

Abstract
Among breast cancers, 10 to 15% of cases would be due to hereditary risk. In these familial cases, mutations in BRCA1 and BRCA2 are found in only 15% to 20%, meaning that new susceptibility genes remain to be found. Triple-negative breast cancers represent 15% of all breast cancers, and are generally aggressive tumours without targeted therapies available. Our hypothesis is that some patients with triple negative breast cancer could share a genetic susceptibility different from other types of breast cancers. We screened 36 candidate genes, using pyrosequencing, in all the 50 triple negative breast cancer patients with familial history of cancer but no BRCA1 or BRCA2 mutation of a population of 3000 families who had consulted for a familial breast cancer between 2005 and 2013. Any mutations were also sequenced in available relatives of cases. Protein expression and loss of heterozygosity were explored in tumours. Seven deleterious mutations in 6 different genes (RAD51D, MRE11A, CHEK2, MLH1, MSH6, PALB2) were observed in one patient each, except the RAD51D mutation found in two cases. Loss of heterozygosity in the tumour was found for 2 of the 7 mutations. Protein expression was absent in tumour tissue for 5 mutations. Taking into consideration a specific subtype of tumour has revealed susceptibility genes, most of them in the homologous recombination DNA repair pathway. This may provide new possibilities for targeted therapies, along with better screening and care of patients.
Keywords: Triple-negative breast cancer, familial non-BRCA1/2 breast cancer, genetic susceptibility, candidate genes, pyrosequencing, DNA repair genes
Introduction
Among all breast cancers, one in ten is associated with hereditary risk. However, mutations in the two major predisposing genes, BRCA1 [MIM: 113705] and BRCA2 [MIM: 600185], are found in only 15% to 20% of hereditary breast cancer families [1]. Other genes have emerged, such as TP53 [MIM: 191170], PTEN [MIM: 601728], STK11 [MIM: 602216] or CDH1 [MIM: 192090] that are known to cause familial syndromes in which breast cancer (BC) incidence is highly increased, or BRIP1 [MIM: 605882], ATM [MIM: 607585], CHEK2 [MIM: 604373] or PALB2 [MIM: 610355] [2], that are associated with a moderate increased risk of breast cancer. But they together represent only about 20% of familial breast cancers. For the vast remaining majority, no mutation is found.
Moreover, breast cancer represents a complex and heterogeneous disease, with different clinical features, response to treatment, histopathology and gene expression profiles. These elements overlap and several large groups of breast with biological and clinical traits in common have been proposed. Because the majority of studies of familial breast cancer grouped all subtypes together, it has been difficult to identify new causal genes, if the causes of different subtypes have different genetic origins. It may thus be pertinent to evaluate genetic predisposition to a specific subtype of breast cancer.
Five types of breast cancer have been identified by gene expression profile [3,4]; two luminal receptor positive subgroups (A and B), a human epidermal growth factor 2 (HER2) overexpressing group, a normal breast-like or unclassified group, and a basal-like group that is largely triple-negative breast cancer (TNBC). Later, a claudin-low group was also described (consisting mainly of TNBC).
Triple-negative breast cancers, defined by no expression of estrogen receptor (ER), progesterone receptor (PR), or human epidermal growth factor receptor 2 (HER2) in tumour material, stand for 10-20% of all breast cancers. TNBC are aggressive tumours, affect frequently young patients and have poor clinical outcome. Treatment of TNBC has been challenging due to the absence of well-defined molecular targets.
Genetic predisposition to triple-negative breast cancers has been little studied, except for the two main genes BRCA1 and BRCA2 mutations. For TNBC, studies have shown that 9-14% overall and ~20% of cases diagnosed under age 50 harbour germline BRCA1 mutations [5]. This observation opened new potential therapeutic approaches, such as the anti-PARP1. Mutations in BRCA2 are associated to a lesser extent with TNBC, with 3.9% of triple-negative tumours patients having a germline mutation in this gene [6]. Few other breast cancer susceptibility genes have been evaluated for mutations in TNBC [7,8].
Our hypothesis is that some patients with familial TNBC could share a genetic susceptibility to cancer different from other types of breast cancer. To explore this idea, we analysed thirty-six candidate genes known or suspected to be part of the DNA repair process, potentially involved in breast cancer genesis in general or in TNBC specifically.
Materials and methods
Patients
Among 3000+ families screened for a familial breast cancer between 2005 and 2013 at the oncogenetics consultation of the Centre Jean Perrin (France), we collected the samples of all the triple negative breast cancer patients with familial history of cancer but no BRCA1 or BRCA2 mutation. In this population, a familial context of cancer is defined as a first degree relative affected with breast cancer or a second degree relative if related by a man, or belonging to a family with a Manchester score >12 (score weighted on anatomopathology) [9]. We included as cases all the patients with a triple negative breast cancer, tested negative for mutations and large genomic rearrangements in BRCA1 and BRCA2, which represent 50 TNBC patients. The control group was composed of 51 samples of DNA from women free of neoplasia [10], matched on age with cases. With this population size and a one-sided alpha =0.05, we reached a power of 70% (Poisson’s law of small numbers [11]).
The mean age at breast cancer diagnosis was 48.6 years old (Table 1). Genomic DNA from these patients, their clinical and pathological information, genealogical trees and tissue samples were provided by the oncogenetic department and the anatomopathology department, at the Centre Jean Perrin (Clermont-Ferrand, France).
Table 1.
Characteristics of the studied population
| Characteristics | Patients, n=50 | Controles, n=51 |
|---|---|---|
| Mean age at breast cancer diagnostic (patients) or at sample collection (controls) | 48.6 | 49.6 |
| Age at diagnosis (or collection for controls) | ||
| ≤40 years | 15 | 14 |
| between 41 and 50 years | 11 | 10 |
| >50 years | 24 | 27 |
| Family history of cancer | ||
| Family with breast cancer only | 4 | na |
| Breast and ovarian family | 5 | na |
| Breast and other cancers | 46 | na |
| Breast cancer family history | ||
| 1 breast cancer ≤35 years | 4 | na |
| 2 breast cancers | 19 | na |
| ≥3 breast cancers | 27 | na |
na: not applicable.
Each mutation discovered in a proband was sought in all her relatives with available DNA.
All subjects signed an informed consent sheet that was approved by the Regional Ethics Committee (Auvergne, France).
Enrichment
We used the solution-based NimbleGen® Sequence Capture EZ Choice (Roche®) to enrich genomic DNA in the coding sequence of the 36 candidate genes. For each gene, all exons and intron-exon boundaries were covered by the capture.
