterations in cardiac performance, exclusively the level of RyR2 increased following DAU treatment, corresponding with a slight, but significant increase of its mRNA levels. Presumably, this upregulation of RyR2 can alter channel stoichiometry dramatically because the expressions of other proteins in the complex formed by RyR2 (triadin and calsequenstrin) remained unchanged. Changes in RyR2 complex stoichiometry could attenuate interactions among RyR2 accessory proteins, reducing the control of RyR2 from the luminal site of SR, and consequently increasing the open probability of the RyR2 and it thus may have detrimental consequences for cardiomyocytes function by triggering abnormal Ca2+ release and/or Ca2+ leak [21,22].
Similarly, cardiac expression of FK506 binding protein (FKBP12), a stabilizer of RyR2 [24] which prevents aberrant activation of the channel during the resting phase of the cardiac cycle [33], also remained unaltered in our study. Importantly, the absent compensatory upregulation of FKBP12 can lead to defective RyR2 function. Additionally, expression of SERCA2a and its regulator phospholamban remained unaltered as well, suggesting that the possible Ca2+ overload remained without compensatory changes in expression of these proteins of SR. Presumably, the unbalanced upregulation of RyR2 suggests that several RyR2 units could be unbound to FKBP12, and not being correctly regulated on the surface of the SR. Thus, the associated persistent Ca2+ leak and consequent Ca2+ overload are the most plausible explanation of development of anthracycline cardiomyopathy in our study.
Interestingly, isolated cardiomyocytes from DAU hearts exhibited abnormal increase of cell shortening in our study. Although anthracyclines typically lead to depressed cardiac function in situ and in vivo [16,18,26], controversial findings of increased performance of cardiac preparations were found also by others. In vitro, anthracyclines cause both positive as well as negative inotropic effect, likely depending on concentration of used drug and/or experimental settings [17,19,20]. Presumably, the augmented cardiomyocytes shortening observed in our study might be an effect of complex modulation of cardiomyocytes by both, immediate direct and/or indirect action of daunorubicin on RyR2 as well as compensatory changes in RyR2 expression in the early stage of cardiomyopathy.
In addition to an abnormal Ca2+ release, our finding of an increased cell shortening might be related also to other observed alterations in our left ventricular samples. In agreement with other studies using anthracyclines [34,35], we found an increase of β-myosin heavy chain (β-MHC) expression on mRNA level (Myh7, Myh7b genes) concomitantly with only non-significantly reduced a-myosin heavy chain level (±-MHC; Myh6 gene). These changes were independent of changes in mRNA expressions of thyroid hormone receptors [36] or components of RAAS axis [37]. ²-MHC is, in comparison to ±-MHC, characterized by a lower ATPase activity and lower filament sliding velocity, but it can generate cross-bridge force with a higher economy of energy consumption [38]. Therefore, a shift to enhanced ²-MHC expression could lead to better preservation of intracellular energy and could be viewed as a compensatory alteration tending to improve force-generating ability [39]. On the other hand, studies using ²-MHC transgenic mice reported that shift from ±- to ²-MHC may be rather maladaptive and promotes cardiac disease progression presumably due to the decreased contractile performance of cardiac myocytes lasting also under beta-adrenergic stimulation [40]. Thus, the absent response to isoproterenol stimulation in DAU cardiomyocytes could be associated with the shift from ±- to ²-MHC.
With regard to the increased RyR2 expression with an abnormal cell shortening, we next further analyzed the cytoskeleton proteins (sarcomeric skeleton protein α-actinin and microtubular protein α-tubulin) due to their undoubted role in normal myocyte function. Cytoskeletal microtubules transmit mechanical and chemical stimuli within and between cells, contribute substantially to cell stability by anchoring subcellular structures, modulate cardiomyocyte function [41] and play a role in energy metabolism [42]. In this vein, microtubule disruption was associated with an altered Ca2+ handling [43] and a reduced myocyte stiffness with an increment in sarcomere motion [44]. It has been shown that anthracyclines reduce β-tubulin [34]. Similarly, we found a reduction in α-tubulin protein content that could diminish viscous intracellular load and reduce cardiomyocytes stiffness [45] and help to enhance cell shortening and reduce times to myocyte relaxation. Furthermore, in association with increased RyR2 expression and upregulation of β-MHC, the reduction in α-tubulin could contribute to the increased shortening of isolated cells from DAU hearts.
