

Abstract
Plasmodium vivax infections often recur due to relapse of hypnozoites from the liver. In malaria-endemic areas, tools to distinguish relapse from reinfection are needed. We applied amplicon deep sequencing to P. vivax isolates from 78 Cambodian volunteers, nearly one-third of whom suffered recurrence at a median of 68 days. Deep sequencing at a highly variable region of the P. vivax merozoite surface protein 1 gene revealed impressive diversity—generating 67 unique haplotypes and detecting on average 3.6 cocirculating parasite clones within individuals, compared to 2.1 clones detected by a combination of 3 microsatellite markers. This diversity enabled a scheme to classify over half of recurrences as probable relapses based on the low probability of reinfection by multiple recurring variants. In areas of high P. vivax diversity, targeted deep sequencing can help detect genetic signatures of relapse, key to evaluating antivivax interventions and achieving a better understanding of relapse-reinfection epidemiology.
Keywords: amplicon sequencing, deep sequencing, genetic diversity, hypnozoite, malaria, microsatellite, multiplicity of infection, Plasmodium vivax, pvmsp1, relapse
In recent years, there has been an increased appreciation that global malaria elimination efforts cannot succeed without a better understanding of Plasmodium vivax, the leading cause of malaria outside Africa [1–3]. In particular, P. vivax's ability to cause periodic relapse poses a major barrier to malaria elimination, because hypnozoites, the parasite stages in the liver that reactivate to cause relapse, are not killed by traditional blood-stage drugs [4, 5].
In Southeast Asia, P. vivax relapses are common and frequent: up to two-thirds of individuals not treated with antirelapse therapy suffer 1 or more relapses, approximately 3–4 weeks after plasma levels of antimalarials wane [6–11]. However, because individuals can also become reinfected, it is difficult to determine the true relapse rate and to distinguish when treatment failures are due to relapse. Molecular genotyping aimed at distinguishing relapses from reinfections has been confounded by the frequent finding of genetically different parasites at relapse, even in the setting of known relapse [12–15]. Thus, tools to assess interventions targeting P. vivax in clinical studies are missing.
We and others have previously shown that P. vivax populations in Thailand and Cambodia exhibit great genetic diversity despite relatively low-level transmission: many alleles circulate on a population level, and individuals commonly harbor multiple genetic variants at once [16–18]. With this diversity in mind, we applied targeted deep sequencing to a malaria cohort in Cambodia in which one-third of individuals suffered recurrent P. vivax infections. We hypothesized that the within-host diversity unveiled by deep sequencing at a highly polymorphic molecular marker would expose genotypic patterns suggestive of relapse. We found that enhanced detection of minority variants revealed patterns of variant overlap between initial and recurrent parasite isolates within individuals. This finding, combined with population-based characterization of haplotypes, provide a statistical framework for determining the probability of reinfection and relapse. Our findings shed light on the nature of P. vivax hypnozoite activation and represent important steps toward identifying genotypic signatures of relapse.
MATERIALS AND METHODS
Study Population
We studied parasites collected from 78 P. vivax–infected adults, aged 18 to 49, enrolled in a malaria cohort and treatment study conducted from September 2010 to March 2011 in Oddar Meanchay province in northern Cambodia [19]. Of the 220 cohort volunteers, most were military personnel with frequent travel to forested areas. The first 80 subjects who developed malaria (either falciparum or vivax) were treated with dihydroartemisinin-piperaquine, after which vivax patients were treated with chloroquine per national Cambodian treatment guidelines. Subjects were followed for recurrence with weekly blood smears for 6 weeks, then by clinical symptoms thereafter with a monthly blood smear. Those with P. vivax were treated with primaquine to clear liver-stage hypnozoites at the conclusion of their participation in the cohort study, which ranged from 2 to 6 months’ duration. Ethical approval for the study was granted by the institutional review boards of the University of North Carolina, Walter Reed Army Institute of Research, and the National Ethics Committee for Health Research in Cambodia.
