

Abstract
Tumors often consist of hypoxic regions which are resistant to chemo- and radiotherapy. Evofosfamide (also known as TH-302), a 2-nitroimidazole triggered hypoxia-activated prodrug, preferentially releases the DNA cross-linker bromo-isophosphoramide mustard in hypoxic cells. The intracellular kinase mTOR plays a key role in multiple pathways which are important in cancer progression. Here we investigated the enhanced efficacy profile and possible mechanisms of evofosfamide in combination with mTOR inhibitor (mTORi) everolimus or temsirolimus in renal cell carcinoma (RCC) xenograft models. The antitumor activities of the mTORi everolimus or temsirolimus alone, evofosfamide alone, or the combination were investigated in the 786-O and Caki-1 RCC cells in vitro and in vivo xenograft models. Two schedules were tested in which evofosfamide was started on the same day as the mTORi or 1 week after. Combination mechanisms were investigated by measuring a panel of pharmacodynamic biomarkers by immunohistochemistry. Antitumor efficacy in both RCC xenograft models was enhanced by the combination of evofosfamide and mTORi. Evofosfamide reduced the increased hypoxia induced by mTORi. Combination treatment induced increased DNA damage, decreased cell proliferation, and decreased survivin. Addition of mTORi did not change evofosfamide-mediated cytotoxicity in 786-O or Caki-1 cells in vitro which might suggest cell non-autonomous effects, specifically increased tumor hypoxia, are important for the in vivo combination activity. Taken together, evofosfamide potentiates the antitumor efficacy of mTOR inhibitors and inhibits the increased tumor hypoxia caused by mTOR inhibition. These studies provide a translational rationale for combining evofosfamide with mTOR inhibitors in clinical studies.
Keywords: Evofosfamide, hypoxia-activated prodrug, mTOR inhibitor, pharmacodynamics biomarker, renal cell carcinoma, TH-302, xenograft
Introduction
Renal cell carcinoma (RCC) accounts for approximately 3% of adult malignancies and close to 90% of all renal neoplasms [1]. Over the last decade, biologic response modifiers have been widely explored and developed in the treatment of RCCs, particularly for patients with metastatic and/or unresectable disease. These options include molecularly targeted therapy, biological therapy, and combinations [2]. Primary and metastatic RCCs are angiogenesis-dependent and hypoxia-driven malignancies [3,4]. Agents targeting tumor angiogenesis have been investigated for the treatment of RCC, for example, inhibitors of vascular endothelial growth factor (VEGF) or mammalian target of rapamycin (mTOR) pathways [5]. mTOR is downstream in the PI3K/AKT signaling pathway and is a critical regulator of cell proliferation, metabolism, and protein synthesis [6]. mTOR functions as a sensor of nutritional/metabolic stress during cell development and promotes protein synthesis and cell growth during nutrient or energy rich periods [6]. In addition, mTOR inhibition sensitizes tumor cells to DNA-damage induced apoptosis by inhibiting p53-induced p21 expression [7]. As a result, mTOR inhibition increases the cytotoxicity of drugs that damage DNA [7]. Furthermore, mTOR inhibitors exhibit antiangiogenic activity mainly via two mechanisms: decreasing synthesis and release of angiogenic growth factors from the cancer cells, and blocking growth and proliferation of vascular cells [8-10]. Therapeutics targeting angiogenesis deprive the tumor of oxygen and nutrients, resulting in growth inhibition and increased hypoxia [11]. Increased hypoxia is associated with tumor progression, metastasis and resistance to therapy [12-14]. Thus, the simultaneous targeting of tumor hypoxia may improve the efficacy of antiangiogenic mTOR-targeted therapy.
Inhibition of mTOR has shown antitumor effects in both preclinical [15,16] and clinical [17,18] settings. Two mTOR inhibitors, temsirolimus and everolimus, have been approved for FDA for the treatment of RCC. Although mTOR inhibitors have shown clinical benefit in RCC, such treatment rarely results in a cure and patients frequently develop resistance to the drugs. This class of agents is largely cytostatic, resulting in relatively few tumor regressions. Thus, cytotoxic effects achieved by combing in an mTOR inhibitor and other targeted agents would be desirable [19]. A number of reports have identified potent combination effects from combining conventional cytotoxic agents and mTOR inhibitors in experimental settings [7,20,21].
Evofosfamide, previously known as TH-302, (1-methyl-2-nitro-1H-imidazole-5-yl) methyl N,N’-bis (2-bromoethyl) phosphorodiamidate is a nitroimidazole-linked prodrug of a brominated version of isophosphoramide mustard (Br-IPM). Evofosfamide is reduced at the nitroimidazole site of the prodrug by intracellular reductases and leads to the release of Br-IPM when exposed to hypoxic conditions. Br-IPM can then act as a DNA crosslinking agent [22]. In vitro cytotoxicity and clonogenic assays employing human cancer cell lines demonstrate that evofosfamide has little cytotoxic activity under normoxic conditions and greatly enhanced cytotoxic potency under hypoxic conditions [23]. Evofosfamide has demonstrated broad antitumor activities in preclinical models [24,25] and has showed encouraging activity in multiple clinical trials [26,27]. Evofosfamide has shown the ability to selectively kill hypoxic cells and reduce the hypoxic compartment fraction in a number of xenograft models. In addition, preclinical data suggest that after activation, the activated moiety of evofosfamide may diffuse to areas outside the hypoxic regions, demonstrating a “bystander” effect [23,24].
Here we investigated the therapeutic potential of evofosfamide when in combination with the mTOR inhibitors everolimus or temsirolimus in preclinical RCC models.
Materials and methods
Compounds
Evofosfamide was manufactured by Syngene (Bangalore, India). It was formulated in saline (0.9% NaCl) at the maximal solubility of 10 mg/ml. The solution was filtered through 0.2 µm filter prior to animal dosing. Everolimus and Temsirolimus, purchased from Ontario Chemical (Guelph, Canada), were formulated in 5% DMSO, 5% Tween 80, and 90% D5W (5% dextrose in water).
Cell lines
786-O (VHL-/- PTEN-/-), and Caki-1 (VHL+/+ PTEN+/+) cell lines were obtained from the American Type Culture Collection (ATCC, Rockville, MD) and were genetically authenticated by using microsatellite marker analysis (Idexx Bioresearch, Columbia, MO). Cells were cultured in the ATCC-suggested media with 10% fetal bovine serum added and maintained in a 5% CO2, humidified environment at 37°C.
Xenograft models
Specific pathogen-free homozygous female nude mice (Nu-Foxn 1nu NU/NU, Charles River Laboratories, Wilmington, MA, USA) were used for the xenograft models. Mice were given food and water ad libitum and housed in microisolator cages. Four- to 6-week-old animals were tagged with microchips (Locus Technology, Manchester, MD) for identification. All animal studies were approved by the Institutional Animal Care and Use Committee of Threshold Pharmaceuticals, Inc, and animals were maintained in accordance to guidelines of the American Association of Laboratory Animal Care. Determined by the specific tumor growth kinetics, 5 x 106 786-O or Caki-1 cells, mixed 1:1 with Matrigel (BD Bioscience, San Jose, CA) in a total volume of 0.2 ml were implanted in the subcutaneous area of the flank of the mouse.
In vivo antitumor activity
Tumor growth and body weight were measured twice-a-week after cell implantation. Tumor volume was calculated as (length x width2)/2. When the mean value of tumor volume was approximately 100-150 mm3 mice were randomized into 10 mice per group and the treatment started (Day 1). Antitumor activity was assessed by tumor growth kinetics and Tumor Growth Inhibition (TGI). TGI was defined as (1-ΔT/ΔC) x 100, where ΔT/ΔC is the ratio of the change in mean tumor volume of the treated group (ΔT) and of the control group (ΔC). Animals were euthanized when individual tumor size was over 2000 mm3 or individual tumor size was over 1000 mm3 if mean tumor volume in the group exceeded 1000 mm3. In the 786-O xenograft model, due to the slow tumor growth and tumor regression in the vehicle group when tumor volumes were over 600-700 mm3, TGI was determined when the average tumor size in the vehicle group reached 600 mm3. TGI in Caki-1 xenograft model was determined on the last measurement when all the animals in the vehicle group survived. Tumor Growth Delay to 1000 mm3 (TGD1000) in Caki-1 model was determined as the increased time (in days) for the treated groups’ tumor size on average to reach 1000 mm3 as compared to the vehicle group using the tumor growth curves. Data are expressed as the mean ± SEM. One-way analysis of variance with Dunnett’s test (GraphPad PRISM 4, La Jolla, CA) was used for analysis. A P level < 0.05 was considered statistically significant.