These genes were known or suspected to be part of the DNA repair process (POLB [MIM: 174760], ERCC1 [MIM: 126380], APC [MIM: 611731], EPCAM [MIM: 185535], MLH1 [MIM: 120436], MLH3 [MIM: 604395], MSH2 [MIM: 609309], MSH6 [MIM: 600678], MUTYH [MIM: 604933], PMS1 [MIM: 600258], PMS2 [MIM: 600259], PTEN, TP53, ATM, AURKA [MIM: 603072], CHEK2, CDH1, NRIP1 [MIM: 602490], FGFR2 [MIM: 176943], STK11, WRN [MIM: 604611]), or to be actor in the BRCA1 pathway (BRIP1, BARD1 [MIM: 601593], COBRA1 [MIM: 611180], BRAP [MIM: 604986], BRCC3 [MIM: 300617], PALB2 [MIM: 610355], BAP1 [MIM: 603089], BLM [MIM: 604610], MRE11A [MIM: 600814], NBN [MIM: 602667], RAD50 [MIM: 604040], RAD51D [MIM: 602948], RAD51C [MIM: 602774], RAD51D [MIM: 602954]), the most implicated in TNBC so far. We also studied one gene, BABAM1 [MIM: 612766], located at locus 19p13.1 identified in genome wide association studies to be associated specifically with TNBC (and not with breast cancer in general) [12].
Sample preparation and next generation sequencing
DNA was extracted from peripheral blood with Macherey Nucleo Spin blood X kit, before storage in a DNA Bank.
We used the Roche® Rapid Library Preparation method for sequencing on GS-FLX 454 Roche®. DNA was fragmented by nebulization, ligated to Roche 454 adaptators and purified with AmpureXP beads. The quality of the library was assessed with a Bioanalyzer 2100 (Agilent) and an Infinite 200 plate reader (Tecan). Libraries were then amplified by ligation-mediated PCR (LM-PCR), in which three indexed samples were combined equimolarly. The enrichment process was done with the NimbleGen SeqCap EZ Library LR method, with a double capture. Em-PCR targeting 2 copies per beads was performed prior to sequencing on a GS-FLX 454 Roche®, with Titanium kits.
Bioinformatic analysis
GS Reference Mapper v2.8 (Roche) was used to ensure demultiplexing of the samples, read alignment against the reference genome (hg19 of March 2012) and variant calling. The output files were then filtered by homemade software to sort variants according to minimum coverage (>5 reads) and frequency (>20%). Variant annotation was performed using Alamut Batch software v1.1.2 (Interactive Biosoftware, Rouen, France).
Non-synonymous missense substitutions
Population allele frequencies were extracted from the Exome Variant Server (http://evs.gs.washington.edu/EVS), 1000 Genomes (http://www.1000genomes.org), and dbSNP (version 137; http://www.ncbi.nlm.nih.gov/projects/SNP). Predicted deleterious missense mutations were selected using algorithms (SIFT, MAPP and AlignGVGD) [13] (http://agvgd.iarc.fr). When two algorithms out of three predicted a variation to be deleterious, it was retained for further analysis. We also confirmed prediction for retained variants with PolyPhen2 [14].
Insertion or deletion
The Roche 454 pyrosequencing technology is known to generate numerous homopolymer-associated single base errors [15], which create artifactual insertions or deletions. All in/del variations found within a homopolymer were filtered out with a spectrum based error corrector for 454 sequencing data, named HECTOR [16].
Splicing variants
The potential impact of variants located on splicing junctions was evaluated using SpliceSiteFinder, MaxEntScan, and GeneSplicer prediction software [17,18]. Variants predicted to alter splicing by at least one of these software was retained for further analysis.
Confirmation of variants
SNV (Single Nucleotide Variation) and indels detected using pyrosequencing within the coding sequence or intron/exon boundaries (-20 to +6) were further filtered, excluding variants recorded as polymorphisms. Variants predicted as deleterious in silico were confirmed by Sanger sequencing, using AmpliTaq DNA® Polymerase (Life Technologies) and BigDye® Terminator v3.1 (Life Technologies). The same primers and reagents were used to search for the confirmed variants in the control group and to analyse each proband’s mutation in their relatives.
Loss of heterozygosity
Tumour tissue from mutation carriers was macrodissected from formalin-fixed paraffin-embedded tissue, and DNA was extracted from the collected cells using the Maxwell® 16 system (Promega) according to the manufacturer’s instructions for formalin-fixed, paraffin-embedded samples. Primers were designed to produce small PCR products spanning the variants of interest. The mutation from each carrier was sequenced directly from a PCR product, generated using AmpliTaq DNA® Polymerase (Life Technologies), by dideoxy-sequencing using the BigDye® Terminator v3.1 and an ABI 3130XL sequencer (Life Technologies). The presence or absence of LOH (loss of heterozygosity) in tumours was determined by visual comparison of the relative size of the peaks, observed in the blood (all variants were heterozygous) and in the tumour traces.
Immunohistochemistry
Immunohistochemical detection of proteins coded by the mutated genes was performed on 4 µm-thick formalin-fixed, paraffin-embedded tissue sections of breast tumours found in the mutation carriers. Non-malignant breast tissue from a patient without any mutation was used as a control. Epitopes were retrieved by CC1 (Ventana, Tuscon, AZ) and the sections further incubated with the following antibodies: rabbit monoclonal anti-APC (LS-C49666), rabbit polyclonal anti-RAD51D ((Ala152), LS-C177138) and anti-PALB2 (LS-C288058), mouse monoclonal anti-MRE11A (LS-C53370) (all from LifeSpan Biosciences, Seattle, WA), mouse monoclonal MLH1 (clone M1, 790-4535) et MSH6 (clone 44, 790-4455) (Ventana). Antigen-antibody reaction was visualised by ultraView DAB or AP (Ventana). All procedures were performed in a fully automated immunohistochemical stainer (Benchmark XT, Ventana).
Results
Variant analysis
Seven heterozygous missense substitutions were observed in six patients, predicted as deleterious by bioinformatic analysis. All seven mutations were absent from the control group. These mutations were in six different genes: CHEK2 NM_001005735.1:c.1214G>A (p.Cys405Tyr); MLH1 NM_000249.3:c.199G>T (p.Gly67Trp); MRE11A NM_005591.3:c.901C>G (p.Leu301Val); MSH6 NM_000179.2:c.431G>T (p.Ser144Ile); PALB2 NM_024675.3:c.2897T>C (p.Ile966Thr); RAD51DNM_001142571.1:c.853G>A (p.Gly285Arg) (two cases). One, MLH1 c.199G>T, has been described as pathogenic, two (MSH6 c.431G>T; RAD51D c.853G>A) were known in genomic international databases but very rare in the general population (MAF<0.1%) and not previously associated with breast cancer; and the three others (CHEK2 c.1214G>A; MRE11A c.901C>G; PALB2 c.2897T>C) have not been described. One patient carried two variants, one in MLH1 and another in PALB2 (Table 2).
Table 2.
