

Abstract
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKI) have been used as a powerful targeting therapeutic agent for treatment of lung adenocarcinoma for years. Nevertheless, the efficacy of TKI was hampered by the appearance of acquired TKI-resistance. In the present study, we aimed to search, predict, and screen the agents that can overcome the acquired TKI-resistance of lung adenocarcinoma by using the expression profiles of differentially expressed genes (DEGs) and Connectivity map (CMAP). The profiles of DEGs were obtained by searching GEO microarray database, and then, they were submitted to CMAP for analysis in order to predict and screen the agent that might reverse the TKI-resistance of lung cancer cells. Next, the effects of the selected agent on TKI-resistant cancer cells were tested and the possible signaling pathways were also evaluated. As a result, valproic acid (VPA) was selected. Then, we used a low-concentration of VPA that has little effect on the cell growth for analysis. Interestingly, the results showed that treatment with a combination of VPA and Erlotinib significantly led to a decrease in cell viability and an increase in cell apoptosis for TKI-resistant HCC827-ER cells, relative to those treated with VPA or Erlotinib alone. Further experiments confirmed that inhibition of MAPK and AKT might be involved in this process. Analyzing the DEGs through the CMAP is a good strategy for exploitation of anti-tumor agents. VPA might markedly increase the sensitivity of TKI-resistant lung adenocarcinoma cells to Erlotinib, thus reversing the acquired TKI-resistance of cancer cells and raising VPA as a potential agent for TKI-resistant lung cancer therapy.
Keywords: Lung adenocarcinoma, TKI-resistance, valproic acid, signaling pathways, reversion
Introduction
Lung cancer, a malignant lung tumor characterized by uncontrolled cell growth in tissues of the lung, is one of the leading causes of cancer-related mortality worldwide, with most cases refractory to surgical resection because 40% of which is in an advanced stage at the time of diagnosis [1]. Thus, the five-year survival rate for this disease is often low.
Non-small cell lung cancer (NSCLC) accounts for about 80% of all lung cancer cases, of which lung adenocarcinoma has replaced squamous cell carcinoma as the most common subtype for years. Although the treatment for early-stage lung adenocarcinomas is primarily surgical, chemotherapy has been applied as the main treatment approach for lung cancers in their late-stages [2]. Conventional chemotherapy including application of cisplatin in combination with other anti-tumor agents has been the first treatment plan for advanced lung cancer. However, the efficacy of both treatments has been weakened by the occurrence of chemoresistance in most cases. Cancer biotherapy has attracted much attention and been recently proven to be a promising treatment strategy.
Epidermal growth factor receptor (EGFR) has been regarded as a target for cancer therapy for several years because EGF has been indicated to stimulate the proliferation and metastasis of cancer cells through complex mechanisms [3]. The discovery of mutations of EGFR in 15-20% of lung adenocarcinomas has provided a successful way for treatment of high-stage adenocarcinomas through EGFR-targeting therapeutic methods [2]. EGFR-targeted treatment can induce DNA damage and abrogate G2/M phase of the cell cycle, leading to lung cancer cell apoptosis and tumor growth suppression in vitro and in vivo [4]. EGFR tyrosine kinase inhibitors (TKI), Gefitinib and Erlotinib, have clinically been used for treatment of a variety of malignant disorders, especially EGFR-mutated lung carcinoma [5]. They have been proven to be effective for lung adenocarcinoma patients harboring mutated EGFR but not wild-type EGFR [6]. Moreover, a recent meta-analysis has revealed that the TKIs could significantly prolong the overall survival time of the lung cancer patients relative to those of the controls [7].
Notably, the success of EGFR-targeted therapy is hampered by the threat of drug resistance in spite of the effectiveness of EGFR TKIs for lung cancer patients. Clinically, a proportion of patients exhibit de novo resistance to EGFR TKIs and are refractory to targeted therapy that is supposed to be effective based on the biology and characteristic of the cancer, and some patients who initially respond to therapy obviously developed acquired resistance to the drug treatment [8]. The mechanisms underlying the acquired TKI-resistance remain unclear. Reports showed that loss of PTEN, a cancer suppressor, resulted from activation of PI3K/AKT signaling pathways, increases cancer cell TKI-resistance [9]. Also, activation of MAPK pathways might play an important role in the TKI-resistance of EGFR-mutant cancer cells [10]. Moreover, over-expression of a receptor tyrosine kinase, AXL, might contribute to TKI-resistance of EGFR-mutant lung cancer cells and thus might be regarded as a potential target for reversion of TKI-resistance [11]. Therefore, to find a new way abrogating or relieving the TKI-resistance is required for lung adenocarcinoma therapy.
Cancer as a multi-gene disease requires powerful tools such as large-scale gene expression analysis provided by technological advances rather than traditional single gene studies for an understanding. The utilization of such analysis may be useful in identifying new biomarkers that will help reverse the drug-resistance of cancer cells. Gene expression profiling is used as a powerful tool for demonstrating disease-specific molecular mechanisms such as biological pathways, and predicting drug response or resistance [12-14]. Connectivity Map (CMAP) is a collection of genomewide searchable database from thousands of gene-expression signatures of various cultured cancer cells exposed to a large collection of small molecule compounds [15] and thus has previously been used for discovery of the unexplored connections among small molecules, diseases and signaling pathways [16,17]. Evidence shows that CMAP was used to identify LY294002, a PI3K inhibitor, a modulator of glucocorticoid resistance in infant acute lymphoblastic leukemia and implementation of LY294002 might improve glucocorticoid response and the prognosis of the disease [18]. For lung cancer, a phenothiazine-like antipsychotic drug, trifluoperazine [19], and a third generation tyrosine kinase inhibitor, bosutinib [20], were identified by CMAP to overcome and reverse EGFR-TKI resistance.