Drug treatment
Maximum tolerated dose (MTD) for both monotherapy and combination therapy was defined as the highest dose resulting in no animal deaths, less than 20% weight loss for any one animal in an experimental group, no significant changes in general clinical signs, and no abnormal gross anatomical findings after necropsy. General clinical signs included: respiratory rate, behavior, and response to normal stimuli [28]. The doses of compounds used in all studies were no higher than MTD. In the studies to investigate the antitumor activity of evofosfamide in combination with mTOR inhibitor, everolimus was administered at 5 mg/kg, QDx19, po; temsirolimus was dosed at 20 mg/kg, QDx19, ip; and evofosfamide was given at 50 mg/kg, 5 days per week, ip; everolimus or temsirolimus treatment in both monotherapy and combination groups started at Day 1. Evofosfamide was either administered from Day 1 (same day as the initiation of mTOR inhibitor treatment, Concurrent Schedule, CS) for 3 weeks, or from Day 8 (one week after mTOR inhibitors’ treatment start, Delayed Schedule, DS) for 2 weeks, in both monotherapy and combination therapy groups. In the studies to investigate the pharmacological mechanisms and changes of pharmacodynamics (PD) biomarkers, 5-8 animals per group were treated with vehicle, 5 mg/kg of everolimus in the 786-O xenograft model or 20 mg/kg of temsirolimus in the Caki-1 xenograft model daily for 8 days, evofosfamide-alone at 150 mg/kg, ip once, or 8 days’ mTOR inhibitors treatment followed by one dose of evofosfamide 150 mg/kg, respectively. For these PD studies, tumors were harvested 4 hrs after the last treatment of the mTOR inhibitors in the monotherapy groups or 72 hrs after evofosfamide treatment in both evofosfamide monotherapy and combination groups.
Histology and immunohistochemistry
The exogenous hypoxia marker, pimonidazole hydrochloride (Hypoxyprobe, Burlington, MA) at 60 mg/kg was ip injected one hour before animal was euthanized. Tumors were harvested, fixed in 10% neutral buffered formalin, and embedded in paraffin. Five-micrometer thick paraffin sections were cut and adhered to poly--lysine-coated glass microscope slides. After deparaffinization and rehydration of the slides, antigen retrieval was conducted by incubating the slides with Heat Induced Epitope Retrieval Buffer (Biocare Medical, Concord, CA) in a Decloaker Chamber (Biocare Medical). Endogenous peroxidase was quenched by Peroxidaze 1 (Biocare Medical) and non-specific binding was blocked by Background Sniper (Biocare Medical). Slides were incubated with the primary antibodies for 1 hr at RT followed by secondary HRP-conjugated anti-rabbit IgG (Abcam, Cambridge, MA). The primary antibodies included rabbit polyclonal anti-pimonidazole (Hypoxyprobe, 1:400), rabbit monoclonal anti-γH2AX (Abcam, 1:3000), rabbit monoclonal anti-Ki67 (Abcam, 1:2000), and rabbit polyclonal anti-CD31 (Abcam, 1:30), rabbit polyclonal anti-survivin (Abcam, 1:250) and rabbit monoclonal anti-Glut-1 (Abcam, 1:2500), rabbit polyclonal anti-Phospho-S6 Ribosomal Protein (Cell Signaling Technology, Danvers, MA, 1:25).
Image analysis
At 20x magnification, digital images of pimonidazole-stained sections were captured and assembled to compose a whole tumor image. Pimonidazole positive regions were extracted using Image-Pro Plus v6.0 (MediaCybernetics, Rockville, MD). To compare the volume of the hypoxic compartment in the tumors after evofosfamide treatment, the hypoxic fraction (HF) was evaluated as pimonidazole-positive hypoxic area in the whole tumor. All images were captured under consistent illumination and exposure for their respective stains. No image post-processing was done. Custom-made scripts were developed in Image Pro-Plus to analyze the target signals using color and morphologic segmentation tools. For semi-quantification of CD31, Glut-1, and pS6-positive areas, point counting was performed in the fields containing viable tumor cells. A total of 10-15 fields per section at 400x magnification were counted in each animal on a 1-cm2 eyepiece graticule with 10 equidistant grid lines. The percentage of fractional area (percentage of positive area per total area counted of the section) was calculated using the following formula: percentage of positive fractional area = number of grid intersections with positive staining/total number of grid intersections multiplied x 100%. γH2AX, survivin, or Ki67 positive cells were counted at 400x magnification. Ten fields per section were used. The percentage of positive cells was calculated as number of positive cells/number of total cells x 100%. Statistical analysis was used in all instances with a p value <0.05 considered significant. One-way analysis of variance with Dunnett’s test (GraphPad PRISM 4) was used to compare the significance of the multiple groups. The Student’s t-test was used to find the significance between two groups.
In vitro cytotoxicity assays
786-O or Caki-1 were seeded into 24-well plates (20,000 cells/ well) and incubated overnight. Cells were pre-treated with indicated concentrations of either everolimus or temsirolimus for 1 h, followed by evofosfamide treatment for 2 h under either normoxia (21% O2) or hypoxia (N2). Drug containing medium was removed and cells were washed twice. Everolimus or temsirolimus was added back to the cells and incubated for additional 3 days in an atmosphere of 5% CO2, 95% air and 100% relative humidity. On day 3, the Alamar Blue assay was performed to quantify viable cells. IC50 values were calculated using GraphPad Prism4 software.
Results
Evofosfamide potentiates the efficacy of mTOR inhibitors in the RCC xenograft models
The antitumor effects of evofosfamide in combination with mTOR inhibitors were assessed in the 786-O and Caki-1 xenograft models. 786-O exhibits a genotype of VHL-/- and PTEN-/-, and Caki-1 exhibits VHL+/+ and PTEN+/+. When the average tumor size was 150 mm3, ten tumor-bearing animals in each group were treated with vehicle, mTOR inhibitor-alone, evofosfamide-alone, or mTOR inhibitor and evofosfamide in combination. Two schedules of evofosfamide treatment were employed in the studies: Concurrent Schedule (CS) in which evofosfamide was given from Day 1 for 3 weeks, and Delayed Schedule (DS) in which evofosfamide was given from Day 8 for 2 weeks. The detailed study schemes are presented in Figure 1A. Both everolimus and temsirolimus were tested in the 786-O xenograft model. Treatment with everolimus alone resulted in a dramatic tumor reduction compared with vehicle treatment (P<0.05). Consistent with previous results, evofosfamide monotherapy did not show antitumor activity in this model. Combination therapy of evofosfamide and everolimus significantly increased the antitumor activity, compared with vehicle, evofosfamide monotherapy, or everolimus monotherapy group (P<0.05) (Figure 1B and Table 1). Both CS and DS combination groups exhibited similar antitumor activity. When temsirolimus was tested in the 786-O xenograft model, temsirolimus-alone yielded TGI of 113%. In the combination of evofosfamide and temsirolimus treatment group, TGIs were 137% and 142% in CS and DS, respectively (P<0.01 vs. temsirolimus monotherapy or evofosfamide monotherapy treated animals). There was no statistical difference between the CS and DS combination groups (Figure 1C).
Figure 1.