In addition to the observed RyR2 upregulation, we also detected elevated mRNA expression of Scn5a (gene encoding Nav1.5, a sodium ion channel protein) and Kcnq1 (gene encoding Kv7.1, a potassium channel protein). These changes could contribute not only to abnormal electrogenesis, manifesting as prolonged QT, but also to an abnormal balance of key ions involved in the excitation-contraction coupling and thus influencing also the process of cardiac contraction [46,47].
Importantly, we found a suspicious discrepancy between an enhanced function of ventricular cardiomyocytes in vitro and a depressed contractility of the heart during closed-chest preparation (before and after β-AR stimulation) in the DAU group. We expect the involvement of several possible mechanisms: i) structural abnormalities of ventricular tissue like expanded extracellular space, e.g. interstitial edema [48] that could impair the signal transduction between myocytes, ii) compensatory systemic neurohumoral activation/inhibition that is irrelevant in isolated ventricular myocytes, and/or iii) loss of cardiomyocytes due to apoptosis or necrosis [16,49]. Importantly, expression of all observed genes related to the cardiac hypertrophy and heart failure (Acta1, Nppb, Nppa, Ctf1) were augmented in DAU group. Altogether, this points to cardiac dysfunction with dilated cardiomyopathy rather than hypertrophy, in agreement with other studies [50].
Generally, an increased oxidative (as well as nitrosative) stress is believed to be a trigger mechanism for anthracycline cardiomyopathy [5,51]. However, we found no alteration in proteins involved in activation of oxidative stress or pro/antioxidant factors in DAU group when compared to CON. Similarly, we did not observed any alterations in lipid peroxidation using TBARS experiment between experimental groups. Lack of oxidative injury after anthracyclines, in particular in acute studies, was reported also by others [52]. Supportingly, single observed difference in NO signalling was the slightly but significantly increased protein level of negative eNOS modulators - cav-1 and cav-3 in DAU group. As well accepted, eNOS may cause tissue damage by generating reacting nitrogen species what has been described also in case of anthracyclines medication [51]. Uncoupling of eNOS by cav-1 deficiency has been proposed for anthracycline-induced cardiomyopathy [53]. Contrariwise, upregulation of caveolins will rather decrease eNOS activity. This finding rather refuses the possibility of nitrosative stress in hearts of DAU rats. Altogether, our findings suggest that oxidative as well as nitrosative stress play rather a minor role in early daunorubicine cardiomyopathy.
Conclusion
Taking together, short-term DAU administration impaired the performance of the heart in situ but, on the cellular level, single cardiomyocytes displayed an enhanced function. We identified three potential mechanisms responsible for the exaggerated performance. In particular, increased expression of RyR2, without compensatory changes in other Ca2+ regulatory proteins, was identified as the responsible component resulting into development of cardiomyopathy presumably via increased cytosolic Ca2+ overload, transiently stimulating muscle contraction, manifesting by enhanced cardiomyocyte shortening in our study. Also, decreased levels of α-tubulin can contribute to controversial performance by reducing cell stiffness and so facilitating cardiomyocyte shortening and, particularly, relaxation. Additionally, a compensatory upregulation of β-MHC could theoretically contribute to observed cardiomyocytes function. However, whether the isoform shift from α- to β-MHC not only maintains but could even increase the contraction amplitude must be proven in additional studies.