In the present study, we aimed to discover agents that might overcome the acquired TKI-resistance of EGFR-mutated lung adecarcinoma cells. We screened and ranked the genes differentially expressed in TKI-sensitive versus TKI-resistant lung cancer cells. The ranked gene list (denoted as signature) was then submitted to the CMAP database for analysis and identification of molecules or drugs. Among the candidate compounds found, Valproic acid (2-n-propylpentanoic acid, VPA) was selected as a potential therapeutic agent for TKI-resistant lung cancer cells. In the subsequent validation experiments, the mechanisms of VPA reversing TKI-resistance were also evaluated.
Materials and methods
Gene expression and Identification of differentially expressed genes (DEGs)
Data were obtained from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/; accession number: GSE38310), comprising information about gene expression profile of TKI-sensitive lung adenocarcinoma cell HCC827 and its TKI-resistant clones that originated from HCC827 exposed to Erlotinib for a period of time from cell culture.
The original data classified as HCC827 and its Erlotinib-resistant clone cell lines (HCC827-ER) were analyzed using Welch’s t-test by dChip Software (Version: 2010.01) [21]. Then, 1.5-fold change with a p value less than 0.05 was used as the cut-off criterion for selecting genes that were differentially expressed in the HCC827-ER group relative to the HCC827 group. DEGs were screened and selected for further analysis.
Identification of small molecules
The CMAP database (http://www.broad.mit.edu/cmap/) contains whole genomic expression profiles for small active molecular inferences, including 6,100 classes of small molecular interference experiments and 7,056 expression profiles. The DEGs, divided into up- and down-regulated groups and converted into a probe set on the HG-U133A platform, were submitted to CMAP for GSEA (Gene Set Enrichment Analysis). Finally, a correlation score for each perturbagen was calculated, ranging from -1 to +1. According to the method in the literature [22], compounds with negative connectivity scores, which imply a mode of action by the matched compounds to reverse the expression direction of query genes in Erlotinib-resistant cells, were recorded as potential therapeutic agents for Erlotinib resistance.
Compounds and cell culture
The human lung cancer cell line, HCC827 cell line, was obtained from the American Type Culture Collection. The cells were cultured in DMEM medium that contained 10% fetal bovine serum in a humidified atmosphere with 5% CO2 at 37°C.
The HCC827-ER cell line was established by culturing HCC827 cells in 10% FBS culture media containing Erlotinib. Cells were initially maintained at an Erlotinib concentration of 0.02 µM and the dose was gradually increased over a period of 18 weeks until the final concentration of Erlotinib was 15 µM. Then, single-cell cloning techniques were used by which only actively dividing cells were chosen (indicating resistance). As a result, HCC827-ER cells were established. Then, HCC827-ER cells were maintained in 10% FBS in DMEM containing the final established Erlotinib concentration of 15 µM.
Cell viability assay
For quantitative viability assays, the cells were plated in 96-well plates (1 × 104 cells/well). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays were performed to assess the cell viability. Sterile MTT dye (200 µl; 5 mg/ml; Sigma, USA) was added. After the cells were incubated for 4 h at 37°C in 5% CO2, the MTT medium mixture was removed and 200 µl of dimethyl sulfoxide was added to each well. Absorbance was determined at 490 nm by using a multi-well spectrophotometer (Thermo Electron, Andover, USA).
Cell apoptosis analysis
Apoptotic cells were evaluated using an annexin V-FITC kit (Beyotime, China). The cells were scraped and stained with annexin V-FITC and propidium iodide according to the manufacturer’s protocol. In brief, the cells were washed with PBS. After 195 µl of the binding buffer was added, 5 µl of FITC-labeled annexin V was added and incubated for 10 min at 25°C. The cells were then incubated with 10 µl propidium iodide for 10 min in an ice bath in the dark and the apoptotic cells were determined by flow cytometry (FACS) analysis.
Western blot analysis
The cells were harvested, pelleted by centrifugation, washed with ice-cold PBS, and lysed with RIPA buffer [150 mM NaCl, 50 mM Tris base (pH 8.0), 1 mM EDTA, 0.5% sodium deoxycholate, 1% NP-40, 0.1% sodium dodecyl sulfate (SDS), 1 mM DTT, 1 mM PMSF, and 1 mM Na3VO4] that was supplemented with a protease and phosphatase inhibitor. The proteins were separated by 10% SDS-polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane (Life Technologies, Gaithersburg, MD). The blots were then incubated in a fresh blocking solution with an appropriate dilution of the primary antibody at 4°C for 24 h.