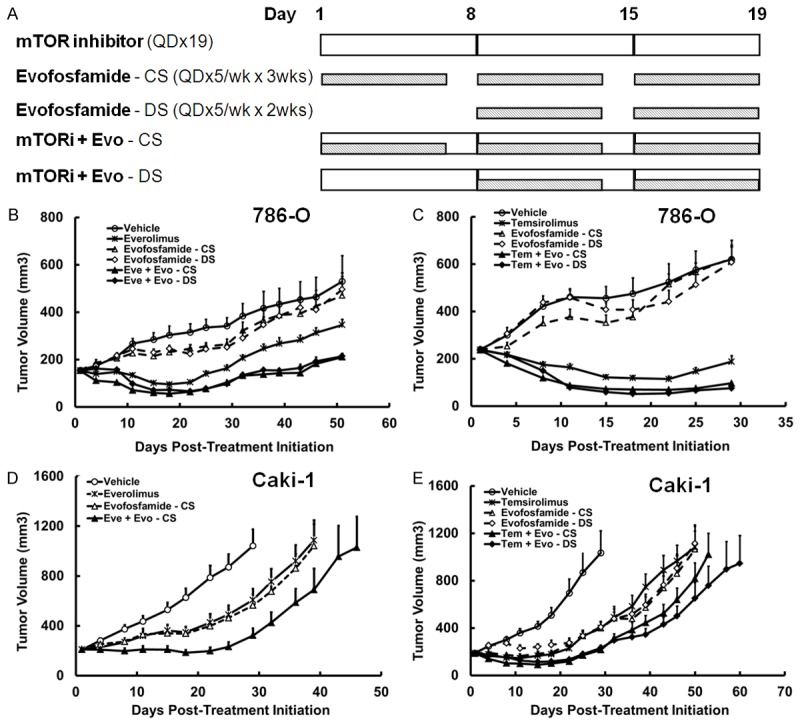
Antitumor efficacy and safety profile of evofosfamide in combination with mTOR inhibitors in 786-O and Caki-1 RCC xenograft models. A. Treatment schedule and experimental design; B-E. Tumor growth of evofosfamide in combination with mTOR inhibitors in the 786-O and Caki-1 xenograft models. Animals were monitored daily and tumor growth was quantified twice a week. Data are expressed as Mean ± SEM of 10 animals per group. Evo, evofosfamide; mTORi, mTOR inhibitor; Eve, everolimus; Tem, temsirolimus; CS, concurrent schedule; and DS, delayed schedule.
Table 1.
Summary of antitumor activity and safety profile of evofosfamide monotherapy, everolimus or temsirolimus monotherapy, and evofosfamide in combination with mTOR inhibitors in the 786-O and Caki-1 RCC xenograft models
| 786-O (mTORi: Eve) | 786-O (mTORi: Tem) | Caki-1 (mTORi: Eve) | Caki-1 mTORi: Tem) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||
| TGI % | MBL % | TGI % | MBL % | TGI % | TGD1000 | MBL % | TGI % | TGD1000 | MBL % | |
| mTORi | 48* | 0 | 113* | 3 | 52* | 10 | 1 | 74* | 17 | 4 |
| Evofosfamide CS | 15 | 0 | 0 | 7 | 57* | 10 | 0 | 75* | 19 | 2 |
| mTORi + Evo CS | 84*,a,b | 0 | 136*,a,b | 9 | 87*,a,b | 24 | 4 | 96*,a,b | 24 | 9 |
| Evofosfamide DS | 25 | 0 | 14 | 0 | 81* | 19 | 0 | |||
| mTORi + Evo DS | 83*,a,b | 0 | 142*,a,b | 8 | 94*,a,b | 32 | 7 | |||
TGI, Tumor Growth Inhibition; TGD1000, Tumor Growth Delay to Vehicle reaching the size of 1000 mm3; MBL, maximal body weight loss due to drug treatment as compared with the first day of treatment; Evo, evofosfamide; mTORi, mTOR inhibitor; Eve, everolimus; Tem, temsirolimus; CS, concurrent schedule in which evofosfamide was given from Day 1 for 3 weeks; DS, delayed schedule in which evofosfamide was given from Day 8 for 2 weeks;
P<0.05 compared with Vehicle.
P < 0.05 as compared to mTOR inhibitor monotherapy.
P < 0.05 as compared to evofosfamide monotherapy.
Evofosfamide and everolimus was tested in the Caki-1 xenograft model only with CS schedule. Enhanced efficacy was observed in the combination treatment group compared to vehicle or the monotherapy groups (Figure 1D). Evofosfamide in combination with temsirolimus was tested in the Caki-1 xenograft model as well. Evofosfamide monotherapy showed moderate antitumor efficacy with a TGI of 75-81%. The combination treatment significantly increased the antitumor activity, compared with vehicle, temsirolimus monotherapy or evofosfamide monotherapy treatment (P<0.05). The two schedules of CS and DS worked similarly by using TGI as a readout (97 and 95%, respectively, Figure 1E). TGD1000 in DS combination group reached 32 days, and in CS group was 24 days. In addition, all drug treatments were well tolerated, as no significant body weight loss was observed (Supplementary Figure 1).
Evofosfamide reduces increased hypoxia induced by mTOR inhibitors in RCC models
786-O or Caki-1 tumor-bearing animals were treated with vehicle, mTOR inhibitor alone, evofosfamide alone, or evofosfamide in combination with mTOR inhibitor when the tumor size reached 300-500 mm3. Evofosfamide was given as a single dose of 150 mg/kg, ip, everolimus at 5 mg/kg, QDx8, po, in the 786-O xenograft model, and temsirolimus at 20 mg/kg, QDx8, ip in the Caki-1 xenograft model. In the combination group, 4 hrs after the last dose of mTOR inhibitor evofosfamide 150 mg/kg was ip administered. The tissue harvest schedule is presented in Figure 2A. CD31 antibody was employed to label the endothelial cells of the blood vessels. As a defining feature of RCC, both 786-O and Caki-1 xenograft tumors were well-vascularized. 786-O had a higher microvessel density (MVD) in vehicle treated tumors compared to Caki-1 (13.3% vs. 8.5%). As monotherapy, everolimus or temsirolimus alone significantly reduced intratumoral vasculature (Figure 2B-D) compared with vehicle treatment (P<0.05). Evofosfamide did not exhibit antiangiogenic effects in the Caki-1 xenograft tumors, but reduced the MVD in the 786-O model. When tumors were treated with the combination therapy of evofosfamide and mTOR inhibitor, there was no significant change on MVD compared with mTOR inhibitor monotherapy alone.
Figure 2.