Mutations identified and personal and familial cancer history of the proband
| Family | Cancer history and age of proband at cancer | Number of breast cancers in the family | Breast cancers HR and HER2 status | Other cancers in relatives | Gene affected | Mutation |
|---|---|---|---|---|---|---|
| 1 | TN breast cancer, 35 yrs | 1 | - TN | 1 pancreas, 1 thyroid cancer | RAD51D | NM_001142571.1:c.853G>A |
| 2 | TN breast cancer, 37 yrs | 2 | - TN- HR-HER- | 1 ENT | RAD51D | NM_001142571.1:c.853G>A |
| 3 | TN breast cancer, 40 yrs | 2 | - TN- HR+HER- | 1 CNS | MSH6 | NM_000179.2:c.431G>T |
| 4 | TN breast cancer, 64 yrs | 3 | - TN- PR+ER-HER-- NA | 1 ovarian cancer, 1 lung cancer, 1 stomach cancer | MRE11A | NM_005591.3:c.901C>G |
| 5 | TN breast cancer, 51 yrs; contralateral TN breast cancer, 63 yrs; endometrial 63 yrs, colon 68 yrs | 3 | - TN- TN- NA | 1 endometrial, 5 colon, 1 ovarian cancer | MLH1 | NM_000249.3:c.199G>T |
| PALB2 | NM_024675.3:c.2897T>C | |||||
| 6 | TN breast cancer, 51 yrs thyroid cancer, 42 yrs; bladder cancer, 50 yrs | 3 | - TN- NA- NA | 1 colon, 1 astrocytome, 1 retinal melanoma? | CHEK2 | NM_001005735.1:c.1214G>A |
NA: Not available; TN: Triple-negative breast cancer; HR: Hormone Receptor (ER+PR); ER: Estrogen Receptor; PR: Progesterone receptor; CNS: Central Nervous System; ENT: Ear, Nose and Throat; yrs: years.
The RAD51D missense mutation c.853G>A (p.Gly285Arg) was found in two patients of different families. This amino-acid is located at the C-terminus of the AAA+ ATPase domain of the RAD51D protein, which contributes significantly to nucleotide binding [19].
We found one missense substitution in MRE11A (or MRE11), c.901C>G (p.Leu301Val). The amino-acid 301 is highly conserved and is located in the major functional domains of the protein: the DNA repair exonuclease domain, and the DNA-binding region.
We report a new CHEK2 germline missense substitution, c.1214G>A (p.Cys405Tyr), affecting the Ser-Thr/Tyr-protein kinase catalytic domain. This amino-acid is highly conserved and there is a large physico-chemical gap between the two amino-acids. In silico prediction classifies this variant as disease causing. We previously observed that in silico predictions and in vitro analysis correlate well in this region of CHEK2 [20].
PALB2 c.2897T>C missense substitutions is not localised on an identified protein activity domain, but was predicted as deleterious in silico.
The MSH6 c.431G>T substitution affects PWWP and DNA mismatch repair domains of the protein. This mutation has been already reported with an allele frequency <0.01, but its significance is unknown.
The MLH1 c.199G>T mutation is known to be pathogenic.
No splicing variants, truncating variants or in/dels after correction with the HECTOR software [16] were observed in the 36 genes panel.
Analysis of mutations in relatives
Following mutations in relatives of cases was possible for three of the six families. We only found a mutation in an unaffected first-degree relative of the RAD51D c.853G>A carrier.
A similar pattern of incomplete segregation in affected relatives has been observed for susceptibility alleles that confer modest increased risk, and reported for variants in CHEK2, ATM, BRIP1 and PALB2 [21-24].
Loss of heterozygosity analysis
Two of six mutations showed partial or complete loss of the wild-type allele in the tumour. Tumour material was not sufficient to perform this analysis for one of the two RAD51D c.853G>A carrier. Tumour DNA of the individual with the MRE11A c.901C>G germline mutation showed complete loss of the wild-type allele (Figure 1A). This complete loss of heterozygosity in the tumour suggests that this variant could be a very early event in the oncogenesis of this patient’s tumour. The carrier of the RAD51D c.853G>A variant that could be tested showed partial loss of the wild type allele in her tumour (Figure 1B).
Figure 1.
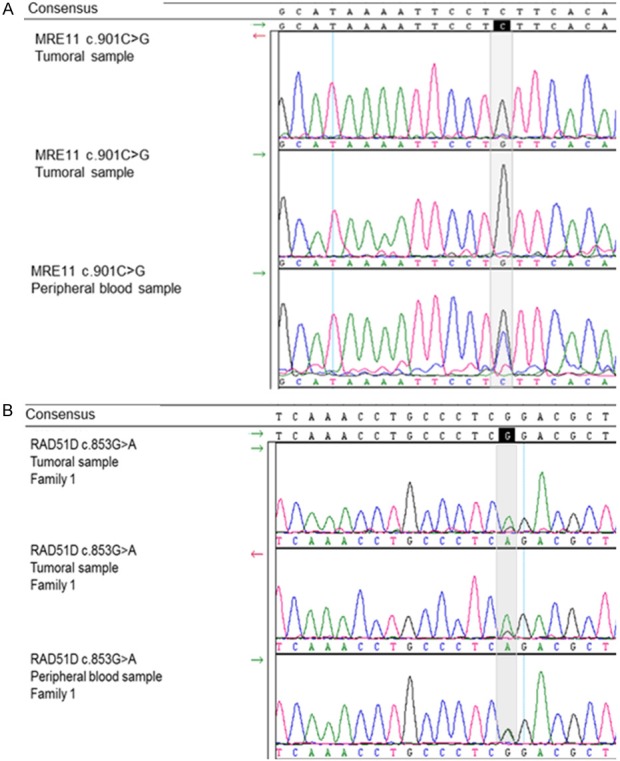
Sanger electropherograms representing DNA sequences of the two breast tumours with loss of the wild-type allele. A. In the peripheral blood, the patient carry the mutation MRE11 c.901C>G in the heterozygous state (last trace): 2 peaks, one blue (nulcleotide C) and one black (nulcleotide G). In the tumour (the 2 upper traces), there is no more allele with a C (blue trace) at position 901, the wild-type allele has disappeared. B. In the peripheral blood, the patient carry the mutation RAD51D c.853G>A in the heterozygous state (last trace). In the tumour, there is almost no more G at position 853. MRE11 c.901C>G and RAD51D c.853G>A loss of heterozygosity in the tumour.
These loss of heterozygosity support that MRE11A c.901C>G and RAD51D c.853G>A are involved in tumour development according to the two-hits model of tumour suppressor genes.
Immunochemistry analysis
RAD51D protein was absent in tumour cells of both mutated probands while it was present in tumour-infiltrating lymphocytes as well as in control normal breast cells (Figure 2). No expression of MLH1 nor PALB2 was observed in the tumour cells nuclei of mutated cases, as compared to the control breast tissue (Figure S1). MSH6 and MRE11A staining was identical in mutated tumour and control tissues. Expression of CHEK2 could not be tested due to the small quantity of tumour tissue available.
Figure 2.