Clinical implications
Despite tremendous research and testing of thousands of potentially protective, predominantly oxidative stress reducing agents, leading to disappointing clinical results with classical antioxidants, only a single drug has been approved for use in clinical practice: dexrazoxane [5]. According to the generally accepted hypothesis, this drug exerts its cardioprotective effects by prevention of site-specific iron-catalyzed ROS production [54]. However, studies with chelators that are stronger and more selective for iron yielded negative or, at best, mixed outcomes suggesting that other mechanism may be better bases for designing approaches to achieve efficient and safe cardioprotection [7]. In parallel, dexrazoxan chelates also other ions, including calcium [55] and an experimental study showed that dexrazoxan protection from chronic DAU cardiomyopathy was accompanied by prevention of chronic calcium overload of the myocardial tissue, without any significant effect on the myocardial level of iron [7]. In this context, also our findings highlight the prevention of calcium dysregulation, particularly its overload, as the most promising candidate approach for cardioprotection against anthracycline injury.
Acknowledgements
Grant APVV-0887-11 Molecular aspects of drug induced heart failure and ventricular arrhythmias from the Slovak Research and Development Agency, grants 1/0564/13 and 1/0294/15 from the Science Grant Agency (VEGA), Slovak Republic and Grant of European Regional Development Fund-Project FNUSA-ICRC (No.CZ.1.05/1.1.00/02.0123) and the grant of Slovak Society of Cardiology 2007.
Disclosure of conflict of interest
Authors declare no conflict of interest.
References
- 1.Chen B, Peng X, Pentassuglia L, Lim CC, Sawyer DB. Molecular and cellular mechanisms of anthracycline cardiotoxicity. Cardiovasc Toxicol. 2007;7:114–121. doi: 10.1007/s12012-007-0005-5. [DOI] [PubMed] [Google Scholar]
- 2.Roziakova L, Mistrik M, Batorova A, Kruzliak P, Bojtarova E, Dubrava J, Gergel J, Mladosievicova B. Can We Predict Clinical Cardiotoxicity with Cardiac Biomarkers in Patients After Haematopoietic Stem Cell Transplantation? Cardiovasc Toxicol. 2015;15:210–6. doi: 10.1007/s12012-014-9286-7. [DOI] [PubMed] [Google Scholar]
- 3.Georgakopoulos P, Roussou P, Matsakas E, Karavidas A, Anagnostopoulos N, Marinakis T, Galanopoulos A, Georgiakodis F, Zimeras S, Kyriakidis M, Ahimastos A. Cardioprotective effect of metoprolol and enalapril in doxorubicin-treated lymphoma patients: a prospective, parallel-group, randomized, controlled study with 36-month follow-up. Am J Hematol. 2010;85:894–896. doi: 10.1002/ajh.21840. [DOI] [PubMed] [Google Scholar]
- 4.Lipshultz SE, Lipsitz SR, Sallan SE, Simbre VC 2nd, Shaikh SL, Mone SM, Gelber RD, Colan SD. Long-term enalapril therapy for left ventricular dysfunction in doxorubicin-treated survivors of childhood cancer. J. Clin. Oncol. 2002;20:4517–4522. doi: 10.1200/JCO.2002.12.102. [DOI] [PubMed] [Google Scholar]
- 5.Berthiaume JM, Wallace KB. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol Toxicol. 2007;23:15–25. doi: 10.1007/s10565-006-0140-y. [DOI] [PubMed] [Google Scholar]
- 6.van Dalen EC, Caron HN, Dickinson HO, Kremer LC. Cardioprotective interventions for cancer patients receiving anthracyclines. Cochrane Database Syst Rev. 2011:CD003917. doi: 10.1002/14651858.CD003917.pub3. [DOI] [PubMed] [Google Scholar]