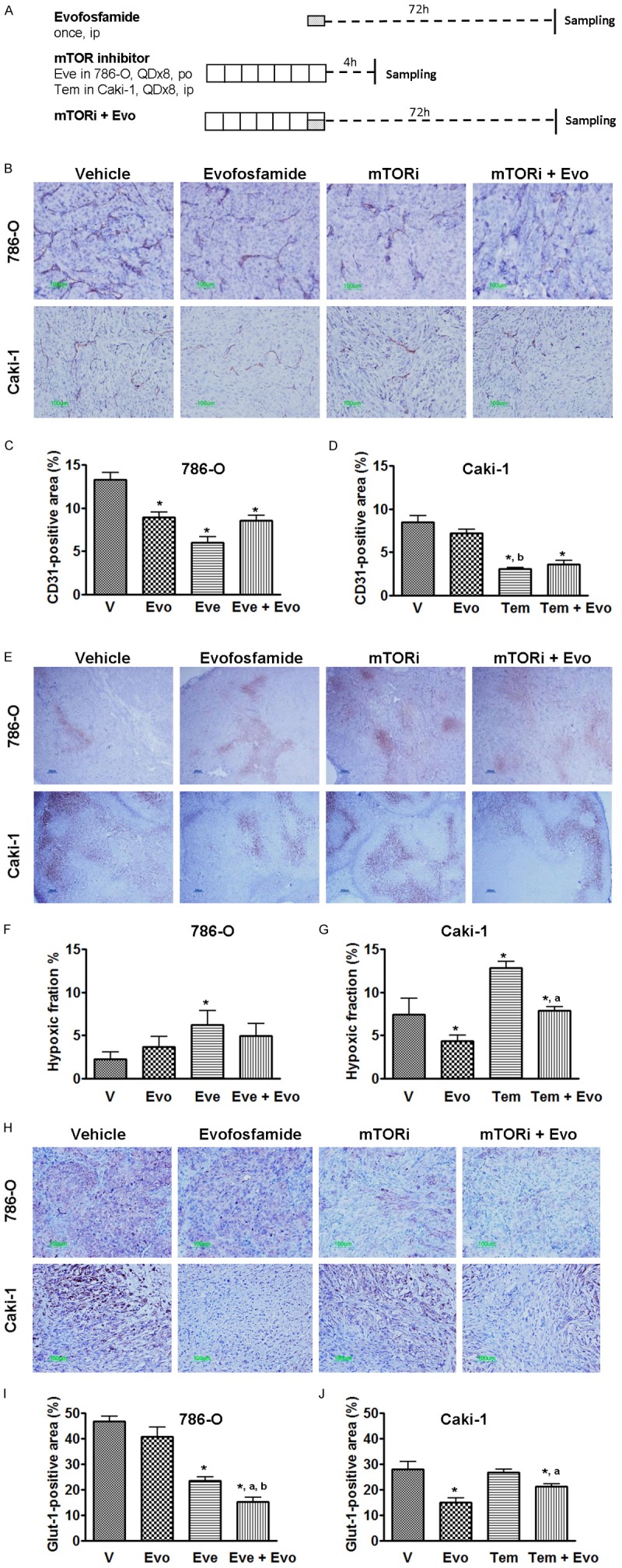
Effect of evofosfamide in combination with mTOR inhibitors on tumor angiogenesis and hypoxia. A. Treatment schedule and experimental design; Tumor-bearing animals received evofosfamide, 150 mg/kg, ip, everolimus 5 mg/kg, po in the 786-O xenografts; and temsirolimus 20 mg/kg, ip in the Caki-1 xenografts. B, E and H. Representative images of CD31, pimonidazole and Glut-1 immunostaining, respectively; C and D. Morphometric analysis of tumor microvessel density; F and G. Morphometric analysis of tumor hypoxic fraction. I and J. Morphometric analysis of percentage of Glut-1 expression inside the tumor. *, P < 0.05 as compared to Vehicle. a, P < 0.05 as compared to mTOR inhibitor monotherapy. Each bar represents Mean ± SEM of 5-8 animals per group. Evo, evofosfamide; mTORi, mTOR inhibitor; Eve, everolimus; and Tem, temsirolimus.
Consistent with the observed decrease of vessel density, tumor hypoxia, detected by pimonidazole immunostaining, was significantly increased after mTOR inhibitor treatment (Figure 2E-G). The hypoxic fraction (HF) in the vehicle-treated 786-O xenograft tumors was only 2.3 ± 0.8%. Evofosfamide alone did not change the hypoxia level with a resulting HF of 3.7 ± 1.3%. After 8 days treatment with everolimus, morphometric analysis of the hypoxic compartments showed that the HF was significantly increased to 6.3 ± 1.6% compared to vehicle treatment. When everolimus and evofosfamide were combined, the HF was decreased to 5.0 ± 1.4%. In the Caki-1 model, a significant increase of tumor hypoxia was observed in the temsirolimus monotherapy group. After temsirolimus treatment, the tumor hypoxic fraction was 12.8 ± 0.7%, as compared to 7.5 ± 1.9% in vehicle treated group (P<0.05). The tumor hypoxic fraction in the evofosfamide monotherapy group was 4.3 ± 0.7%. When evofosfamide was administered in combination with temsirolimus the hypoxic fraction was 7.9 ± 0.5%, significantly decreased from that observed in the temsirolimus monotherapy group (P<0.05).
We used glucose transporter-1 (Glut-1) as an endogenous hypoxia biomarker as well as a biomarker to evaluate effects on cancer metabolism by the different treatments [29-31]. As shown in Figure 2H-J, everolimus-alone significantly reduced the Glut-1 expression in 786-O xenograft tumors, however temsirolimus-alone did not change Glut-1 expression in Caki-1 xenografts. Interestingly, evofosfamide significantly decreased the levels of Glut-1 in the Caki-1 model. In both models, Glut-1 expression was further reduced in the combination therapy compared with mTOR inhibitor-alone.
Combination of evofosfamide and mTOR inhibitor enhances the inhibition of tumor cell proliferation, increases the induction of DNA damage, and further suppresses the level of survivin expression
To assess the effect of the combination of evofosfamide and mTOR inhibitor on tumor cell proliferation, DNA damage, and apoptosis, Ki67, γH2AX, and survivin, respectively, were used as biomarkers in immunohistochemistry. Both evofosfamide and mTOR inhibitor monotherapy significantly decreased the percentage of Ki67-positve proliferating cells compared to the vehicle treatment (P<0.05). Combination treatment further reduced tumor cell proliferation compared with evofosfamide-alone (P<0.05) or mTOR inhibitor-alone (P<0.05, Figure 3A-C). γH2AX foci formation was employed to evaluate the treatment-induced DNA damage. In the vehicle-treated xenograft tumor, few γH2AX positive cells were present and were mainly located near the necrotic regions. Similar to published results [24], evofosfamide monotherapy significantly increased the percentage of γH2AX-positive cells which were evenly distributed throughout the tumor. mTOR inhibitor-alone significantly induced DNA damage (P<0.05 vs. vehicle) as well, but to a less extent compared with evofosfamide monotherapy treatment in both models. When the combination therapy was applied, compared with evofosfamide monotherapy a significant increase of DNA damage cells was observed in the 786-O xenograft tumors but not in Caki-1 xenografts (Figure 3D-F). Survivin functions as a cell cycle-regulated apoptosis inhibitor [32,33]. A significant suppression of survivin level were observed after the monotherapy treatment (P<0.05 vs. vehicle). In both models, combination therapy further reduced the expression of survivin compared with vehicle or either monotherapy group (P<0.05, Figure 3G-I). Taken together, after the combination treatment, the necrotic fraction was enlarged with commonly observed focal hemorrhage, as showed by H & E histology staining (Figure 3J).
Figure 3.