The sources of antibodies were as follows: GAPDH mouse polyclonal antibody (Santa Cruz); p-MAPK (Thr202/Tyr204), MAPK, and p-AKT (Ser473); and AKT rabbit monoclonal antibody (Cell Signaling), Bax, Bcl-2, cytochrome C mouse monoclonal antibody, caspase-3 rabbit polyclonal antibody (Santa Cruz). After the blots were extensively washed, the membranes were incubated with horseradish peroxidase-coupled secondary antibody (1:2000, Zhongshan Biotech Company, China) at 25°C for 1 h. The bands were visualized and quantified using the Image-Pro Plus 5.0 software (Media Cybernetics). p-MAPK and p-AKT band intensities were normalized to MAPK and AKT band intensities, respectively. Bax, Bcl-2, cytochrome C and caspase-3 were adjusted by the GAPDH band intensities.
Statistical analysis
Data were expressed as mean value ± SD. Differences between groups were analyzed using ANOVA or a t-test. These analyses were performed on SPSS version 18.0 for Windows (SPSS Inc., Chicago, IL). P-value < 0.05 was considered statistically significant.
Results
Identification of DEGs
The t-test in the dChip Software was used to analyze the gene expression profiles of HCC827 cells and their TKI-resistant clones and identify the DEGs in TKI-resistant cells. A P value of less than 0.05 was used as the significant threshold for DEGs. According to the criteria, 1054 gene were shown to have an altered expression, including 483 up-regulated and 571 down-regulated genes.
Identification of related active small molecules or agent
The DEGs, involving up-regulated and down-regulated gene groups, were submitted to CMAP for analysis that could identify small molecules reversing TKI-resistance for HCC827-ER cells. The negatively-correlated gene expression patterns associated with drug-treated cancer cells were selected and listed according to the negative connectivity scores. As a result, among the sorted molecules, valproic acid (VPA) was selected because its connectivity scores was -1, indicating that VPA, a histone deacetylase (HDAC) inhibitor, might have the potential to overcome Erlotinib resistance in lung cancer cells (Table 1).
Table 1.
Top 10 predicted small molecule agents with potential abilities to overcome Erlotinib resistance of lung cancer
| CMAP name | Dose | Cell | Score |
|---|---|---|---|
| valproic acid | 200 µM | PC3 | -1.000 |
| hexamethonium bromide | 10 µM | PC3 | -0.947 |
| PF-00562151-00 | 10 µM | PC3 | -0.888 |
| ticlopidine | 13 µM | PC3 | -0.884 |
| cimetidine | 16 µM | MCF7 | -0.869 |
| ciprofloxacin | 11 µM | PC3 | -0.843 |
| PHA-00816795 | 10 µM | PC3 | -0.839 |
| norethisterone | 13 µM | PC3 | -0.837 |
| naphazoline | 16 µM | PC3 | -0.835 |
| retrorsine | 11 µM | PC3 | -0.834 |
Development and characterization of TKI-resistant HCC827-ER and HCC827 cancer cells
TKI-resistant HCC827-ER was obtained by gradual increase of Erlotinib in cell culture. To evaluate the characterization of both cells, we tested the IC50 of Erlotinib. As expected, the IC50 of Erlotinib was 19.6 nM (0.02 μM) for HCC827, and 98.3 μM for HCC827-ER cells, indicating that the TKI-resistant lung adenocarcinoma cells were well-established. Evidence suggests that MAPK and AKT might be involved in TKI-resistance of cancer cells [23], and therefore, we tested the phosphorylated protein expression of MAPK and AKT by immunoblotting analysis. Consequently, the data showed an increase in p-MAPK and p-AKT for HCC827-ER in contrast to those for HCC827, implying that the MAPK and AKT pathways were activated in HCC827-ER (Figure 1).
Figure 1.
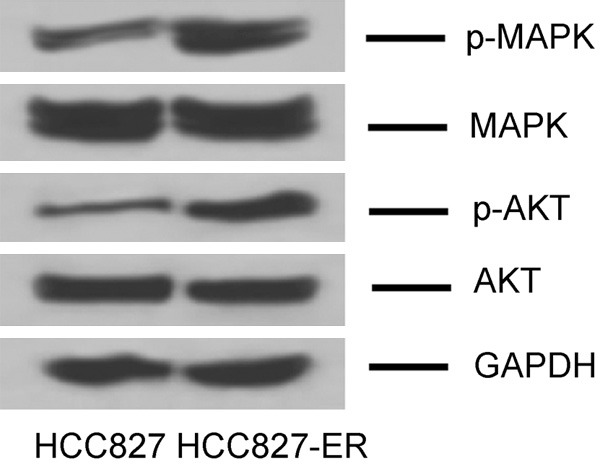
Expression of phosphor-MAPK and phosphor-AKT proteins in HCC827 and HCC827-ER by immunoblotting analysis.
Effects of VPA on HCC827-ER and HCC827 cancer cell growth
To test whether VPA has an effect on cancer cell growth, we divided the two types of cancer cells into two groups and treated them with various concentrations (0, 0.1, 0.2, 0.4, 0.8, 1.6, 3.2 mM) of VPA for 48 h, respectively. Then, the cell viability and apoptosis were evaluated. The results showed that the cell viability gradually decreased and cell apoptosis increased with the elevation of VPA concentration for each group, respectively (Figure 2), suggesting that VPA could inhibit the cell growth in a dose-dependent manner. The IC50 values of VPA for both cells were assessed and the results were 1.6 mM and 2.5 mM for HCC827 and HCC827ER, respectively. However, VPA was not likely to affect cell viability in each group when the VPA concentration was less than or equal to 0.2 mM, indicating that VPA might inhibit cancer cell viability at a relatively high concentration level, while might not directly influence cancer cell viability at a low level. Therefore, we used 0.2 mM as a candidate for further evaluation in order to reduce the interference of its cell viability suppression.