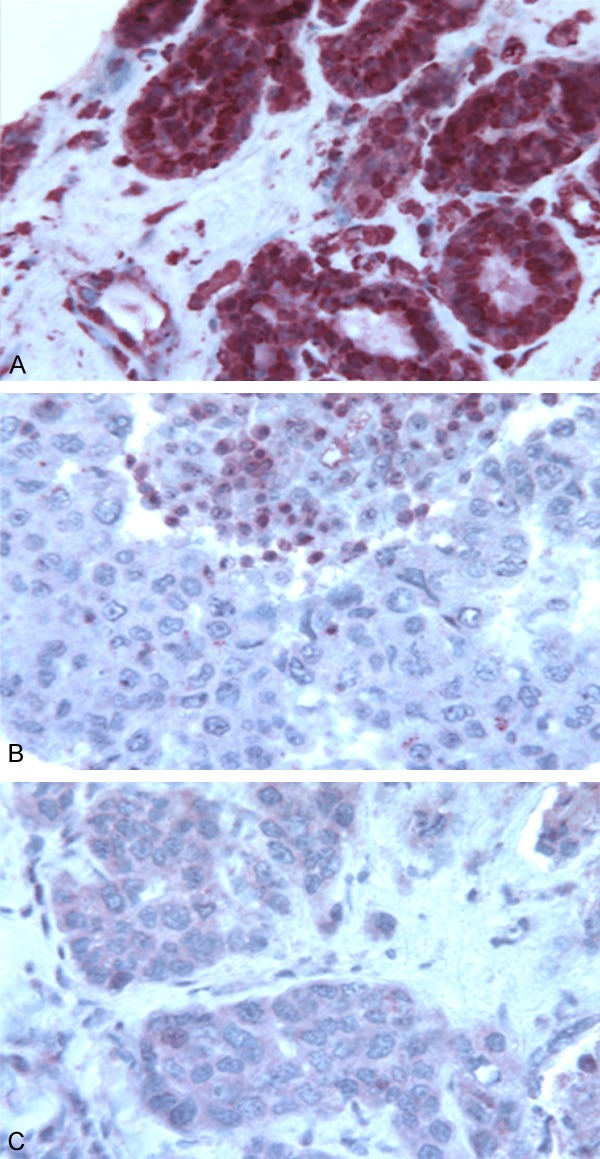
Immunohistochemical detection of the RAD51D protein. Formalin-fixed, paraffin-embedded tissue sections of breast tumours of the two RAD51D mutation carriers. A. The RAD51D protein is present as in control normal breast cells (red color in cytoplasm and nuclei). B and C. The RAD51D protein is absent in tumour cells of both mutated probands. Immunohistochemical detection of the RAD51D protein in non-malignant control breast and in tumoural tissue of mutation carriers.
Discussion
Germline mutations in specific genes could be responsible for triple negative breast cancer predisposition. We found seven missense mutations in 50 patients, within six different genes: RAD51D (two cases), MRE11A, CHEK2, MLH1, PALB2, MSH6. There is no previous study on familial non-BRCA1/2 triple-negative breast cancer. Literature concerns either non-familial triple-negative breast cancer [8] or familial breast cancer in general [25].
Because of a stringent selection on two criteria (familial history plus triple-negative subtype of breast cancer), our population is of limited size. However, the strength of the study has been reinforced by the homogeneity of the population.
The high number of mutations found in our work can also be explained by the choice we made to study a highly selected population as part of an initial population of familial breast cancers of 3000+ patients. Indeed, if a gene is frequently implicated in one subtype of breast cancer, and never in other subtypes, analysing the population of breast cancer in general, with all subtypes, will lead to conclude that mutations in this gene are rare or absent; however this gene could be an important predisposition factor for the small and specific subgroup of tumours where it is more frequent.
Mutations in homologous recombination DNA repair genes
We show evidence for an implication in TNBC predisposition of three genes coding important proteins of the homologous recombination (HR) DNA repair pathway, RAD51D, MRE11 and PALB2.
RAD51D c.853G>A mutation is located on a key activity domain of the protein. We observed loss of heterozygosity in the tumour of one of the two carriers (the other was unavailable). Consistent with this, the RAD51D protein was absent from tumour cells of both mutated probands. RAD51D c.853G>A mutation was found in two individuals, and both presented a similar phenotype, with triple-negative breast cancer at a very young age, 35 and 37 years, and with few cancers in the family (breast, thyroid and pancreatic cancers) and no ovarian cancer in the family (see Figures Family 1 and 2).
In mammalian cells, the RAD51 paralogs are involved in homologous recombination and in maintaining genomic integrity by telomere protection [26,27]. Mutations in RAD51D were mainly associated with increased risk of hereditary ovarian cancer and, although they have been observed in breast and ovarian cancer families, the association with breast cancer alone remains unclear [28-33]. A large scale study focused on triple negative breast cancer suggested that mutations in RAD51D could confer a risk of triple negative and basal subtypes of breast cancer specifically [8]. Finally, a case report described a 36-year old woman with a high-grade, triple negative invasive ductal carcinoma who carried a pathogenic mutation in RAD51D (c.556C>T; p.Arg186*) detected by 18-gene panel testing [34]. Her (non-affected) mother and her maternal aunt (medullary breast cancer at 54 years) harbored the same mutation. The phenotype of this patient was very similar to that of the two carriers of the RAD51D mutation from our study.
Like what we showed for the RAD51D mutation, the implication of MRE11 c.901C>G mutation in TNBC predisposition is supported by complete loss of the wild-type allele in the tumour. Immunohistochemistry revealed normal expression of MRE11A in the proband’s tumour tissue. However, the epitope recognized by the antibody does not cover the mutation site, so mutated protein cannot be discriminated from wild type MRE11A. Because the mutation is in a major functional domain, the mutated protein is most likely non-functional, although expressed in normal quantity. Only functional studies could confirm this hypothesis. In our study, the individual carrying the MRE11A c.901C>G mutation had bilateral triple negative breast cancer. Her sister had ovarian cancer, her brother thyroid cancer, one nephew lung cancer and two nieces breast cancer (see Figure Family 4).
MRE11A forms a protein complex with RAD50 and NBN. This complex plays key roles in DNA double-strand break (DSB) repair, meiotic recombination, and cell cycle checkpoints, and MRE11A is also involved in maintenance of telomeres [35-37]. Mutations in MRE11A have been linked to Ataxia-Telangectasia-Like Disorder (ATLD) [MIM 604391]. One of the first reports of a suspected association of malignancy and mutations in MRE11A was about two brothers who died of pulmonary adenocarcinoma before the age of 20 [38]. One MRE11A mutation was also found in one case of ovarian cancer among 151 families with ovarian and breast cancer [39]. In breast cancer, evaluation of protein-truncating variants and rare missense substitutions falling in the key functional domains of the MRN complex proteins suggested a two- to three-fold increased risk of breast cancer [40]. However there is no study on MRE11 and breast cancer of triple-negative type, but some element suggest that it could be linked. Aberrantly reduced expression of MRE11A was reported in 7% of 1000 breast tumours [41]. Moreover, among breast tumours with aberrant expression of MRE11A, 30% were triple negative. Two heterozygous mutations in MRE11A were found in eight patients with tumours with no or reduced expression of the three MRN complex proteins [41]. Experimentally, MRE11A-mutated mice exhibit extensive oncogene-induced mammary hyperplasia with frequent progression to invasive breast cancer. In addition, these mammary tumours showed largely absent ER staining and the presence of the basal-like breast cancer markers EGFR and CK5/6 in all tumours. They also exhibited hallmarks of aggressive disease, including high histopathological grade, elevated chromosomal instability and frequent development of lung metastases [42].