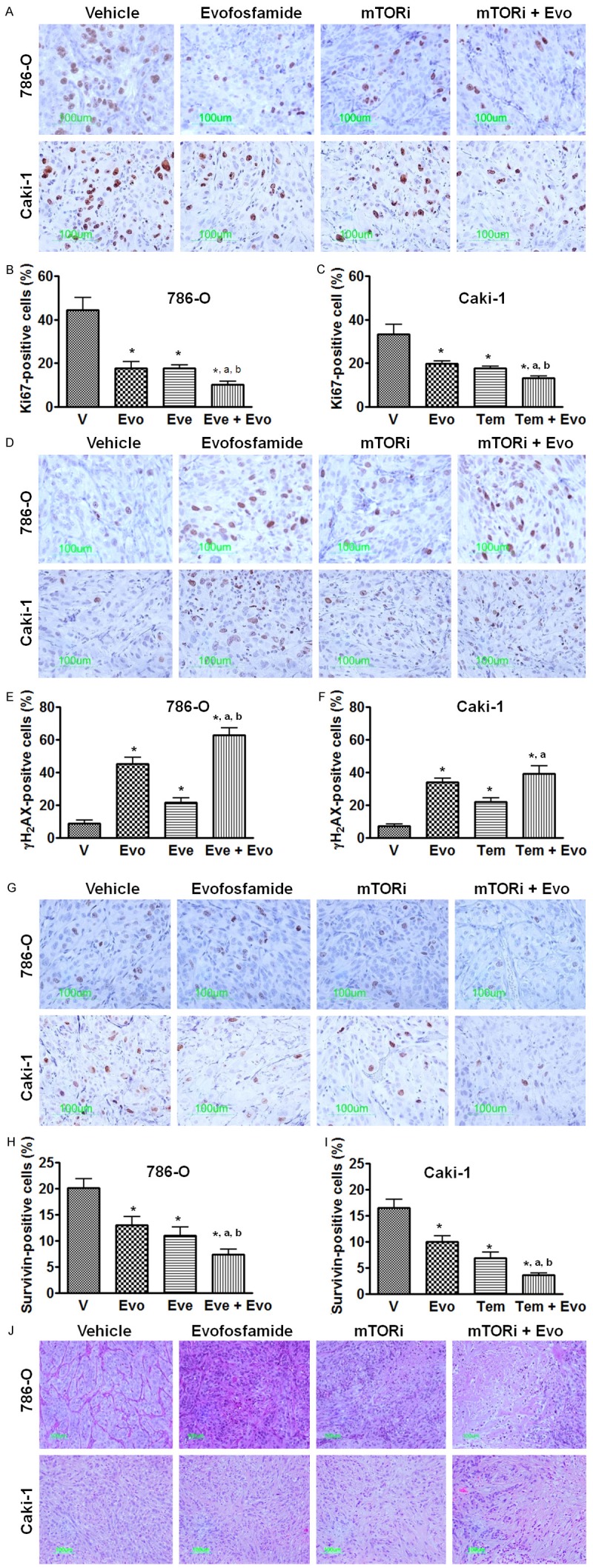
Effect of evofosfamide in combination with mTOR inhibitor on cell proliferation, DNA damage, and survivin expression. Everolimus and temsirolimus were used in the 786-O and Caki-1 xenografts, respectively. A. Representative images of Ki67 immunostaining, a marker of cell proliferation; B and C. Morphometric analysis of percentage of Ki67 positive cells inside the tumor; D. Representative images of γH2AX immunostaining, a marker of DNA damage; E and F. Morphometric analysis of percentage of γH2AX positive tumor cells; G. Representative images of survivin immunostaining; H and I. Morphometric analysis of percentage of survivin positive cells inside the tumor. J. Representative images of H. & E. Histology staining. *, P < 0.05 as compared to Vehicle. a, P < 0.05 as compared to mTOR inhibitor monotherapy; b, P < 0.05 as compared to evofosfamide monotherapy. Each bar represents Mean ± SEM of 5-8 animals per group. Evo, evofosfamide; mTORi, mTOR inhibitor; Eve, everolimus; and Tem, temsirolimus.
Phosphorylated S6 ribosomal protein (pS6) expression was inhibited by mTOR inhibitors
Phosphorylated S6 ribosomal protein (pS6) is considered a reliable phospho-protein biomarker of mTOR activity [34]. As shown in Figure 4, pS6 is widely expressed in vehicle treated 786-O and Caki-1 xenograft tumors. Four hours after mTOR inhibitor’s treatment, the expression of pS6 was significantly reduced (P<0.05 vs. vehicle treatment). 72 hrs after evofosfamide treatment, pS6 levels were not changed compared to the vehicle control in both models. Similar results were obtained on phospho-mTOR expression, another biomarker of mTOR pathway [34] (Supplementary Figure S2).
Figure 4.
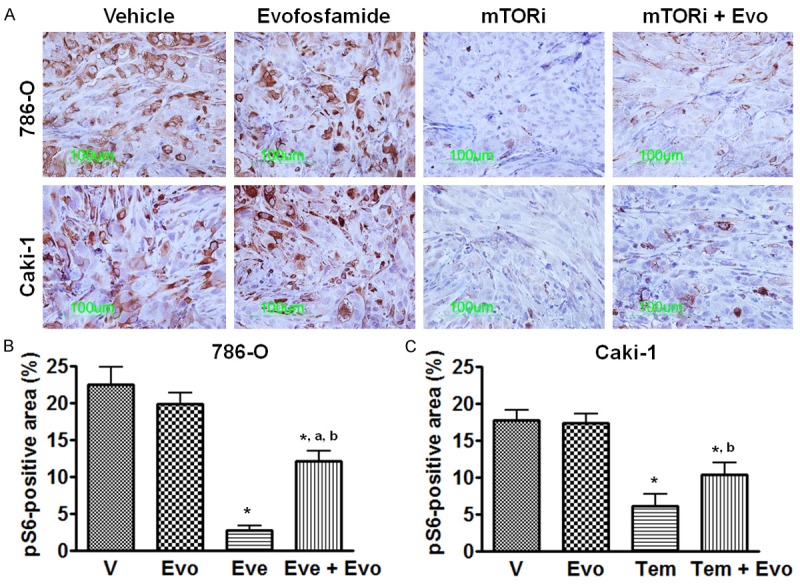
Effect of mTOR inhibitor on phosphorylated S6 ribosomal protein (pS6) expression. Everolimus and temsirolimus were used in the 786-O and Caki-1 xenografts, respectively. A. Representative images of pS6 immunostaining; B and C. Morphometric analysis of percentage of pS6 positive area inside the tumor. *, P < 0.05 as compared to Vehicle. a, P < 0.05 as compared to mTOR inhibitor monotherapy; b, P < 0.05 as compared to evofosfamide monotherapy. Each bar represents Mean ± SEM of 5-8 animals per group. Evo, evofosfamide; mTORi, mTOR inhibitor; Eve, everolimus; and Tem, temsirolimus.
Evofosfamide exhibits little additivity with mTOR inhibitors in in vitro cytotoxicity assays
Cells were incubated with either evofosfamide alone or combined with either everolimus or temsirolimus at the indicated concentration. IC10-20 of either everolimus or temsirolimus for each cell type was chosen for the combination study. As shown in Figure 5 and Table 2, evofosfamide alone exhibited hypoxia selective- and concentration-dependent cytotoxicity. Addition of either everolimus or temsirolimus did not significantly change evofosfamide-mediated cytotoxicity in both cell types with either mTOR inhibitor.
Figure 5.
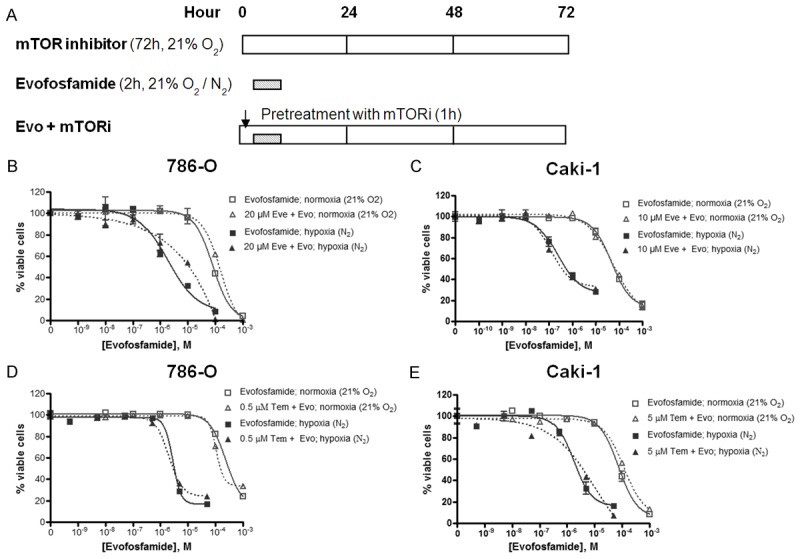
In vitro cytotoxicity of evofosfamide in combination with everolimus or temsirolimus in 786-O and Caki-1 RCC cells. A. Treatment schedule and experiment design. B and C. Evofosfamide alone, or combined with everolimus in 786-O and Caki-1 cells under either air (21% O2) or hypoxia (N2), respectively; D and E. Evofosfamide alone, or combined with temsirolimus in 786-O and Caki-1 cells under either air (21% O2) or hypoxia (N2). The viable cells were detected using AlamarBlue and quantified with microspectrofluorometry. The data are the representative of two independent experiments. Evo, evofosfamide; Eve, everolimus; and Tem, temsirolimus.
Table 2.