Figure 2.
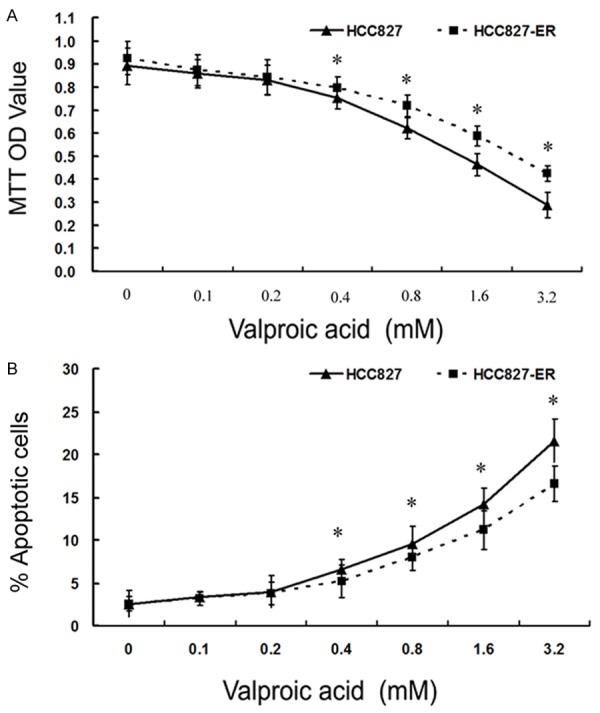
Cell viability and apoptosis of cells treated with various concentrations of VPA assessed by MTT (A) and apoptosis assay (B). (*P < 0.05 vs Control).
Effects of VPA and Erlotinib on HCC827-ER and HCC827 cancer cells
To learn whether VPA could reverse TKI-resistance of HCC827-ER to any extent, we conducted further experiments. Cells were divided into two groups according to the cell type, namely, HCC827-ER and HCC827. Each group was divided into four subgroups and treated with 0.2 mM VPA, 15 μM Erlotinib, 0.2 mM VPA+15 μM Erlotinib, and DMEM as a control for 48 h, respectively. Then, cell proliferation and apoptosis were tested.
As shown in Figure 3A, 3B, single use of VPA did not significantly affect cell growth in either HCC827-ER or HCC827 cancer cells as compared with those in the control group. Conversely, single use of Erlotinib could markedly repress cell proliferation of TKI-sensitive HCC827 but not HCC827-ER cells. Interestingly, when Erlotinib was combined with VPA, an increase in cell apoptosis and a decrease in cell viability were observed, indicating that VPA might overturn the acquired TKI-resistance of HCC827-ER cells.
Figure 3.
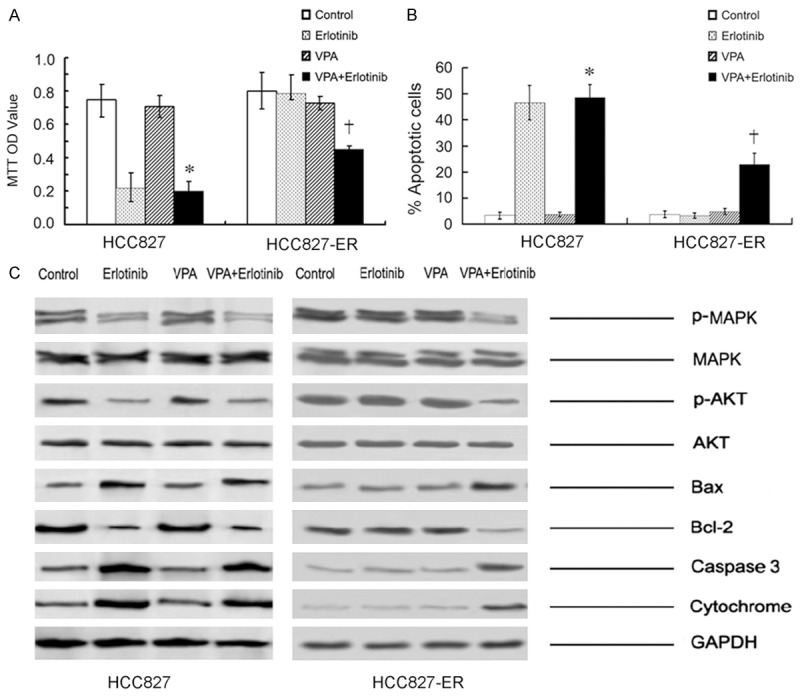
Cell viability (A) and apoptosis (B) in HCC827 and HCC827-ER assessed by MTT and apoptosis assay. (HCC827: *P > 0.05 vs Erlotinib; *P < 0.05 vs Control or VPA; HCC827-ER: †P < 0.05 vs Control or Erlotinib or VPA). (C) Expression of the MAPK, AKT and mitochondrial pathway-related proteins assessed by immunoblotting.