Amplicon Deep Sequencing of pvmsp1
DNA from filter paper blood spots was extracted using the Invitrogen Pro 96 Genomic DNA kit (Invitrogen, Carlsbad, California). A 117–base pair (bp) variable portion of the 33-kDa subunit of the 42-kDa region of pvmsp1 (Figure 1A) was chosen for deep sequencing based on previous work showing great nucleotide diversity across this region [20]. Amplicons were generated using nested polymerase chain reaction (PCR) using the primers and conditions listed in the Supplementary Methods. Forward primers used in the second round were modified for Ion Torrent sequencing by inclusion of a multiplex identifier (MID) sequence. Each sample was amplified in duplicate, using unique MIDs. If amplification failed, the cycle number in both rounds was increased to 35 cycles. This was necessary in approximately one-quarter of the samples. Amplicons were cleaned and normalized to 1–2 ng/µL concentration using the SequalPrep Normalization Plate Kit (Life Technologies, Carlsbad, California), then pooled and sequenced on the Ion Torrent platform from Life Technologies.
Figure 1.

Ultradeep sequencing of pvmsp1. A, The target amplicon contained a 117-bp variable region of the 42-kDa fragment of pvmsp1. B, DNA alignment of all 67 unique pvsmp1 variants detected in the 108 isolates from 78 individuals. Each concentric ring represents a unique sequence. Nucleotides are represented by different colors (adenine, red; thymine, blue; cytosine, green; and guanine, yellow). C, Frequency of unique pvmsp1 haplotypes within the study population (out of 108 isolates). Only haplotypes that appeared in more than 1 isolate are shown. The red portions of the columns represent the proportion that occurred as a minority variant (existing at 0.5%–20% frequency within the individual isolate). D, Multiplicity of infection among initial (blue) and recurrent (green) isolates. Abbreviation: bp, base pair.
Haplotype Determination From Deep Sequencing
Haplotypes of pvmsp1 variants were determined by an in-house bioinformatics pipeline that uses a clustering method to construct the most likely haplotypes within a patient while removing false haplotypes due to PCR or sequencing error (http://baileylab.umassmed.edu/seekdeep). In brief, raw sequence reads were separated on the basis of MIDs from the pooled data into amplicon-specific data, then filtered on read length, overall quality scores, and presence of primer sequences. Amplicon reads were trimmed of MIDs, tags, and primers and organized into unique clusters based on their sequence. Then, for each sample, sequence clusters differing only by indels of 1 and 2 bases or sequences harboring low-quality mismatches or low k-mer frequency errors (k-mer occurring less than 0.2%) were collapsed together. Low quality was defined as either a mismatching base Q < 20 or any Q < 15 within an 11 bp region centered on the mismatch, as has been applied previously to rigorous single-nucleotide polymorphism discovery from shotgun data [21]. Next, potential PCR chimeras within patients were identified and removed based on the presence of both potential parental frequencies existing at higher frequencies with the patient. Finally, for each patient, haplotype clusters that were present in 2 independent duplicate PCR samples at ≥0.5% frequency were counted as unique variants. In this way, consensus haplotype determination was performed across the combined haplotypes from all individuals. Final haplotypes for analysis were each assigned a unique population identifier (CAM.00–CAM.66).
Microsatellite Genotyping
A subset of 65 samples were also genotyped at 3 microsatellite markers using previously published primers: PvMS7 and PvMS10 on chromosomes 2 and 5 [22], respectively, and MS10 on chromosome 13 (referred to here as MS10.13) [23]. PvMS7 and MS10.13 have been recommended as microsatellite markers of highest priority based on their balanced representation of diversity and frequent use [24], and MS10.13 was previously shown to exhibit high diversity in Cambodia [17]. PCR was performed using the same conditions for all 3 markers (Supplementary Text), except that hemi-nested PCR was used in samples where MS10.13 had poor amplification. Fragments were sized on a 3730 × L DNA Analyzer with results analyzed using Gene Mapper 4.1 software (Applied Biosystems). Peaks above a threshold of 100 units of relative fluorescent intensity and distinct above background noise were considered true amplification products, while peaks that were less than one-third the intensity of the strongest peak or visually appeared to be stutter peaks were excluded [25]. Alleles were grouped into bins of 3 bp for PvMS7 and MS10.13 and 4 bp for PvMS10 based on the expected repeat size for each microsatellite.