Finally, we found a mutation in PALB2, c.2897T>C (Ile966Thr), which is an unreported missense mutation predicted to be deleterious in silico. The individual carrying this mutation also harbour a MLH1 pathogenic mutation, and presented with a TNBC at 51 years, a contralateral TNBC and endometrial cancer at 63 years, and colon cancer at 68 years. The MLH1 mutation was present in the familial branch presenting several Lynch-syndrome cancers (5 colon cancers, 1 ovarian cancer and 1 endometrial cancer) but not the PALB2 mutation (see Figure Family 5). The proband’s multiple tumours may be caused by her double-heterozygote status. This hypothesis is reinforced by the absence of both PALB2 and MLH1 expression in the breast tumour. No LOH for PALB2 was found, but LOH in PALB2 breast tumours does not seem to be a common feature of PALB2 tumour genesis [43]. Partner and localizer of BRCA2 (PALB2) is a BRCA2-interacting protein that is crucial for key BRCA2 genome caretaker functions. Monoallelic PALB2 mutations are associated with increased risk of breast and pancreatic cancer and recent studies have shown that PALB2 also interacts with BRCA1 [44,45]. Studies performed in European populations suggest overrepresentation of triple negative (basal-like) tumours in PALB2-related breast cancers [43,46,47]. Heikkinen et al. [47] reported that 54.5% of breast tumours of PALB2 c.1592delT mutation carriers presented the triple negative phenotype versus 12.2% of other familial or 9.4% of sporadic breast cancer patients. Prevalence of PALB2 germline mutations in individuals with TNBC was around 1%, similar to that in familial non-BRCA1/2 breast cancer cohorts [7]. In contrast, studies in Australasian breast cancer families did not find any association between PALB2 mutations and hormone-receptor negative breast tumours [48,49].
These results together suggest RAD51D mutation may predispose specifically to triple negative breast cancer. Taking into consideration a specific subtype of tumour could have revealed a susceptibility allele specific to TNBC, that need to be further explored.
There is growing evidence that MRE11A is a breast cancer susceptibility gene, and some data suggest that it could be particularly associated to triple negative/basal-like breast cancers.
Consistent with the fact that PALB2 is a major partner of BRCA1, and like literature begin to suggest it [43,46,47], our results support an implication of this gene in TNBC predisposition.
There is evidence that a substantial fraction of TNBC has HR DNA repair pathway defects [50,51]. TNBC are characterized by extensive genomic instability [52], and deficiency in HR DNA repair is a source of genomic instability. BRCA1-mutated breast tumours and basal-like breast cancers show the same profile of aberrant aCGH profiles [51]. As they are HR deficient, BRCA1-mutated breast tumours can be targeted with DNA-damaging agents or PARP inhibitors. The heterogeneity of triple negative breast tumours may explain why PARP inhibitors show weak efficiency in some clinical assays[53]. Therefore treatments that seem efficient in BRCA1-mutated tumours should be tested in triple negative/basal-like breast tumours with germline mutation in the HR pathway (like RAD51D or MRE11 genes).
Mutations in other genes
We found three mutations in three other genes, one in CHEK2 and two in genes of the MMR (mismatch repair) group, MSH6 and MLH1.
The new germline CHEK2 missense substitution, c.1214G>A (Cys405Tyr) was not associated with LOH in the tumour, but it has been reported that tumour-specific loss of the wild-type allele is not characteristic for CHEK2-associated breast cancer [54]. CHEK2 functions as a homodimer: one deleteriou s allele is sufficient to produce a majority of non-functional complexes. The individual carrying this mutation presented with a multi-site personal history of cancer, with thyroid cancer at the age of 42, bladder cancer at 50, and triple negative breast cancer at 51. Her family history included another triple negative breast cancer affecting her mother at 56, a possible retinal melanoma in her maternal uncle, whose son presented with a colon cancer at 59, and another maternal cousin with an astrocytoma at 48 (see Figure Family 6). Checkpoint kinase 2 (CHEK2) is an important signal transducer of cellular responses to DNA damage and that acts as a tumour suppressor gene. Studies have provided evidence that CHEK2-truncating and/or missense mutations are associated with increased risk of breast, prostate, thyroid, colon and kidney cancers [55,56].
This CHEK2 mutation, c.1214G>A, could predispose to breast cancer, and, consistent with literature, could also be associated with a multi-site cancer predisposition. However the association of CHEK2 mutations with triple-negative breast cancer have never been reported.
We observed a missense substitution in MSH6, predicted to be deleterious in silico, but the literature is controversial. In our study there is no strong evidence for a causal role in the breast cancer presented by the carrier: the tumour displayed no LOH and expressed MSH6 at a normal level. Although the family of the carrier is heavily affected with cancer, none belonging to the Lynch spectrum was reported. The maternal branch showed breast cancer and central nervous system (CNS) cancer (see Figure Family 3).
Several studies suggest an aside role of MSH6 among MMR genes. Mutations of MSH6 have been reported to be the most associated with extra-colonic cancers [57,58].
The MLH1 c.199G>T was found in the individual also carrying the PALB2 c.2897T>C mutation. This MLH1 mutation is known as pathogenic, causing Lynch Syndrome, and cancers of this mutation carrier and her family correspond to Lynch Syndrome cancer spectrum. However, TNBC could be associated with Lynch Syndrome. Immunohistochemistry of four TNBC (4/226, 1.8%) showed loss of MMR proteins (3 lost MLH1 and PMS2, and 1 lost MSH2 and MSH6); whereas none of the 90 non-triple-negative carcinomas showed the same loss [59]. Moreover, triple-negative tumours and MMR deficient colon tumours share a high genomic instability that other breast cancer subtypes don’t show.
Whether breast cancer is part of Lynch syndrome tumour spectrum is controversial. This individual breast cancer could be unrelated with his MLH1 mutation, or could suggest to further explore the implication of TNBC, and not breast cancer in general, in the Lynch Syndrome cancer spectrum.
Conclusion
Familial non-BRCA1/2 TNBC seems to be associated with rare mutations in a diversity of genes, in contrast to the major-gene model typified by genes like BRCA1 and BRCA2.
We report here that mutations in RAD51D, MRE11 and PALB2 confer a risk of triple-negative breast cancer, which could be much higher in this subtype.
This observation goes hand in hand with new approaches in breast cancer predisposition screening, especially with the recent possibilities of large-scale gene screening with high-throughput sequencing.
Identification of genes involved in triple-negative breast cancer genesis could also improve care and treatment of affected families. An approach based on genomic abnormalities starts to emerge as a substitute to the current organ-based strategy.
Looking for these mutations could be of great interest to guide therapeutic choice, and the fraction of breast cancer that should benefit from specific treatments, like PARP-inhibitor, would be larger, better identified and targeted.
Our findings have to be confirmed worldwide on larger populations of triple negative familial non-BRCA1/BRCA2 breast cancer, and it would probably be of interest to add basal markers to forthcoming studies.