To explore the status of signaling pathways, we further tested the pathway proteins by western blot analysis. As shown in Figure 3C, combination of VPA and Erlotinib might lead to a decrease in the expression of p-MAPK and p-AKT protein. Accordingly, an increase in caspase-3 and a decrease in bcl-2 were also observed in this subgroup, indicating that VPA might reverse TKI-resistance of cancer cells via inactivation of MAPK and AKT pathways that in turn initiate mitochondrial apoptotic pathway.
Partial involvement of MAPK and AKT pathways in TKI-resistance reversion
To shed new light on the roles of MAPK and AKT pathways in the TKI-resistance, we conducted further research. HCC827-ER cells were divided into five groups as I, II, III, IV and V, respectively. Group I was treated with DMEM for 48 h and used as a control group. For group II and IV, cells were treated with 15 μM Erlotinib for 48 h. Nevertheless, cells in group IV were pretreated with a combination of 10 µM U0126 (a specific MAPK inhibitor, Cellsignal) and 10 µM MK-2206 (a specific AKT inhibitor, Selleckchem) for 2 h. Cells in group III were treated with only the MAPK and AKT inhibitors for 2 h. In groups V, cells were treated with a combination of 0.2 mM VPA and 15 μM Erlotinib (VPA+Erlotinib) for 48 h. Cell viability and apoptosis were assessed by MTT and apoptosis assays, respectively.
As shown in Figure 4, the data showed that single treatment with the inhibitors (Group III: U0126+MK-2206) slightly induced cell apoptosis and suppressed cell viability relative to those of the control group (Group I: DMEM) without significance. Interestingly, pretreatment of the inhibitors in combination with Erlotinib (Group IV: Erlotinib+U0126+MK-2206) could markedly induce cell apoptosis, compared with those in Group III; however, this effect seemed to be weaker than that in Group V (Group V: Erlotinib+VPA), indicating that inhibition of MAPK and AKT might only play partial roles in the acquired TKI-resistance for HCC827-ER cells.
Figure 4.
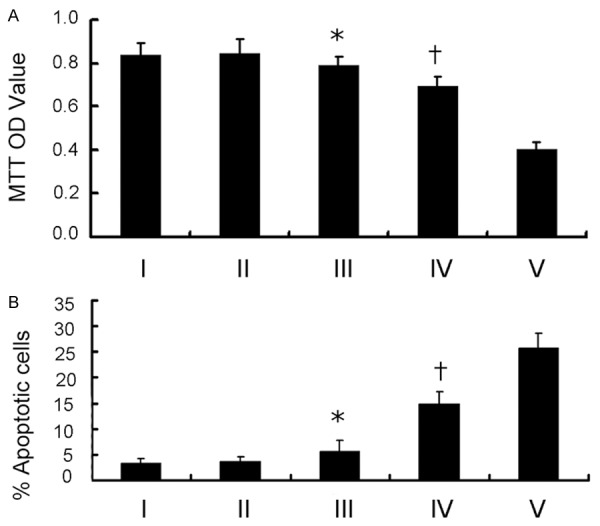
Cell viability (A) and apoptosis (B) of HCC827-ER cells treated with DMEM (I), Erlotinib (II), U0126+MK-2206 (III), Erlotinib+U0126+MK-2206 (IV), Erlotinib+VPA (V) assessed by MTT and apoptosis assay, respectively. (*P > 0.05 vs I or II; *P < 0.05 vs IV or V; †P < 0.05 vs III or V).
Discussion
In the present study, we found that VPA might be a potential agent that might overcome the EGFR-TKI resistance of lung adenocarcinoma cells by using GEO database and CMAP analysis. Then, we have tested the biological effects of VPA on TKI-resistant cancer cells. The results showed that VPA could reverse the TKI-resistance of cancer cells at a low concentration, and inhibition of MAPK and AKT signaling pathways might be involved in the process.
VPA is a common anti-epileptic agent that is often used for treatment of epilepsy and nonepileptic central nervous system (CNS) disorders through modulation of γ-aminobutyric acid (GABA) and glutamatergic neurotransmission [24]. Moreover, VPA can alter gene expression and modulate cell proliferation and apoptosis because it is reported to be an inhibitor of class I histone deacetylases (HDAC) [25]. Therefore, it has been thought to act as a potential anti-tumor agent due to its HDAC inhibitory activity. The anti-tumor effects of VPA are complicated. Primarily, VPA has a direct anti-cancer effect [26,27], in accordance with our results that VPA might inhibit cancer cell viability at a relatively high concentration level but has little effects on cell proliferation at a low concentration. Moreover, VPA inhibits cancer growth by cell cycle arrest, induction of differentiation, and inhibition of tumor vascularization [28]. Thus, VPA could inhibit cancer cell growth via multi-pathways. Reports showed that VPA could enhance the effect of herpes simplex virus type I thymidine kinase (HSV-TK) on glioma [29] and inhibit invasion and migration of breast cancer by regulating NM23H1 expression [10]. Besides, VPA might sensitize cancer cells to chemotherapy agents [26]. Use of clinically achievable doses of VPA could significantly increase the sensitivity of thyroid cancer cells to a tyrosine-kinase inhibitor imatinib [30], and a ligand of death receptors, TRAIL [31]. It also can overcome acquired Everolimus-resistance for renal cell carcinoma by down-regulating cdk2/cyclin A expression [32]. In the present study, the results showed that low concentration of VPA could overcome the TKI-resistance of lung cancer cells. Nevertheless, the precise mechanisms are still unclear.