Summary of IC50 of evofosfamide alone, everolimus or temsirolimus alone, or in combination in 786-O and Caki-1 cells
| 786-O | Caki-1 | |||
|---|---|---|---|---|
|
|
||||
| Normoxia (21% O2) | Hypoxia (N2) | Normoxia (21% O2) | Hypoxia (N2) | |
|
|
||||
| IC50 (µM) | IC50 (µM) | IC50 (µM) | IC50 (µM) | |
| Evofosfamide | 84 | 2.5 | 63 | 0.5 |
| Everolimus | 81 | 48 | ||
| Temsirolimus | 18 | 16 | ||
| Evo + Eve | 140 | 11 | 71 | 0.4 |
| Evo + Tem | 150 | 2.8 | 120 | 3.9 |
Evo, evofosfamide; Eve, everolimus; Tem, temsirolimus.
Discussion
In this study we demonstrated the antitumor effect of combining evofosfamide, a hypoxia-activated prodrug, and mTOR inhibitors in preclinical models of RCC. mTOR inhibitors have antiangiogenic effects, and as observed in many preclinical studies the use of antiangiogenic agents usually leads to an increase of intratumoral hypoxia [11,12,35]. Evofosfamide exhibits hypoxia-selective cytotoxic activity in both in vivo and in vitro settings. This provides a basis for mechanism-based combination therapy of evofosfamide combined with mTOR inhibitor in the preclinical studies to support the potential clinical evaluation of the combination.
We employed two RCC cell line models, 786-O (VHL-/- PTEN-/-) and Caki-1 (VHL+/+ PTEN+/+), in the study. The major consequence of loss-of-function VHL is continuous activation of HIF-1α, resulting in increased angiogenesis and cell growth, and adaption to low-oxygen, low-pH, and low-nutrient environments [36,37]. Similarly, loss of PTEN increases cell survival in adverse tumor microenvironments and stimulates angiogenic gene expression as well as other essential genes required to survive under low oxygen conditions [38]. As expected, the VHL-/-, PTEN-/- 786-O cell line yielded higher level of angiogenesis and lower tumor hypoxia levels compared to the VHL+/+ and PTEN+/+ Caki-1 as characterized by CD31 and pimonidazole immunostaining, respectively. In addition, PTEN-deficient cells are sensitive to growth inhibition by pharmacologic mTOR blockade [39]. In this study, at the same dose and regimen, temsirolimus yielded superior tumor growth inhibition in 786-O xenografts compared to Caki-1 xenografts. On the other hand, as a monotherapy, evofosfamide exhibited better antitumor activity in Caki-1 xenografts due to their higher hypoxic fraction compared to 786-O xenografts. However, regardless of which model was profiled, the combination of evofosfamide and an mTOR inhibitor exhibited an enhanced antitumor activity compared to the monotherapy regimens. It might suggest this combination approach may be effective independent of VHL or PTEN status.
Our data suggests that inhibition of angiogenesis by mTOR inhibitors play a key role in the enhanced activity observed by combination treatment with evofosfamide in xenograft RCC models. Two different treatment schedules were used in the present study, the Concurrent Schedule in which evofosfamide and mTOR inhibitor treatment started on the same day; and the Delayed Schedule in which evofosfamide treatment started seven days after mTOR inhibitor treatment started. In all three studies, including two models, 786-O and Caki-1, and two mTOR inhibitors, everolimus and temsirolimus, Concurrent Schedule and Delayed Schedule yielded very similar combination efficacy profiles. It appears that the specific schedule had little effect on the resulting antitumor activity. However, in Delayed Schedule the treatment period of evofosfamide was only 2 weeks compared with 3 weeks in Concurrent Schedule, and the total exposure of evofosfamide in Delayed Schedule was only 2/3 of that in Concurrent Schedule (500 mg/kg vs. 750 mg/kg). Body weight change, as a safety readout, indicated that the treated animals well-tolerated the combination regimens. Concurrent Schedule resulted in slightly more body weight loss compared to Delayed Schedule, but no statistical difference was observed. It is important to note that the significant difference between Concurrent Schedule and Delayed Schedule is that at the time when evofosfamide treatment was initiated. The latter regimen is accompanied with a higher level of intratumoral hypoxia induced by an mTOR inhibitor.
An antiangiogenic effect of mTOR inhibitors leads to increased hypoxia was observed in the present study by pimonidazole immunohistochemistry. On one hand, increased hypoxia is associated with the treatment failure, a more aggressive, invasive, metastatic phenotype and is one of the mechanisms of resistance to mTOR inhibitors [40]; on the other hand, increased hypoxia could provide an increased activation of the hypoxia-activated prodrug evofosfamide in the combination setting. Interestingly, evofosfamide exhibited a different activity pattern in the Caki-1 xenograft model compared to the 786-O xenograft model. In the Caki-1 xenograft, evofosfamide significantly reduced the hypoxic fraction, as observed previously in other xenograft models profiled [24]. But in the 786-O xenograft model, evofosfamide did not change the volume of the hypoxic fraction, and even showed a trend leading to an increase in hypoxic fraction. This latter observation may be related to the significantly decreased microvessel density observed after evofosfamide treatment in the 786-O xenograft model as characterized by CD31 staining. Evofosfamide may have interfered with hypoxia-induced HIF-1α-dependent pathways such as VEGF-related angiogenesis leading to subsequent decreased blood vessel density [41]. Alternatively, the effect of evofosfamide on tumor vasculature may exhibit xenograft model specific-dependency. It has been previously shown that tirapazamine, also a hypoxia-activated prodrug, causes extensive central vascular dysfunction in the HCT-116 xenograft tumors within 24 hours of dosing [42]. The mechanisms underlying these distinct responses between the models remain unclear. However, in both models we studied, evofosfamide consistently reduced the tumor hypoxia induced by mTOR inhibitor, as seen in the combination group. The hypoxia selective targeting of evofosfamide is further supported by the schedule-dependent enhanced antitumor activity with the combination treatment.
Both mTOR inhibitors and evofosfamide have been reported to induce cell cycle arrest [23,43-45]. This anti-proliferative effect of mTOR inhibitors may be attributed to their ability to down-regulate synthesis of essential cell cycle proteins, including Cyclin D1, HIF-1α, and c-Myc [43,44]. Evofosfamide causes cell-cycle arrest in a concentration-dependent and hypoxia-selective manner [23,45]. Consistent with these reports, both types of agents reduce tumor cell proliferation as monotherapies in the RCC xenograft models. When evofosfamide was added to mTOR inhibitors, the number of proliferating cells, detected by Ki67 staining, was significantly decreased, suggesting an additive cytostatic effect. On the other hand, γH2AX staining, a marker of DNA damage, was significantly increased by the combination treatment in both models, consistent with the additive cytotoxic effect observed with the combination regimens. In addition, the mTOR inhibitors may sensitize tumor cells to the DNA alkylating agent treatment as reported previously for other alkylator combinations [6,20,21]. Survivin is a member of the inhibitor of apoptosis protein (IAP) family that is selectively overexpressed in most common types of human cancers, and has been implicated in the control of cell division, inhibition of apoptosis, and tumor cell resistance to chemotherapy [32,33]. The present results showed that co-treatment of evofosfamide and mTOR inhibitors resulted in a significant reduction in survivin levels compared to the effect of either single agent. Similar to levels of tumor hypoxia, survivin is also an unfavorable prognostic marker correlating with decreased overall survival in many malignancies, including RCC [46]. We hypothesize that patients treated with the combination of evofosfamide and an mTOR inhibitor may yield a more favorable outcome than treatment with either agent alone. The enhanced DNA damage and apoptosis after the combination regimen observed in the study is consistent with a mechanistic basis for the complementarity of the two agents.
Another mechanism for mTOR inhibitors inhibition on tumor growth is by reducing bioenergetic efficiency [5]. Cancer cells primarily rely on glycolysis to provide ATP for sustained growth. mTOR increases expression of amino acid and glucose transporters. Here we showed that everolimus significantly decreased the level of the Glut-1 glucose transporter in the 786-O xenograft model, however temsirolimus did not change Glut-1 levels in the Caki-1 xenograft model. Glut-1 is often considered as an endogenous hypoxia biomarker; however, the decrease of Glut-1 expression by everolimus treatment is not consistent with the increased pimonidazole positivity. It might suggest that the decreased Glut-1 expression is not due to changes in oxygen levels or tumor hypoxia but points to a metabolism inhibitor effect linked to mTOR inhibition. Similar finding was reported by Preze et al. in a syngeneic rat chondrosarcoma model [47]. Conversely, evofosfamide significantly reduced Glut-1 expression in the Caki-1 model. This latter result might be related to the selective hypoxic cell cytotoxicity mediated by evofosfamide, given that Glut-1 is upregulated by hypoxia-induced HIF-1 transcription factor [48]. Correspondingly, significant reduction in Glut-1 expression was observed after the combination treatments in both models.