Acknowledgements
We gratefully acknowledge the Centre Jean Perrin CRB biobank (BB-0033-00075) for handling and providing tissue material. We thank Dr Manon Sourdeix for constructive discussions.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Cui J, Antoniou AC, Dite GS, Southey MC, Venter DJ, Easton DF, Giles GG, McCredie MR, Hopper JL. After BRCA1 and BRCA2-what next? Multifactorial segregation analyses of threegeneration, population-based Australian families affected by female breast cancer. Am J Hum Genet. 2001;68:420–431. doi: 10.1086/318187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Melchor L, Benitez J. The complex genetic landscape of familial breast cancer. Hum Genet. 2013;132:845–863. doi: 10.1007/s00439-013-1299-y. [DOI] [PubMed] [Google Scholar]
- 3.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lonning PE, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 5.Robertson L, Hanson H, Seal S, Warren-Perry M, Hughes D, Howell I, Turnbull C, Houlston R, Shanley S, Butler S, Evans DG, Ross G, Eccles D, Tutt A, Rahman N. BRCA1 testing should be offered to individuals with triple-negative breast cancer diagnosed below 50 years. Br J Cancer. 2012;106:1234–1238. doi: 10.1038/bjc.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalez-Angulo AM, Timms KM, Liu S, Chen H, Litton JK, Potter J, Lanchbury JS, Stemke-Hale K, Hennessy BT, Arun BK, Hortobagyi GN, Do KA, Mills GB, Meric-Bernstam F. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin Cancer Res. 2011;17:1082–1089. doi: 10.1158/1078-0432.CCR-10-2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong-Brown MW, Avery-Kiejda KA, Bowden NA, Scott RJ. Low prevalence of germline PALB2 mutations in Australian triple-negative breast cancer. Int J Cancer. 2014;134:301–305. doi: 10.1002/ijc.28361. [DOI] [PubMed] [Google Scholar]
- 8.Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, Olson JE, Godwin AK, Pankratz VS, Olswold C, Slettedahl S, Hallberg E, Guidugli L, Davila JI, Beckmann MW, Janni W, Rack B, Ekici AB, Slamon DJ, Konstantopoulou I, Fostira F, Vratimos A, Fountzilas G, Pelttari LM, Tapper WJ, Durcan L, Cross SS, Pilarski R, Shapiro CL, Klemp J, Yao S, Garber J, Cox A, Brauch H, Ambrosone C, Nevanlinna H, Yannoukakos D, Slager SL, Vachon CM, Eccles DM, Fasching PA. Inherited Mutations in 17 Breast Cancer Susceptibility Genes Among a Large Triple-Negative Breast Cancer Cohort Unselected for Family History of Breast Cancer. J. Clin. Oncol. 2015;33:304–11. doi: 10.1200/JCO.2014.57.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonaiti B, Alarcon F, Bonadona V, Pennec S, Andrieu N, Stoppa-Lyonnet D, Perdry H, Bonaiti-Pellie C. [A new scoring system for the diagnosis of BRCA1/2 associated breast-ovarian cancer predisposition] . Bull Cancer. 2011;98:779–795. doi: 10.1684/bdc.2011.1397. [DOI] [PubMed] [Google Scholar]
- 10.Delort L, Satih S, Kwiatkowski F, Bignon YJ, Bernard-Gallon DJ. Evaluation of breast cancer risk in a multigenic model including low penetrance genes involved in xenobiotic and estrogen metabolisms. Nutr Cancer. 2010;62:243–251. doi: 10.1080/01635580903305300. [DOI] [PubMed] [Google Scholar]
- 11.Gail M. Power computations for designing comparative Poisson trials. Biometrics. 1974;30:231–237. [Google Scholar]
- 12.Stevens KN, Fredericksen Z, Vachon CM, Wang X, Margolin S, Lindblom A, Nevanlinna H, Greco D, Aittomaki K, Blomqvist C, Chang-Claude J, Vrieling A, Flesch-Janys D, Sinn HP, Wang-Gohrke S, Nickels S, Brauch H, Ko YD, Fischer HP, Schmutzler RK, Meindl A, Bartram CR, Schott S, Engel C, Godwin AK, Weaver J, Pathak HB, Sharma P, Brenner H, Muller H, Arndt V, Stegmaier C, Miron P, Yannoukakos D, Stavropoulou A, Fountzilas G, Gogas HJ, Swann R, Dwek M, Perkins A, Milne RL, Benitez J, Zamora MP, Perez JI, Bojesen SE, Nielsen SF, Nordestgaard BG, Flyger H, Guenel P, Truong T, Menegaux F, Cordina-Duverger E, Burwinkel B, Marme F, Schneeweiss A, Sohn C, Sawyer E, Tomlinson I, Kerin MJ, Peto J, Johnson N, Fletcher O, Dos Santos Silva I, Fasching PA, Beckmann MW, Hartmann A, Ekici AB, Lophatananon A, Muir K, Puttawibul P, Wiangnon S, Schmidt MK, Broeks A, Braaf LM, Rosenberg EH, Hopper JL, Apicella C, Park DJ, Southey MC, Swerdlow AJ, Ashworth A, Orr N, Schoemaker MJ, Anton-Culver H, Ziogas A, Bernstein L, Dur CC, Shen CY, Yu JC, Hsu HM, Hsiung CN, Hamann U, Dunnebier T, Rudiger T, Ulmer HU, Pharoah PP, Dunning AM, Humphreys MK, Wang Q, Cox A, Cross SS, Reed MW, Hall P, Czene K, Ambrosone CB, Ademuyiwa F, Hwang H, Eccles DM, Garcia-Closas M, Figueroa JD, Sherman ME, Lissowska J, Devilee P, Seynaeve C, Tollenaar RA, Hooning MJ, Andrulis IL, Knight JA, Glendon G, Mulligan AM, Winqvist R, Pylkas K, Jukkola-Vuorinen A, Grip M, John EM, Miron A, Alnaes GG, Kristensen V, Borresen-Dale AL, Giles GG, Baglietto L, McLean CA, Severi G, Kosel ML, Pankratz VS, Slager S, Olson JE, Radice P, Peterlongo P, Manoukian S, Barile M, Lambrechts D, Hatse S, Dieudonne AS, Christiaens MR, Chenevix-Trench G, Beesley J, Chen X, Mannermaa A, Kosma VM, Hartikainen JM, Soini Y, Easton DF, Couch FJ. 19p13.1 is a triple-negative-specific breast cancer susceptibility locus. Cancer Res. 2012;72:1795–1803. doi: 10.1158/0008-5472.CAN-11-3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stone EA, Sidow A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 2005;15:978–986. doi: 10.1101/gr.3804205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013 doi: 10.1002/0471142905.hg0720s76. Chapter 7: Unit7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo C, Tsementzi D, Kyrpides N, Read T, Konstantinidis KT. Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS One. 2012;7:e30087. doi: 10.1371/journal.pone.0030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wirawan A, Harris RS, Liu Y, Schmidt B, Schroder J. HECTOR: a parallel multistage homopolymer spectrum based error corrector for 454 sequencing data. BMC Bioinformatics. 2014;15:131. doi: 10.1186/1471-2105-15-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pertea M, Lin X, Salzberg SL. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res. 2001;29:1185–1190. doi: 10.1093/nar/29.