- 7.Simunek T, Sterba M, Popelova O, Adamcova M, Hrdina R, Gersl V. Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep. 2009;61:154–171. doi: 10.1016/s1734-1140(09)70018-0. [DOI] [PubMed] [Google Scholar]
- 8.Egom EE, Kruzliak P, Rotrekl V, Lei M. The effect of the sphingosine-1-phosphate analogue FTY720 on atrioventricular nodal tissue. J Cell Mol Med. 2015;19:1729–1734. doi: 10.1111/jcmm.12549. doi: 10.1111/jcmm.12549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagner R, Piler P, Gabbasov Z, Maruyama J, Maruyama K, Nicovsky J, Kruzliak P. Adjuvant cardioprotection in cardiac surgery: update. Biomed Res Int. 2014;2014:808096. doi: 10.1155/2014/808096. doi: 10.1155/2014/808096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braunwald E. Heart failure. JACC Heart Fail. 2013;1:1–20. doi: 10.1016/j.jchf.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Hanna AD, Janczura M, Cho E, Dulhunty AF, Beard NA. Multiple actions of the anthracycline daunorubicin on cardiac ryanodine receptors. Mol Pharmacol. 2011;80:538–549. doi: 10.1124/mol.111.073478. [DOI] [PubMed] [Google Scholar]
- 12.Hanna AD, Lam A, Tham S, Dulhunty AF, Beard NA. Adverse effects of doxorubicin and its metabolic product on cardiac RyR2 and SERCA2A. Mol Pharmacol. 2014;86:438–449. doi: 10.1124/mol.114.093849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boucek RJ Jr, Dodd DA, Atkinson JB, Oquist N, Olson RD. Contractile failure in chronic doxorubicin-induced cardiomyopathy. J Mol Cell Cardiol. 1997;29:2631–2640. doi: 10.1006/jmcc.1997.0494. [DOI] [PubMed] [Google Scholar]
- 14.Boucek RJ Jr, Miracle A, Anderson M, Engelman R, Atkinson J, Dodd DA. Persistent effects of doxorubicin on cardiac gene expression. J Mol Cell Cardiol. 1999;31:1435–1446. doi: 10.1006/jmcc.1999.0972. [DOI] [PubMed] [Google Scholar]
- 15.Kim SY, Kim SJ, Kim BJ, Rah SY, Chung SM, Im MJ, Kim UH. Doxorubicin-induced reactive oxygen species generation and intracellular Ca2+ increase are reciprocally modulated in rat cardiomyocytes. Exp Mol Med. 2006;38:535–545. doi: 10.1038/emm.2006.63. [DOI] [PubMed] [Google Scholar]
- 16.Soga M, Kamal FA, Watanabe K, Ma M, Palaniyandi S, Prakash P, Veeraveedu P, Mito S, Kunisaki M, Tachikawa H, Kodama M, Aizawa Y. Effects of angiotensin II receptor blocker (candesartan) in daunorubicin-induced cardiomyopathic rats. Int J Cardiol. 2006;110:378–385. doi: 10.1016/j.ijcard.2005.08.061. [DOI] [PubMed] [Google Scholar]
- 17.Matsushita T, Okamato M, Toyama J, Kodama I, Ito S, Fukutomi T, Suzuki S, Itoh M. Adriamycin causes dual inotropic effects through complex modulation of myocardial Ca2+ handling. Jpn Circ J. 2000;64:65–71. doi: 10.1253/jcj.64.65. [DOI] [PubMed] [Google Scholar]
- 18.Cernecka H, Ochodnicka-Mackovicova K, Kucerova D, Kmecova J, Nemcekova V, Doka G, Kyselovic J, Krenek P, Ochodnicky P, Klimas J. Enalaprilat increases PPARbeta/delta expression, without influence on PPARalpha and PPARgamma, and modulate cardiac function in sub-acute model of daunorubicin-induced cardiomyopathy. Eur J Pharmacol. 2013;714:472–477. doi: 10.1016/j.ejphar.2013.06.040. [DOI] [PubMed] [Google Scholar]
- 19.Kelso EJ, Geraghty RF, McDermott BJ, Cameron CH, Nicholls DP, Silke B. Characterisation of a cellular model of cardiomyopathy, in the rabbit, produced by chronic administration of the anthracycline, epirubicin. J Mol Cell Cardiol. 1997;29:3385–3397. doi: 10.1006/jmcc.1997.0563. [DOI] [PubMed] [Google Scholar]