Cell-based in vitro studies employing the same two cell lines 786-O and Caki-1 did not show any enhanced cytostatic or cytotoxic activity when evofosfamide was combined with everolimus or temsirolimus. The lack of concordance between in vivo and in vitro models was also observed in other studies when everolimus was combined with a series of cytotoxic antitumor agents by O’Reily and colleagues [20]. The combination mechanisms may be multi-factorial and complex in this setting, which may include cell intrinsic effects, such as increased DNA damage, increased apoptosis, or effects that require the tumor microenvironment, for instance, inhibition of tumor hypoxia. But the lack of in vitro combination activity may suggest the complementary pharmacological effects observed in the xenograft model setting are at the tumor microenvironmental level rather than cell autonomous effects.
Ribosomal protein S6 is a major downstream target and effector of the mTOR pathway and is considered the most reliable phospho protein marker for mTOR activities [34]. Following activation by the ribosomal protein S6 kinase, phosphorylated pS6 (pS6) participates in the regulation of cell proliferation, cell growth, and protein synthesis [49]. In the present study, mTOR inhibitors caused a striking inhibition of phosphorylated S6 ribosomal protein expression, while evofosfamide treatment did not change pS6 expression. This result might suggest a minimal effect of evofosfamide on mTOR signaling.
In summary, our findings demonstrate that evofosfamide in combination with mTOR inhibitor exhibits antitumor activity in preclinical RCC models. The additive effect is consistent with the mTOR inhibition increasing the tumor hypoxic fraction, and evofosfamide’s selective targeting of the hypoxic compartment. The combination efficacy profile may also be due to the combined anti-proliferative, cytostatic, and cytotoxic activity of the agents. These observations provide a translational rationale for the design of clinical trials to evaluate the efficacy and safety profile of evofosfamide in combination with mTOR inhibitors in RCC.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Campbell SC, Lane BR. Malignant renal tumors. In: Wein A, Kavoussi L, Novick A, Partin A, Peters C, editors. Campbell-Walsh Urology. 10th edition. Philadelphia: Elsevier Saunders; 2012. pp. 1413–74. [Google Scholar]
- 3.Baldewijns MM, van Vlodrop IJ, Vermeulen PB, Soetekouw PM, van Engeland M, de Bruïne AP. VHL and HIF signaling in renal cell carcinogenesis. J Pathol. 2010;221:125–38. doi: 10.1002/path.2689. [DOI] [PubMed] [Google Scholar]
- 4.Lainakis G, Bamias A. Targeting angiogenesis in renal cell carcinoma. Curr Cancer Drug Targets. 2008;8:349–58. doi: 10.2174/156800908785133132. [DOI] [PubMed] [Google Scholar]
- 5.Su D, Stamatakis L, Singer EA, Srinivasan R. Renal cell carcinoma: molecular biology and targeted therapy. Curr Opin Oncol. 2014;26:321–7. doi: 10.1097/CCO.0000000000000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010;40:310–22. doi: 10.1016/j.molcel.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beuvink I, Boulay A, Fumagalli S, Zilbermann F, Ruetz S, O’Reilly T, Natt F, Hall J, Lane HA, Thomas G. The mTOR inhibitor RAD001 sensitizes tumor cells to DNA-damaged induced apoptosis through inhibition of p21 translation. Cell. 2005;120:747–759. doi: 10.1016/j.cell.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 8.Karar J, Maity A. PI3K/AKT/mTOR pathway in angiogenesis. Front Mol Neurosci. 2011;4:51. doi: 10.3389/fnmol.2011.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Del Bufalo D, Ciuffreda L, Trisciuoglio D, Desideri M, Cognetti F, Zupi G, Milella M. Antiangiogenic potential of the Mammalian target of rapamycin inhibitor temsirolimus. Cancer Res. 2006;66:5549–54. doi: 10.1158/0008-5472.CAN-05-2825. [DOI] [PubMed] [Google Scholar]
- 10.Manegold PC, Paringer C, Kulka U, Krimmel K, Eichhorn ME, Wilkowski R, Jauch KW, Guba M, Bruns CJ. Antiangiogenic therapy with mammalian target of rapamycin inhibitor RAD001 (Everolimus) increases radiosensitivity in solid cancer. Clin Cancer Res. 2008;14:892–900. doi: 10.1158/1078-0432.CCR-07-0955. [DOI] [PubMed] [Google Scholar]
- 11.Chang YS, Adnane J, Trail PA, Levy J, Henderson A, Xue D, Bortolon E, Ichetovkin M, Chen C, McNabola A, Wilkie D, Carter CA, Taylor IC, Lynch M, Wilhelm S. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother Pharmacol. 2007;59:561–74. doi: 10.1007/s00280-006-0393-4. [DOI] [PubMed] [Google Scholar]
- 12.Rice C, Huang LE. From antiangiogenesis to hypoxia: current research and future directions. Cancer Manag Res. 2010;3:9–16. doi: 10.2147/CMR.S14812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–31. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–9. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahalingam D, Medina EC, Esquivel JA 2nd, Espitia CM, Smith S, Oberheu K, Swords R, Kelly KR, Mita MM, Mita AC, Carew JS, Giles FJ, Nawrocki ST. Vorinostat enhances the activity of temsirolimus in renal cell carcinoma through suppression of survivin levels. Clin Cancer Res. 2010;16:141–53. doi: 10.1158/1078-0432.CCR-09-1385. [DOI] [PubMed] [Google Scholar]
- 16.Damiano V, Rosa R, Formisano L, Nappi L, Gelardi T, Marciano R, Cozzolino I, Troncone G, Agrawal S, Veneziani BM, De Placido S, Bianco R, Tortora G. Toll-like receptor 9 agonist IMO cooperates with everolimus in renal cell carcinoma by interfering with tumour growth and angiogenesis. Br J Cancer. 2013;108:1616–23. doi: 10.1038/bjc.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, Kovacevic Z, Lesovoy V, Schmidt-Wolf IG, Barbarash O, Gokmen E, O’Toole T, Lustgarten S, Moore L, Motzer RJ Global ARCC Trial. Words of wisdom. Re: Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–81. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 18.Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, Lebwohl D, Ravaud A RECORD-1 Study Group. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–56. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 19.Houghton PJ. Everolimus. Clin Cancer Res. 2010;16:1368–72. doi: 10.1158/1078-0432.CCR-09-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Reilly T, McSheehy PM, Wartmann M, Lassota P, Brandt R, Lane HA. Evaluation of the mTOR inhibitor, everolimus, in combination with cytotoxic antitumor agents using human tumor models in vitro and in vivo. Anticancer Drugs. 2011;22:58–78. doi: 10.1097/CAD.0b013e3283400a20. [DOI] [PubMed] [Google Scholar]
- 21.Cejka D, Preusser M, Fuereder T, Sieghart W, Werzowa J, Strommer S, Wacheck V. mTOR inhibition sensitizes gastric cancer to alkylating chemotherapy in vivo. Anticancer Res. 2008;28:3801–8. [PubMed] [Google Scholar]
- 22.Duan JX, Jiao H, Kaizerman J, Stanton T, Evans JW, Lan L, Lorente G, Banica M, Jung D, Wang J, Ma H, Li X, Yang Z, Hoffman RM, Ammons WS, Hart CP, Matteucci M. Potent and highly selective hypoxia-activated achiral phosphoramidate mustards as anticancer drugs. J Med Chem. 2008;51:2412–20. doi: 10.1021/jm701028q. [DOI] [PubMed] [Google Scholar]