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogura T, Wilkinson AJ. AAA+ superfamily ATPases: common structure--diverse function. Genes Cells. 2001;6:575–597. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- 20.Desrichard A, Bidet Y, Uhrhammer N, Bignon YJ. CHEK2 contribution to hereditary breast cancer in non-BRCA families. Breast Cancer Res. 2011;13:R119. doi: 10.1186/bcr3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, Hollestelle A, Houben M, Crepin E, van Veghel-Plandsoen M, Elstrodt F, van Duijn C, Bartels C, Meijers C, Schutte M, McGuffog L, Thompson D, Easton D, Sodha N, Seal S, Barfoot R, Mangion J, Chang-Claude J, Eccles D, Eeles R, Evans DG, Houlston R, Murday V, Narod S, Peretz T, Peto J, Phelan C, Zhang HX, Szabo C, Devilee P, Goldgar D, Futreal PA, Nathanson KL, Weber B, Rahman N, Stratton MR. Lowpenetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet. 2002;31:55–59. doi: 10.1038/ng879. [DOI] [PubMed] [Google Scholar]
- 22.Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873–875. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 23.Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, Evans DG, Eccles D, Easton DF, Stratton MR. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–167. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffog L, Evans DG, Eccles D, Easton DF, Stratton MR, Rahman N. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38:1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 25.Tung N, Battelli C, Allen B, Kaldate R, Bhatnagar S, Bowles K, Timms K, Garber JE, Herold C, Ellisen L, Krejdovsky J, DeLeonardis K, Sedgwick K, Soltis K, Roa B, Wenstrup RJ, Hartman AR. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer. 2015;121:25–33. doi: 10.1002/cncr.29010. [DOI] [PubMed] [Google Scholar]
- 26.Smiraldo PG, Gruver AM, Osborn JC, Pittman DL. Extensive chromosomal instability in Rad51d-deficient mouse cells. Cancer Res. 2005;65:2089–2096. doi: 10.1158/0008-5472.CAN-04-2079. [DOI] [PubMed] [Google Scholar]
- 27.Tarsounas M, Munoz P, Claas A, Smiraldo PG, Pittman DL, Blasco MA, West SC. Telomere maintenance requires the RAD51D recombination/repair protein. Cell. 2004;117:337–347. doi: 10.1016/s0092-8674(04)00337-x. [DOI] [PubMed] [Google Scholar]
- 28.Loveday C, Turnbull C, Ramsay E, Hughes D, Ruark E, Frankum JR, Bowden G, Kalmyrzaev B, Warren-Perry M, Snape K, Adlard JW, Barwell J, Berg J, Brady AF, Brewer C, Brice G, Chapman C, Cook J, Davidson R, Donaldson A, Douglas F, Greenhalgh L, Henderson A, Izatt L, Kumar A, Lalloo F, Miedzybrodzka Z, Morrison PJ, Paterson J, Porteous M, Rogers MT, Shanley S, Walker L, Eccles D, Evans DG, Renwick A, Seal S, Lord CJ, Ashworth A, Reis-Filho JS, Antoniou AC, Rahman N. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet. 2011;43:879–882. doi: 10.1038/ng.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson ER, Rowley SM, Sawyer S, kConfab , Eccles DM, Trainer AH, Mitchell G, James PA. Analysis of RAD51D in ovarian cancer patients and families with a history of ovarian or breast cancer. PLoS One. 2013;8:e54772. doi: 10.1371/journal.pone.0054772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez-Lopez R, Osorio A, Ribas G, Pollan M, Sanchez-Pulido L, de la Hoya M, Ruibal A, Zamora P, Arias JI, Salazar R, Vega A, Martinez JI, Esteban-Cardenosa E, Alonso C, Leton R, Urioste Azcorra M, Miner C, Armengod ME, Carracedo A, Gonzalez-Sarmiento R, Caldes T, Diez O, Benitez J. The variant E233G of the RAD51D gene could be a low-penetrance allele in high-risk breast cancer families without BRCA1/2 mutations. Int J Cancer. 2004;110:845–849. doi: 10.1002/ijc.20169. [DOI] [PubMed] [Google Scholar]
- 31.Jara L, Dubois K, Gaete D, de Mayo T, Ratkevicius N, Bravo T, Margarit S, Blanco R, Gomez F, Waugh E, Peralta O, Reyes JM, Ibanez G, Gonzalez-Hormazabal P. Variants in DNA double-strand break repair genes and risk of familial breast cancer in a South American population. Breast Cancer Res Treat. 2010;122:813–822. doi: 10.1007/s10549-009-0709-2. [DOI] [PubMed] [Google Scholar]
- 32.Nadkarni A, Furda A, Rajesh C, McInnes C, Ruch RJ, Pittman DL. Functional characterization of the RAD51D E233G genetic variant. Pharmacogenet Genomics. 2009;19:153–160. doi: 10.1097/FPC.0b013e32831db2fd. [DOI] [PubMed] [Google Scholar]
- 33.Nadkarni A, Rajesh P, Ruch RJ, Pittman DL. Cisplatin resistance conferred by the RAD51D (E233G) genetic variant is dependent upon p53 status in human breast carcinoma cell lines. Mol Carcinog. 2009;48:586–591. doi: 10.1002/mc.20545. [DOI] [PubMed] [Google Scholar]
- 34.Baker JL, Schwab RB, Wallace AM, Madlensky L. Breast Cancer in a RAD51D Mutation Carrier: Case Report and Review of the Literature. Clin Breast Cancer. 2015;15:e71–75. doi: 10.1016/j.clbc.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 35.Williams GJ, Lees-Miller SP, Tainer JA. Mre11-Rad50-Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair (Amst) 2010;9:1299–1306. doi: 10.1016/j.dnarep.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22:5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waltes R, Kalb R, Gatei M, Kijas AW, Stumm M, Sobeck A, Wieland B, Varon R, Lerenthal Y, Lavin MF, Schindler D, Dork T. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am J Hum Genet. 2009;84:605–616. doi: 10.1016/j.ajhg.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Uchisaka N, Takahashi N, Sato M, Kikuchi A, Mochizuki S, Imai K, Nonoyama S, Ohara O, Watanabe F, Mizutani S, Hanada R, Morio T. Two brothers with ataxia-telangiectasia-like disorder with lung adenocarcinoma. J Pediatr. 2009;155:435–438. doi: 10.1016/j.jpeds.2009.02.037. [DOI] [PubMed] [Google Scholar]