- 20.Timolati F, Ott D, Pentassuglia L, Giraud MN, Perriard JC, Suter TM, Zuppinger C. Neuregulin-1 beta attenuates doxorubicin-induced alterations of excitation-contraction coupling and reduces oxidative stress in adult rat cardiomyocytes. J Mol Cell Cardiol. 2006;41:845–854. doi: 10.1016/j.yjmcc.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 21.Gyorke S, Terentyev D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc Res. 2008;77:245–255. doi: 10.1093/cvr/cvm038. [DOI] [PubMed] [Google Scholar]
- 22.Kučerová D, Baba HA, Bokník P, Fabritz L, Heinick A, Mát’uš M, Müller FU, Neumann J, Schmitz W, Kirchhefer U. Modulation of SR Ca2+ release by the triadin-to-calsequestrin ratio in ventricular myocytes. Am J Physiol Heart Circ Physiol. 2012;302:H2008–2017. doi: 10.1152/ajpheart.00457.2011. [DOI] [PubMed] [Google Scholar]
- 23.Bers DM. Macromolecular complexes regulating cardiac ryanodine receptor function. J Mol Cell Cardiol. 2004;37:417–429. doi: 10.1016/j.yjmcc.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 24.Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, Kobayashi S, Hisamatsu Y, Yamamoto T, Kohno M, Noguchi N, Takasawa S, Okamoto H, Matsuzaki M. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca(2+) leak through ryanodine receptor in heart failure. Circulation. 2000;102:2131–2136. doi: 10.1161/01.cir.102.17.2131. [DOI] [PubMed] [Google Scholar]
- 25.Matus M, Kucerova D, Kruzliak P, Adameova A, Doka G, Turcekova K, Kmecova J, Kyselovic J, Krenek P, Kirchhefer U, Mueller FU, Boknik P, Klimas J. Upregulation of SERCA2a following short-term ACE inhibition (by enalaprilat) alters contractile performance and arrhythmogenicity of healthy myocardium in rat. Mol Cell Biochem. 2015;403:199–208. doi: 10.1007/s11010-015-2350-1. [DOI] [PubMed] [Google Scholar]
- 26.Arai M, Tomaru K, Takizawa T, Sekiguchi K, Yokoyama T, Suzuki T, Nagai R. Sarcoplasmic reticulum genes are selectively down-regulated in cardiomyopathy produced by doxorubicin in rabbits. J Mol Cell Cardiol. 1998;30:243–254. doi: 10.1006/jmcc.1997.0588. [DOI] [PubMed] [Google Scholar]
- 27.Burke BE, Gambliel H, Olson RD, Bauer FK, Cusack BJ. Prevention by dexrazoxane of down-regulation of ryanodine receptor gene expression in anthracycline cardiomyopathy in the rat. Br J Pharmacol. 2000;131:1–4. doi: 10.1038/sj.bjp.0703538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kmecova J, Klimas J. Heart rate correction of the QT duration in rats. Eur J Pharmacol. 2010;641:187–192. doi: 10.1016/j.ejphar.2010.05.038. [DOI] [PubMed] [Google Scholar]
- 29.Klimas J, Kmecova J, Jankyova S, Yaghi D, Priesolova E, Kyselova Z, Musil P, Ochodnicky P, Krenek P, Kyselovic J, Matyas S. Pycnogenol improves left ventricular function in streptozotocin-induced diabetic cardiomyopathy in rats. Phytother Res. 2010;24:969–974. doi: 10.1002/ptr.3015. [DOI] [PubMed] [Google Scholar]
- 30.Olson RD, Gambliel HA, Vestal RE, Shadle SE, Charlier HA Jr, Cusack BJ. Doxorubicin cardiac dysfunction: effects on calcium regulatory proteins, sarcoplasmic reticulum, and triiodothyronine. Cardiovasc Toxicol. 2005;5:269–283. doi: 10.1385/ct:5:3:269. [DOI] [PubMed] [Google Scholar]