- 23.Meng F, Evans JW, Bhupathi D, Banica M, Lan L, Lorente G, Duan JX, Cai X, Mowday AM, Guise CP, Maroz A, Anderson RF, Patterson AV, Stachelek GC, Glazer PM, Matteucci MD, Hart CP. Molecular and cellular pharmacology of the hypoxia-activated prodrug TH-302. Mol Cancer Ther. 2012;11:740–51. doi: 10.1158/1535-7163.MCT-11-0634. [DOI] [PubMed] [Google Scholar]
- 24.Sun JD, Liu Q, Wang J, Ahluwalia D, Ferraro D, Wang Y, Duan JX, Ammons WS, Curd JG, Matteucci MD, Hart CP. Selective tumor hypoxia targeting by hypoxia-activated prodrug TH-302 inhibits tumor growth in preclinical models of cancer. Clin Cancer Res. 2012;18:758–70. doi: 10.1158/1078-0432.CCR-11-1980. [DOI] [PubMed] [Google Scholar]
- 25.Liu Q, Sun JD, Wang J, Ahluwalia D, Baker AF, Cranmer LD, Ferraro D, Wang Y, Duan JX, Ammons WS, Curd JG, Matteucci MD, Hart CP. TH-302, a hypoxia-activated prodrug with broad in vivo preclinical combination therapy efficacy: optimization of dosing regimens and schedules. Cancer Chemother Pharmacol. 2012;69:1487–98. doi: 10.1007/s00280-012-1852-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chawla SP, Cranmer LD, Van Tine BA, Reed DR, Okuno SH, Butrynski JE, Adkins DR, Hendifar AE, Kroll S, Ganjoo KN. Phase II study of the safety and antitumor activity of the hypoxiaactivated prodrug TH-302 in combination with doxorubicin in patients with advanced soft tissue sarcoma. J. Clin. Oncol. 2014;32:3299–306. doi: 10.1200/JCO.2013.54.3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borad MJ, Reddy SG, Bahary N, Uronis HE, Sigal D, Cohn AL, Schelman WR, Stephenson J Jr, Chiorean EG, Rosen PJ, Ulrich B, Dragovich T, Del Prete SA, Rarick M, Eng C, Kroll S, Ryan DP. Randomized Phase II trial of gemcitabine plus TH-302 versus gemcitabine in patients with advanced pancreatic cancer. J. Clin. Oncol. 2015;33:1475–81. doi: 10.1200/JCO.2014.55.7504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hureaux J, Lagarce F, Gagnadoux F, Rousselet MC, Moal V, Urban T, Benoit JP. Toxicological study and efficacy of blank and paclitaxel-loaded lipid nanocapsules after i. v. administration in mice. Pharm Res. 2010;27:421–30. doi: 10.1007/s11095-009-0024-y. [DOI] [PubMed] [Google Scholar]
- 29.Wang BY, Kalir T, Sabo E, Sherman DE, Cohen C, Burstein DE. Immunohistochemical staining of GLUT1 in benign, hyperplastic, and malignant endometrial epithelia. Cancer. 2000;88:2774–81. [PubMed] [Google Scholar]
- 30.Amann T, Maegdefrau U, Hartmann A, Agaimy A, Marienhagen J, Weiss TS, Stoeltzing O, Warnecke C, Schölmerich J, Oefner PJ, Kreutz M, Bosserhoff AK, Hellerbrand C. GLUT1 expression is increased in hepatocellular carcinoma and promotes tumorigenesis. Am J Pathol. 2009;174:1544–52. doi: 10.2353/ajpath.2009.080596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagase Y, Takata K, Moriyama N, Aso Y, Murakami T, Hirano H. Immunohistochemical localization of glucose transporters in human renal cell carcinoma. J Urol. 1995;153:798–801. [PubMed] [Google Scholar]
- 32.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–21. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 33.Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:980–4. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 34.Márk Á, Hajdu M, Váradi Z, Sticz TB, Nagy N, Csomor J, Berczi L, Varga V, Csóka M, Kopper L, Sebestyén A. Characteristic mTOR activity in Hodgkin-lymphomas offers a potential therapeutic target in high risk disease--a combined tissue microarray, in vitro and in vivo study. BMC Cancer. 2013;13:250. doi: 10.1186/1471-2407-13-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaustad JV, Simonsen TG, Leinaas MN, Rofstad EK. Sunitinib treatment does not improve blood supply but induces hypoxia in human melanoma xenografts. BMC Cancer. 2012;12:388. doi: 10.1186/1471-2407-12-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991;51:3075–9. [PubMed] [Google Scholar]
- 37.Hahn WC, Weinberg RA. Rules for making human tumor cells. N Engl J Med. 2002;347:1593–603. doi: 10.1056/NEJMra021902. [DOI] [PubMed] [Google Scholar]
- 38.Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, Gottschalk AR, Ryan HE, Johnson RS, Jefferson AB, Stokoe D, Giaccia AJ. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000;14:391–6. [PMC free article] [PubMed] [Google Scholar]
- 39.Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, Frost P, Gibbons JJ, Wu H, Sawyers CL. Enhanced sensitivity of PTENdeficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci U S A. 2001;98:10314–9. doi: 10.1073/pnas.171076798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knaup KX, Jozefowski K, Schmidt R, Bernhardt WM, Weidemann A, Juergensen JS, Warnecke C, Eckardt KU, Wiesener MS. Mutual regulation of hypoxia-inducible factor and mammalian target of rapamycin as a function of oxygen availability. Mol Cancer Res. 2009;7:88–98. doi: 10.1158/1541-7786.MCR-08-0288. [DOI] [PubMed] [Google Scholar]
- 41.von Marschall Z, Cramer T, Hocker M, Finkenzeller G, Wiedenmann B, Rosewicz S. Dual mechanism of vascular endothelial growth factor upregulation by hypoxia in human hepatocellular carcinoma. Gut. 2001;48:87–96. doi: 10.1136/gut.48.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huxham LA, Kyle AH, Baker JH, McNicol KL, Minchinton AI. Tirapazamine causes vascular dysfunction in HCT-116 tumour xenografts. Radiother Oncol. 2006;78:138–45. doi: 10.1016/j.radonc.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 43.Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–62. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 44.Kurmasheva RT, Huang S, Houghton PJ. Predicted mechanisms of resistance to mTOR inhibitors. Br J Cancer. 2006;95:955–60. doi: 10.1038/sj.bjc.6603353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu J, Handisides DR, Van Valckenborgh E, De Raeve H, Menu E, Vande Broek I, Liu Q, Sun JD, Van Camp B, Hart CP, Vanderkerken K. Targeting the multiple myeloma hypoxic niche with TH-302, a hypoxia-activated prodrug. Blood. 2010;116:1524–7. doi: 10.1182/blood-2010-02-269126. [DOI] [PubMed] [Google Scholar]
- 46.Krambeck AE, Dong H, Thompson RH, Kuntz SM, Lohse CM, Leibovich BC, Blute ML, Sebo TJ, Cheville JC, Parker AS, Kwon ED. Survivin and b7-h1 are collaborative predictors of survival and represent potential therapeutic targets for patients with renal cell carcinoma. Clin Cancer Res. 2007;13:1749–56. doi: 10.1158/1078-0432.CCR-06-2129. [DOI] [PubMed] [Google Scholar]
- 47.Perez J, Decouvelaere AV, Pointecouteau T, Pissaloux D, Michot JP, Besse A, Blay JY, Dutour A. Inhibition of chondrosarcoma growth by mTOR inhibitor in an in vivo syngeneic rat model. PLoS One. 2012;7:e32458. doi: 10.1371/journal.pone.0032458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Le QT, Courter D. Clinical biomarkers for hypoxia targeting. Cancer Metastasis Rev. 2008;27:351–62. doi: 10.1007/s10555-008-9144-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Reilly T, McSheehy PM. Biomarker development for the clinical activity of the mTOR inhibitor everolimus (RAD001): processes, limitations, and further proposals. Transl Oncol. 2010;3:65–79. doi: 10.1593/tlo.09277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.