Data Analysis
The final haplotypes were stored and analyzed in Microsoft Excel 2007. Multiplicity of infection (MOI) was defined as the number of unique pvmsp1 haplotypes detected per patient isolate, or in the case of the microsatellite sequencing data, the greatest number of alleles detected in any of the 3 markers. Genetic diversity for each genotyping method was estimated by calculating the virtual heterozygosity (HE) [25]. Agreement between genotyping methods was assessed using the κ coefficient. DNA alignments and figures were generated using MegAlign and GenVison software (DNAStar, Madison, Wisconsin). The median-joining network was created using DNA Alignment v1.2.1.1 and Network v4.6.1.2 (Fluxus Technology, Suffolk, England). Statistical analysis was done using STATA v.12.0 (STATA Corp, College Station, Texas).
RESULTS
P. vivax Genetic Diversity by Amplicon Deep Sequencing
DNA from all 108 P. vivax samples from 78 Cambodian volunteers were successfully amplified in duplicate at a 117-bp-variable region of pvmsp1 and subjected to ultra-deep sequencing using the Ion Torrent platform. After quality filtering, a median of 3237× coverage was achieved per patient sample, and 2 873 657 (97%) of the high-quality reads clustered contributed to utilized haplotypes. The deep coverage enabled detection of variants present at as low as 0.5% within-host frequency, as this threshold translated into an average of approximately 16 sequence reads. In total, 67 unique pvmsp1 haplotypes were detected across the 108 isolates (Figure 1B, Supplementary Figure 1). Overall, these haplotypes displayed 59 variable sites, with the majority displaying nonsynonymous substitutions. Nine common haplotypes appeared in at least 10% of individuals (Figure 1C), while two-thirds (46/67) of haplotypes appeared in only 1 isolate. Virtual heterozygosity at this locus was HE = 0.95, reflecting an average 95% probability that 2 parasite clones taken at random from the population will display different pvmsp1 haplotypes.
Multiplicity of Infection and Detection of Minority Variants
In-host genetic diversity revealed by pvmsp1 deep sequencing was also high. Most initial P. vivax episodes (90%) were composed of polyclonal infections, with an average of 3.6 cocirculating variants, while as many as 10 variants were identified in 1 isolate (Figure 1D). The in-host frequency of each variant, calculated by its proportion of reads within the total reads per individual, demonstrated good concordance between duplicate PCRs done on each isolate using a summed distance metric to quantify noise [26] (Supplementary Figure 2). Minority variants were defined as those existing at <20% in-host frequency [27]. Sixty percent of the 67 identified haplotypes were detected only as minority variants. Some of these minority variant formed part of the mutational path between the more common variants, as depicted in a median joining network based on sequence relatedness, adding support that they are true haplotypes that are not a result of PCR or sequencing error (Figure 2).
Figure 2.

Median joining network of pvmsp1 haplotypes showing all variants detected. Larger nodes depict the common variants. The proportion of samples with the designated haplotype detected in initial and recurrent infections are depicted in white and black, respectively. Nodes representing sequences on the mutational pathway between haplotypes that were not detected are unlabeled. Spatial organization and distance between nodes is arbitrary.
Comparison of Amplicon Deep Sequencing to Microsatellite Genotyping
The subset of 65 isolates also genotyped at 3 P. vivax microsatellite markers similarly revealed high genetic diversity but on average, less genetic complexity within isolates, compared to deep sequencing (Table 1). When results from 3 microsatellite markers were combined, 72% of isolates were polyclonal, compared to 92% by sequencing, and contained an average of 2.1 variants, compared to average of 3.8 variants detected by deep sequencing (P < .001). In a small fraction of isolates (6/65), the combined microsatellite markers revealed a higher MOI than deep sequencing, but there was only 1 case in which a polyclonal infection identified by microsatellite typing was “missed” by deep sequencing at pvmsp1. Overall, the agreement between the 2 genotyping methods for detecting polyclonal infections was fair (κ coefficient = 0.26 [95% confidence interval, .02–.50]).