AKT is a serine/threonine protein kinase that has been thought to be involved in multiple cellular processes, such as cell proliferation, apoptosis, transcription, and cell migration. Evidence shows that activation of AKT pathway might contribute to TKI-resistance of lung cancer cells [23,33]. MAPKs, mainly containing ERK, p38MAPKs, and c-Jun N-terminal kinase and possibly mediating cell proliferation and apoptosis, might also play a role in TKI-resistance of cancer cells [34]. The results of the present study showed that single use of Erlotinib or VPA could hardly induce HCC827-ER apoptosis, with the state of AKT and MAPK signaling pathways rarely changed. However, the combination of clinically achievable concentration of VPA with Erlotinib could markedly induce cell apoptosis, with inactivation of both AKT and MAPKs pathways, confirming that activation of MAPKs and AKT pathways might play a vital role in acquired TKI-resistance of lung cancer cells. During this process, activation of MAPK and AKT signaling pathways provided a survival signal for the TKI-resistant cells, whereas inhibition of both pathways indicated an apoptotic signal. Notably, activation of these two pathways might only be a symbol or play partial roles in the acquired TKI-resistance of HCC827 cells because the effects of the pathway inhibition were less than those of VPA treatment. Therefore, the mechanisms underlying the TKI-resistance reversion by VPA are multi-factorial. In addition to MAPK and AKT, other pathways might also play a role during this process. However, the results suggested the participation of MAPK and AKT signaling pathways in the TKI-resistance of lung adenocarcinoma cells.
CMAP is a tool that can connect therapeutic agents, genes, and disorders through the transitory feature of common gene-expression changes. It also can be used to screen connections among small molecules agents and putative mechanisms of action of unknown molecules. In the present study, VPA was screened out to be a potential agent that reverses the TKI-resistance of lung adenocarcinoma cells. The validation process confirmed the anti-tumor effects of VPA, indicating that CMAP is a reliable tool for exploring molecular mechanisms of drugs.
Several limitations might be involved in the present study. First, only one cell line, HCC827, was used in this experiment. Future studies using other EGFR mutant cell lines, such as H3255, might strengthen the significance of the results. Evidence indicated that exon20-T790M mutation is one of the complicated mechanisms underlying acquired TKI-resistance of lung cancer [35]. However, mutation of exon20-T790M was not found in HCC827-ER, as suggested by recent reports [36,37]. Therefore, the status of exon20-T790M was not detected in our study. In further study, this important mutation might be detected in other cell lines to evaluate the possible effects on this mutation. Second, only a small proportion of mechanisms underlying the TKI-resistance reversion were tested and revealed in this study. Other pathways that might be involved in this process need to be deeply determined in further investigations. Third, the validation experiment was an in vitro study, and further in vivo investigations might make this study clinically relevant.
In conclusion, screening genes in GEO database and using CMAP, we found that VPA might be a potential agent that can reverse EGFR-TKI-resistance of lung cancer cells in the further validation experiments, the results confirmed that clinically achievable concentration of VPA might overcome the acquired TKI-resistance of lung cancer cells, possibly by inactivating AKT and MAPKs pathways.
Acknowledgements
This work was partly supported by the special foundation for the 1130 Project of Xinqiao Hospital of Third Military Medical University (2012).
Disclosure of conflict of interest
None.
References
- 1.Barrow TM, Michels KB. Epigenetic epidemiology of cancer. Biochem Biophys Res Commun. 2014;455:70–83. doi: 10.1016/j.bbrc.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 2.Siegelin MD, Borczuk AC. Epidermal growth factor receptor mutations in lung adenocarcinoma. Lab Invest. 2014;94:129–137. doi: 10.1038/labinvest.2013.147. [DOI] [PubMed] [Google Scholar]