- 31.Charlier HA Jr, Olson RD, Thornock CM, Mercer WK, Olson DR, Broyles TS, Muhlestein DJ, Larson CL, Cusack BJ, Shadle SE. Investigations of calsequestrin as a target for anthracyclines: comparison of functional effects of daunorubicin, daunorubicinol, and trifluoperazine. Mol Pharmacol. 2005;67:1505–1512. doi: 10.1124/mol.104.005728. [DOI] [PubMed] [Google Scholar]
- 32.Zucchi R, Yu G, Ghelardoni S, Ronca F, Ronca-Testoni S. Effect of MEN 10755, a new disaccharide analogue of doxorubicin, on sarcoplasmic reticulum Ca(2+) handling and contractile function in rat heart. Br J Pharmacol. 2000;131:342–348. doi: 10.1038/sj.bjp.0703575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D’Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 34.Cai C, Lothstein L, Morrison RR, Hofmann PA. Protection from doxorubicin-induced cardiomyopathy using the modified anthracycline N-benzyladriamycin-14-valerate (AD 198) J Pharmacol Exp Ther. 2010;335:223–230. doi: 10.1124/jpet.110.167965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Beer EL, Bottone AE, van Rijk MC, van der Velden J, Voest EE. Dexrazoxane pre-treatment protects skinned rat cardiac trabeculae against delayed doxorubicin-induced impairment of crossbridge kinetics. Br J Pharmacol. 2002;135:1707–1714. doi: 10.1038/sj.bjp.0704621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pazos-Moura C, Abel ED, Boers ME, Moura E, Hampton TG, Wang J, Morgan JP, Wondisford FE. Cardiac dysfunction caused by myocardium-specific expression of a mutant thyroid hormone receptor. Circ Res. 2000;86:700–706. doi: 10.1161/01.res.86.6.700. [DOI] [PubMed] [Google Scholar]
- 37.Wiesner RJ, Ehmke H, Faulhaber J, Zak R, Ruegg JC. Dissociation of left ventricular hypertrophy, beta-myosin heavy chain gene expression, and myosin isoform switch in rats after ascending aortic stenosis. Circulation. 1997;95:1253–1259. doi: 10.1161/01.cir.95.5.1253. [DOI] [PubMed] [Google Scholar]
- 38.Holubarsch C, Goulette RP, Litten RZ, Martin BJ, Mulieri LA, Alpert NR. The economy of isometric force development, myosin isoenzyme pattern and myofibrillar ATPase activity in normal and hypothyroid rat myocardium. Circ Res. 1985;56:78–86. doi: 10.1161/01.res.56.1.78. [DOI] [PubMed] [Google Scholar]
- 39.Gupta MP. Factors controlling cardiac myosin-isoform shift during hypertrophy and heart failure. J Mol Cell Cardiol. 2007;43:388–403. doi: 10.1016/j.yjmcc.2007.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krenz M, Robbins J. Impact of beta-myosin heavy chain expression on cardiac function during stress. J Am Coll Cardiol. 2004;44:2390–2397. doi: 10.1016/j.jacc.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 41.Kostin S, Hein S, Arnon E, Scholz D, Schaper J. The cytoskeleton and related proteins in the human failing heart. Heart Fail Rev. 2000;5:271–280. doi: 10.1023/A:1009813621103. [DOI] [PubMed] [Google Scholar]
- 42.Saks V, Kuznetsov AV, Gonzalez-Granillo M, Tepp K, Timohhina N, Karu-Varikmaa M, Kaambre T, Dos Santos P, Boucher F, Guzun R. Intracellular Energetic Units regulate metabolism in cardiac cells. J Mol Cell Cardiol. 2012;52:419–436. doi: 10.1016/j.yjmcc.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 43.Zhang C, Chen B, Guo A, Zhu Y, Miller JD, Gao S, Yuan C, Kutschke W, Zimmerman K, Weiss RM, Wehrens XH, Hong J, Johnson FL, Santana LF, Anderson ME, Song LS. Microtubule-mediated defects in junctophilin-2 trafficking contribute to myocyte transverse-tubule remodeling and Ca2+ handling dysfunction in heart failure. Circulation. 2014;129:1742–1750. doi: 10.1161/CIRCULATIONAHA.113.008452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsutsui H, Tagawa H, Kent RL, McCollam PL, Ishihara K, Nagatsu M, Cooper G 4th. Role of microtubules in contractile dysfunction of hypertrophied cardiocytes. Circulation. 1994;90:533–555. doi: 10.1161/01.cir.90.1.533. [DOI] [PubMed] [Google Scholar]