Table 1.
Comparison of MOI and Diversity by Ion Torrent Amplicon Deep Sequencing Versus Microsatellite Genotyping (N = 65 Samples)
| Deep Sequencing | Microsatellite Genotyping |
||||
|---|---|---|---|---|---|
| Pvmsp1 | PvMS7 | PvMS10 | MS10.13 | MS Markers Combined | |
| Median MOI | 3 | 1 | 1 | 2 | 2 |
| Mean MOI | 3.8 | 1.5 | 1.4 | 1.8 | 2.1 |
| Max MOI | 10 | 3 | 4 | 5 | 5 |
| No. polyclonal | 60 | 27 | 17 | 33 | 47 |
| % polyclonal | 92% | 42% | 26% | 51% | 72% |
| No. alleles | 52 | 19 | 15 | 20 | NA |
| Virtual heterozygosity (HE) | 0.94 | 0.92 | 0.84 | 0.94 | NA |
Abbreviations: MOI, multiplicity of infection; MS, microsatellite; NA, not applicable.
Genotypic Patterns of P. vivax Recurrences
Eight subjects developed P. vivax after treatment for P. falciparum. Of the other 69 subjects who developed P. vivax and were subsequently followed for a median of 115 days, 22 individuals, or approximately one-third of the cohort, developed recurrent P. vivax infection [15]: 16 subjects suffered 1 recurrence, 5 suffered 2 recurrences, and 1 subject suffered 3 recurrences. Therefore, a total of 29 P. vivax recurrences, occurring a median of 68 days (range, 2.5–18 weeks) after treatment with artemisinin-based combination therapy, were available for genotypic analysis. Recurrences were as likely as initial infections to be polyclonal. There was also no difference in the allelic diversity seen in recurrent versus initial infections. Variant CAM.01 appeared to arise more commonly in recurrent infections, while CAM.04 was seen more commonly in initial infections, but neither of these observations was significant when accounting for multiple comparisons (Figure 2).
The relatedness of recurrent pairs was classified according to the degree of overlap between variants found in the recurrent isolate versus the preceding episode (ie, 10R compared to 10, 10RR compared to 10R) (Figure 3). Among the 29 recurrent pairs, approximately one-third or 11/29 were homologous by deep sequencing, exhibiting the same dominant or codominant pvmsp1 haplotypes (Figure 3A and 3B). Of the remaining heterologous (nonhomologous) pairs, over half (10/18) appeared related, displaying both shared (recurring) and novel pvmsp1 variants at recurrence (Figure 3C and 3D). The rest (8/18) displayed completely different genotypes made up of novel variants at recurrence not seen in the preceding infection (Figure 3E). Microsatellite genotyping revealed strikingly different results: of the 21 pairs that were homologous or appeared related by deep sequencing, different alleles were detected at recurrence in at least 1 of the 3 microsatellite markers in all but 2 pairs.
Figure 3.

Representative genotypes of recurrent pairs categorized into homologous or heterologous pairs (A) based on overlap of pvmsp1 variants in the recurrent and preceding infection. Unique haplotypes are represented by specific colors across all samples. B, Homologous pairs were defined as having the same dominant or codominant haplotype at recurrence as seen in the preceding episode. C, In pairs exhibiting minority variant expansion, a minority parasite population existing at <20% in-host frequency in the initial infection reappeared as the dominant variant at recurrence. D, At least 1 shared variant defined our third category, while one-third of heterologous pairs shared no overlap at all (E). Pairs identified as probable relapses based on statistical testing are denoted by an asterisk. A pair that was “indeterminate” by statistical testing but judged as likely relapse based on microsatellite results are denoted by double asterisks. The genotypes of all 29 pairs are depicted in Supplementary Figure 3 (21 depicted here).