- 3.Kasza A. IL-1 and EGF regulate expression of genes important in inflammation and cancer. Cytokine. 2013;62:22–33. doi: 10.1016/j.cyto.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 4.Kuroda S, Tam J, Roth JA, Sokolov K, Ramesh R. EGFR-targeted plasmonic magnetic nanoparticles suppress lung tumor growth by abrogating G2/M cell-cycle arrest and inducing DNA damage. Int J Nanomedicine. 2014;9:3825–3839. doi: 10.2147/IJN.S65990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roskoski R Jr. The ErbB/HER family of proteintyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. doi: 10.1016/j.phrs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Paz-Ares L, Soulieres D, Moecks J, Bara I, Mok T, Klughammer B. Pooled analysis of clinical outcome for EGFR TKI-treated patients with EGFR mutation-positive NSCLC. J Cell Mol Med. 2014;18:1519–1539. doi: 10.1111/jcmm.12278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ameratunga M, Pavlakis N, Gebski V, Broad A, Khasraw M. Epidermal growth factor receptor-tyrosine kinase inhibitors in advanced squamous cell carcinoma of the lung: A metaanalysis. Asia Pac J Clin Oncol. 2014;10:273–278. doi: 10.1111/ajco.12231. [DOI] [PubMed] [Google Scholar]
- 8.Hrustanovic G, Lee BJ, Bivona TG. Mechanisms of resistance to EGFR targeted therapies. Cancer Biol Ther. 2013;14:304–314. doi: 10.4161/cbt.23627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faber AC, Corcoran RB, Ebi H, Sequist LV, Waltman BA, Chung E, Incio J, Digumarthy SR, Pollack SF, Song Y, Muzikansky A, Lifshits E, Roberge S, Coffman EJ, Benes CH, Gomez HL, Baselga J, Arteaga CL, Rivera MN, Dias-Santagata D, Jain RK, Engelman JA. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011;1:352–365. doi: 10.1158/2159-8290.CD-11-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ercan D, Xu C, Yanagita M, Monast CS, Pratilas CA, Montero J, Butaney M, Shimamura T, Sholl L, Ivanova EV, Tadi M, Rogers A, Repellin C, Capelletti M, Maertens O, Goetz EM, Letai A, Garraway LA, Lazzara MJ, Rosen N, Gray NS, Wong KK, Janne PA. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012;2:934–947. doi: 10.1158/2159-8290.CD-12-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, Choi YJ, Choi CM, Kim SW, Jang SJ, Park YS, Kim WS, Lee DH, Lee JS, Miller VA, Arcila M, Ladanyi M, Moonsamy P, Sawyers C, Boggon TJ, Ma PC, Costa C, Taron M, Rosell R, Halmos B, Bivona TG. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–860. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olbryt M, Habryka A, Student S, Jarzab M, Tyszkiewicz T, Lisowska KM. Global gene expression profiling in three tumor cell lines subjected to experimental cycling and chronic hypoxia. PLoS One. 2014;9:e105104. doi: 10.1371/journal.pone.0105104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feldhahn N, Arutyunyan A, Stoddart S, Zhang B, Schmidhuber S, Yi SJ, Kim YM, Groffen J, Heisterkamp N. Environment-mediated drug resistance in Bcr/Abl-positive acute lymphoblastic leukemia. Oncoimmunology. 2012;1:618–629. doi: 10.4161/onci.20249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan Y, Zhou F, Zhang R, Claret FX. Stat3 inhibitor Stattic exhibits potent antitumor activity and induces chemo- and radio-sensitivity in nasopharyngeal carcinoma. PLoS One. 2013;8:e54565. doi: 10.1371/journal.pone.0054565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Xu YH, Lu Y, Ma XP, Chen P, Luo SW, Jia ZG, Liu Y, Guo Y. Identifying differentially expressed genes and small molecule drugs for prostate cancer by a bioinformatics strategy. Asian Pac J Cancer Prev. 2013;14:5281–5286. doi: 10.7314/apjcp.2013.14.9.5281. [DOI] [PubMed] [Google Scholar]
- 17.Siatkowski M, Liebscher V, Fuellen G. CellFateScout - a bioinformatics tool for elucidating small molecule signaling pathways that drive cells in a specific direction. Cell Commun Signal. 2013;11:85. doi: 10.1186/1478-811X-11-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spijkers-Hagelstein JA, Pinhancos SS, Schneider P, Pieters R, Stam RW. Chemical genomic screening identifies LY294002 as a modulator of glucocorticoid resistance in MLLrearranged infant ALL. Leukemia. 2014;28:761–769. doi: 10.1038/leu.2013.245. [DOI] [PubMed] [Google Scholar]
- 19.Yeh CT, Wu AT, Chang PM, Chen KY, Yang CN, Yang SC, Ho CC, Chen CC, Kuo YL, Lee PY, Liu YW, Yen CC, Hsiao M, Lu PJ, Lai JM, Wang LS, Wu CH, Chiou JF, Yang PC, Huang CY. Trifluoperazine, an antipsychotic agent, inhibits cancer stem cell growth and overcomes drug resistance of lung cancer. Am J Respir Crit Care Med. 2012;186:1180–1188. doi: 10.1164/rccm.201207-1180OC. [DOI] [PubMed] [Google Scholar]
- 20.Kim J, Vasu VT, Mishra R, Singleton KR, Yoo M, Leach SM, Farias-Hesson E, Mason RJ, Kang J, Ramamoorthy P, Kern JA, Heasley LE, Finigan JH, Tan AC. Bioinformatics-driven discovery of rational combination for overcoming EGFRmutant lung cancer resistance to EGFR therapy. Bioinformatics. 2014;30:2393–8. doi: 10.1093/bioinformatics/btu323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin M, Wei LJ, Sellers WR, Lieberfarb M, Wong WH, Li C. dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics. 2004;20:1233–1240. doi: 10.1093/bioinformatics/bth069. [DOI] [PubMed] [Google Scholar]