- 45.Hein S, Kostin S, Heling A, Maeno Y, Schaper J. The role of the cytoskeleton in heart failure. Cardiovasc Res. 2000;45:273–278. doi: 10.1016/s0008-6363(99)00268-0. [DOI] [PubMed] [Google Scholar]
- 46.van Hoorn F, Campian ME, Spijkerboer A, Blom MT, Planken RN, van Rossum AC, de Bakker JM, Wilde AA, Groenink M, Tan HL. SCN5A mutations in Brugada syndrome are associated with increased cardiac dimensions and reduced contractility. PLoS One. 2012;7:e42037. doi: 10.1371/journal.pone.0042037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiong Q, Cao Q, Zhou Q, Xie J, Shen Y, Wan R, Yu J, Yan S, Marian AJ, Hong K. Arrhythmogenic cardiomyopathy in a patient with a rare loss-of-function KCNQ1 mutation. J Am Heart Assoc. 2015;4:e001526. doi: 10.1161/JAHA.114.001526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arozal W, Watanabe K, Veeraveedu PT, Ma M, Thandavarayan RA, Sukumaran V, Suzuki K, Kodama M, Aizawa Y. Protective effect of carvedilol on daunorubicin-induced cardiotoxicity and nephrotoxicity in rats. Toxicology. 2010;274:18–26. doi: 10.1016/j.tox.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 49.Dhingra R, Margulets V, Chowdhury SR, Thliveris J, Jassal D, Fernyhough P, Dorn GW 2nd, Kirshenbaum LA. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc Natl Acad Sci U S A. 2014;111:E5537–5544. doi: 10.1073/pnas.1414665111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hydock DS, Lien CY, Hayward R. Anandamide preserves cardiac function and geometry in an acute doxorubicin cardiotoxicity rat model. J Cardiovasc Pharmacol Ther. 2009;14:59–67. doi: 10.1177/1074248408329449. [DOI] [PubMed] [Google Scholar]
- 51.Fogli S, Nieri P, Breschi MC. The role of nitric oxide in anthracycline toxicity and prospects for pharmacologic prevention of cardiac damage. FASEB J. 2004;18:664–675. doi: 10.1096/fj.03-0724rev. [DOI] [PubMed] [Google Scholar]
- 52.Jackson JA, Reeves JP, Muntz KH, Kruk D, Prough RA, Willerson JT, Buja LM. Evaluation of free radical effects and catecholamine alterations in adriamycin cardiotoxicity. Am J Pathol. 1984;117:140–153. [PMC free article] [PubMed] [Google Scholar]
- 53.Polanski AK, Ebner A, Ebner B, Hofmann A, Steinbronn N, Brandt A, Forkmann M, Tausche AK, Morawietz H, Strasser RH, Wunderlich C. Dexrazoxane prevents the development of the impaired cardiac phenotype in caveolin-1-disrupted mice. J Cardiovasc Pharmacol. 2013;61:545–552. doi: 10.1097/FJC.0b013e31828de47c. [DOI] [PubMed] [Google Scholar]
- 54.Lipshultz SE, Sambatakos P, Maguire M, Karnik R, Ross SW, Franco VI, Miller TL. Cardiotoxicity and cardioprotection in childhood cancer. Acta Haematol. 2014;132:391–399. doi: 10.1159/000360238. [DOI] [PubMed] [Google Scholar]
- 55.Hrdina R, Gersl V, Klimtova I, Simunek T, Machackova J, Adamcova M. Anthracycline-induced cardiotoxicity. Acta Medica (Hradec Kralove) 2000;43:75–82. [PubMed] [Google Scholar]