- 39.Heikkinen K, Karppinen SM, Soini Y, Makinen M, Winqvist R. Mutation screening of Mre11 complex genes: indication of RAD50 involvement in breast and ovarian cancer susceptibility. J Med Genet. 2003;40:e131. doi: 10.1136/jmg.40.12.e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Damiola F, Pertesi M, Oliver J, Le Calvez-Kelm F, Voegele C, Young EL, Robinot N, Forey N, Durand G, Vallee MP, Tao K, Roane TC, Williams GJ, Hopper JL, Southey MC, Andrulis IL, John EM, Goldgar DE, Lesueur F, Tavtigian SV. Rare key functional domain missense substitutions in MRE11A, RAD50, and NBN contribute to breast cancer susceptibility: results from a Breast Cancer Family Registry case-control mutation-screening study. Breast Cancer Res. 2014;16:R58. doi: 10.1186/bcr3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bartkova J, Tommiska J, Oplustilova L, Aaltonen K, Tamminen A, Heikkinen T, Mistrik M, Aittomaki K, Blomqvist C, Heikkila P, Lukas J, Nevanlinna H, Bartek J. Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol Oncol. 2008;2:296–316. doi: 10.1016/j.molonc.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gupta GP, Vanness K, Barlas A, Manova-Todorova KO, Wen YH, Petrini JH. The Mre11 complex suppresses oncogene-driven breast tumorigenesis and metastasis. Mol Cell. 2013;52:353–365. doi: 10.1016/j.molcel.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tischkowitz M, Xia B. PALB2/FANCN: recombining cancer and Fanconi anemia. Cancer Res. 2010;70:7353–7359. doi: 10.1158/0008-5472.CAN-10-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A. 2009;106:7155–7160. doi: 10.1073/pnas.0811159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, Yu X. PALB2 links BRCA1 and BRCA2 in the DNAdamage response. Curr Biol. 2009;19:524–529. doi: 10.1016/j.cub.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dansonka-Mieszkowska A, Kluska A, Moes J, Dabrowska M, Nowakowska D, Niwinska A, Derlatka P, Cendrowski K, Kupryjanczyk J. A novel germline PALB2 deletion in Polish breast and ovarian cancer patients. BMC Med Genet. 2010;11:20. doi: 10.1186/1471-2350-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heikkinen T, Karkkainen H, Aaltonen K, Milne RL, Heikkila P, Aittomaki K, Blomqvist C, Nevanlinna H. The breast cancer susceptibility mutation PALB2 1592delT is associated with an aggressive tumor phenotype. Clin Cancer Res. 2009;15:3214–3222. doi: 10.1158/1078-0432.CCR-08-3128. [DOI] [PubMed] [Google Scholar]
- 48.Teo ZL, Park DJ, Provenzano E, Chatfield CA, Odefrey FA, Nguyen-Dumont T, Dowty JG, Hopper JL, Winship I, Goldgar DE, Southey MC. Prevalence of PALB2 mutations in Australasian multiple-case breast cancer families. Breast Cancer Res. 2013;15:R17. doi: 10.1186/bcr3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teo ZL, Provenzano E, Dite GS, Park DJ, Apicella C, Sawyer SD, James PA, Mitchell G, Trainer AH, Lindeman GJ, Shackleton K, Cicciarelli L, Buys SS, Andrulis IL, Mulligan AM, Glendon G, John EM, Terry MB, Daly M, Odefrey FA, Nguyen-Dumont T, Giles GG, Dowty JG, Winship I, Goldgar DE, Hopper JL, Southey MC. Tumour morphology predicts PALB2 germline mutation status. Br J Cancer. 2013;109:154–163. doi: 10.1038/bjc.2013.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han J, Haiman C, Niu T, Guo Q, Cox DG, Willett WC, Hankinson SE, Hunter DJ. Genetic variation in DNA repair pathway genes and premenopausal breast cancer risk. Breast Cancer Res Treat. 2009;115:613–622. doi: 10.1007/s10549-008-0089-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holstege H, Horlings HM, Velds A, Langerod A, Borresen-Dale AL, van de Vijver MJ, Nederlof PM, Jonkers J. BRCA1-mutated and basallike breast cancers have similar aCGH profiles and a high incidence of protein truncating TP53 mutations. BMC Cancer. 2010;10:654. doi: 10.1186/1471-2407-10-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu H, Eirew P, Mullaly SC, Aparicio S. The omics of triple-negative breast cancers. Clin Chem. 2014;60:122–133. doi: 10.1373/clinchem.2013.207167. [DOI] [PubMed] [Google Scholar]
- 53.O’Shaughnessy J, Osborne C, Pippen JE, Yoffe M, Patt D, Rocha C, Koo IC, Sherman BM, Bradley C. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med. 2011;364:205–214. doi: 10.1056/NEJMoa1011418. [DOI] [PubMed] [Google Scholar]
- 54.Suspitsin EN, Yanus GA, Sokolenko AP, Yatsuk OS, Zaitseva OA, Bessonov AA, Ivantsov AO, Heinstein VA, Klimashevskiy VF, Togo AV, Imyanitov EN. Development of breast tumors in CHEK2, NBN/NBS1 and BLM mutation carriers does not commonly involve somatic inactivation of the wild-type allele. Med Oncol. 2014;31:828. doi: 10.1007/s12032-013-0828-9. [DOI] [PubMed] [Google Scholar]
- 55.Cybulski C, Gorski B, Huzarski T, Masojc B, Mierzejewski M, Debniak T, Teodorczyk U, Byrski T, Gronwald J, Matyjasik J, Zlowocka E, Lenner M, Grabowska E, Nej K, Castaneda J, Medrek K, Szymanska A, Szymanska J, Kurzawski G, Suchy J, Oszurek O, Witek A, Narod SA, Lubinski J. CHEK2 is a multiorgan cancer susceptibility gene. Am J Hum Genet. 2004;75:1131–1135. doi: 10.1086/426403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Siolek M, Cybulski C, Gasior-Perczak D, Kowalik A, Kozak-Klonowska B, Kowalska A, Chlopek M, Kluzniak W, Wokolorczyk D, Palyga I, Walczyk A, Lizis-Kolus K, Sun P, Lubinski J, Narod SA, Gozdz S. CHEK2 mutations and the risk of papillary thyroid cancer. Int J Cancer. 2015;137:548–52. doi: 10.1002/ijc.29426. [DOI] [PubMed] [Google Scholar]
- 57.Pande M, Wei C, Chen J, Amos CI, Lynch PM, Lu KH, Lucio LA, Boyd-Rogers SG, Bannon SA, Mork ME, Frazier ML. Cancer spectrum in DNA mismatch repair gene mutation carriers: results from a hospital based Lynch syndrome registry. Fam Cancer. 2012;11:441–447. doi: 10.1007/s10689-012-9534-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonadona V, Bonaiti B, Olschwang S, Grandjouan S, Huiart L, Longy M, Guimbaud R, Buecher B, Bignon YJ, Caron O, Colas C, Nogues C, Lejeune-Dumoulin S, Olivier-Faivre L, Polycarpe-Osaer F, Nguyen TD, Desseigne F, Saurin JC, Berthet P, Leroux D, Duffour J, Manouvrier S, Frebourg T, Sobol H, Lasset C, Bonaiti-Pellie C. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. 2011;305:2304–2310. doi: 10.1001/jama.2011.743. [DOI] [PubMed] [Google Scholar]
- 59.Wen YH, Brogi E, Zeng Z, Akram M, Catalano J, Paty PB, Norton L, Shia J. DNA mismatch repair deficiency in breast carcinoma: a pilot study of triple-negative and non-triple-negative tumors. Am J Surg Pathol. 2012;36:1700–1708. doi: 10.1097/PAS.0b013e3182627787. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.