- 22.Gao L, Zhao G, Fang JS, Yuan TY, Liu AL, Du GH. Discovery of the neuroprotective effects of alvespimycin by computational prioritization of potential anti-parkinson agents. FEBS J. 2014:1110–22. doi: 10.1111/febs.12672. [DOI] [PubMed] [Google Scholar]
- 23.Jeannot V, Busser B, Brambilla E, Wislez M, Robin B, Cadranel J, Coll JL, Hurbin A. The PI3K/AKT pathway promotes gefitinib resistance in mutant KRAS lung adenocarcinoma by a deacetylase-dependent mechanism. Int J Cancer. 2014;134:2560–2571. doi: 10.1002/ijc.28594. [DOI] [PubMed] [Google Scholar]
- 24.Bialer M. Why are antiepileptic drugs used for nonepileptic conditions? Epilepsia. 2012;53(Suppl 7):26–33. doi: 10.1111/j.1528-1167.2012.03712.x. [DOI] [PubMed] [Google Scholar]
- 25.Singh V, Bhatia HS, Kumar A, de Oliveira AC, Fiebich BL. Histone deacetylase inhibitors valproic acid and sodium butyrate enhance prostaglandins release in lipopolysaccharideactivated primary microglia. Neuroscience. 2014;265:147–157. doi: 10.1016/j.neuroscience.2014.01.037. [DOI] [PubMed] [Google Scholar]
- 26.Wang D, Jing Y, Ouyang S, Liu B, Zhu T, Niu H, Tian Y. Inhibitory effect of valproic acid on bladder cancer in combination with chemotherapeutic agents and. Oncol Lett. 2013;6:1492–1498. doi: 10.3892/ol.2013.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai C, Leslie JS, Franko-Tobin LG, Prasnal MC, Yang T, Vienna Mackey L, Fuselier JA, Coy DH, Liu M, Yu C, Sun L. Valproic acid suppresses cervical cancer tumor progression possibly via activating Notch1 signaling and enhances receptor-targeted cancer chemotherapeutic via activating somatostatin receptor type II. Arch Gynecol Obstet. 2013;288:393–400. doi: 10.1007/s00404-013-2762-7. [DOI] [PubMed] [Google Scholar]
- 28.Sidana A, Wang M, Shabbeer S, Chowdhury WH, Netto G, Lupold SE, Carducci M, Rodriguez R. Mechanism of growth inhibition of prostate cancer xenografts by valproic acid. J Biomed Biotechnol. 2012;2012:180363. doi: 10.1155/2012/180363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryu CH, Park KY, Kim SM, Jeong CH, Woo JS, Hou Y, Jeun SS. Valproic acid enhances anti-tumor effect of mesenchymal stem cell mediated HSV-TK gene therapy in intracranial glioma. Biochem Biophys Res Commun. 2012;421:585–590. doi: 10.1016/j.bbrc.2012.04.050. [DOI] [PubMed] [Google Scholar]
- 30.Catalano MG, Pugliese M, Poli R, Bosco O, Bertieri R, Fortunati N, Boccuzzi G. Effects of the histone deacetylase inhibitor valproic acid on the sensitivity of anaplastic thyroid cancer cell lines to imatinib. Oncol Rep. 2009;21:515–521. [PubMed] [Google Scholar]
- 31.Cha HY, Lee BS, Kang S, Shin YS, Chang JW, Sung ES, Kim YS, Choi JW, Kim JH, Kim CH. Valproic acid sensitizes TRAIL-resistant anaplastic thyroid carcinoma cells to apoptotic cell death. Ann Surg Oncol. 2013;20(Suppl 3):S716–724. doi: 10.1245/s10434-013-3232-y. [DOI] [PubMed] [Google Scholar]
- 32.Juengel E, Nowaz S, Makarevi J, Natsheh I, Werner I, Nelson K, Reiter M, Tsaur I, Mani J, Harder S, Bartsch G, Haferkamp A, Blaheta RA. HDAC-inhibition counteracts everolimus resistance in renal cell carcinoma in vitro by diminishing cdk2 and cyclin A. Mol Cancer. 2014;13:152. doi: 10.1186/1476-4598-13-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li B, Ren S, Li X, Wang Y, Garfield D, Zhou S, Chen X, Su C, Chen M, Kuang P, Gao G, He Y, Fan L, Fei K, Zhou C, Schmit-Bindert G. MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. Lung Cancer. 2014;83:146–153. doi: 10.1016/j.lungcan.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 34.Morgillo F, Martinelli E, Troiani T, Orditura M, De Vita F, Ciardiello F. Antitumor activity of sorafenib in human cancer cell lines with acquired resistance to EGFR and VEGFR tyrosine kinase inhibitors. PLoS One. 2011;6:e28841. doi: 10.1371/journal.pone.0028841. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Woo HS, Ahn HK, Lee HY, Park I, Kim YS, Hong J, Sym SJ, Park J, Lee JH, Shin DB, Cho EK. Epidermal growth factor receptor (EGFR) exon 20 mutations in non-small-cell lung cancer and resistance to EGFR-tyrosine kinase inhibitors. Invest New Drugs. 2014;32:1311–1315. doi: 10.1007/s10637-014-0146-x. [DOI] [PubMed] [Google Scholar]
- 36.Suda K, Murakami I, Katayama T, Tomizawa K, Osada H, Sekido Y, Maehara Y, Yatabe Y, Mitsudomi T. Reciprocal and complementary role of MET amplification and EGFR T790M mutation in acquired resistance to kinase inhibitors in lung cancer. Clin Cancer Res. 2010;16:5489–5498. doi: 10.1158/1078-0432.CCR-10-1371. [DOI] [PubMed] [Google Scholar]
- 37.Nanjo S, Yamada T, Nishihara H, Takeuchi S, Sano T, Nakagawa T, Ishikawa D, Zhao L, Ebi H, Yasumoto K, Matsumoto K, Yano S. Ability of the Met kinase inhibitor crizotinib and new generation EGFR inhibitors to overcome resistance to EGFR inhibitors. PLoS One. 2013;8:e84700. doi: 10.1371/journal.pone.0084700. [DOI] [PMC free article] [PubMed] [Google Scholar]