Minority Variant Expansion
In one-third of the heterologous recurrences (6/18), the recurrence genotype displayed a pattern of minority variant expansion, in which a variant existing at <20% in-host frequency in the initial infection reappeared as the dominant variant at recurrence (Figure 3B). This type of pattern is illustrated in patient 81, who in successive recurrences, each 31–35 days after the previous episode, harbored an increasing fraction of the CAM.00 variant until it was the dominant clone in the third recurrence (Figure 4A). This 20-year-old patient denied ever having malaria in the past and was also pvmsp1 antibody–negative upon enrollment (unpublished data), supporting the conclusion that his initial P. vivax episode was a primary mosquito-inoculated infection followed by 3 hypnozoite-induced relapses. Microsatellites genotyping in this patient showed different alleles in at least 1 marker at each transition. However, when minor peaks (distinct from background fluorescence but less than one-third the intensity of the strongest peak) were included for comparison, recurring alleles at each marker also support the impression that the patient suffered multiple relapses rather than new infections (Figure 4B).
Figure 4.
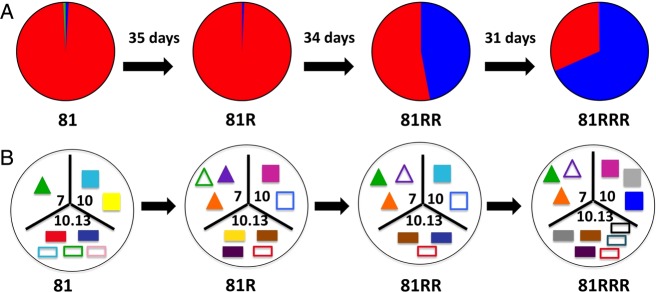
Pictoral representation of pvmsp1 haplotypes and microsatellite alleles MS7, MS10, and MS10.13 found in patient 81 through 4 consecutive P. vivax parasitemic episodes. In Panel A, pie slices reflect the proportion of sequencing reads assigned to each pvmsp1 haplotype variant within each episode (CAM.00 in red, CAM.01 in blue, CAM.51 in yellow). In Panel B, microsatellite alleles for PvMS7, PvMS10, and MS10.13 are depicted as different colored triangles, squares, and rectangles, respectively, within each tripart segment. Alleles that appeared only as minor peaks (less than one-third height of the dominant allele peak) are depicted as hollow shapes.
Classification of Recurrences as Relapses
To determine whether recurrence pairs with shared variants likely represent relapses, we employed a method used previously to distinguish P. falciparum recrudescence and reinfection: we calculated the probability that variants would recur in the same person by chance given their overall population prevalence [28]. This probability is meant to reflect the likelihood that the recurrence genotype represents a new mosquito-inoculated infection. Thus, for the recurrent patient with x variants and sharing a single variant of prevalence y, the binomial probability [29] that this variant is found by chance in a recurrent infection is calculated as 1−(1−y)x.
The probabilities of reinfection by the same variants for the 11 homologous pairs and 10 related pairs by deep sequencing are shown in Table 2. In recurrences where there were multiple shared variants, the combined probability that all shared variants would appear in a reinfection is calculated as the product of the individual probabilities. To classify recurrences as probable relapses arising from hypnozoites within the patient, we employed a cutoff of 10%, where if the probability of reinfection by the shared variant(s) is ≤0.10, the recurrence is classified as a relapse, and otherwise, “indeterminate.” Among the 21 homologous and related pairs, 5 had reinfection probabilities of 10%–20%, placing them in the indeterminate category. We evaluated the microsatellite data in the indeterminate pairs and found that 2 (patients 123 and 151) demonstrated similar alleles at all 3 microsatellite markers tested, making it unlikely that the recurrent infection was an unrelated new infection. These 2 pairs were thus reclassified as probable relapses.
Table 2.
Classification of Recurrence Pairs Based on Number and Prevalence of Shared Clones Between Episodes
| Recurrence Pair | Days to Recurrence | No. of Clones at Recurrence | Shared Clonesa | Shared Clone Prevalence | Probability of Reinfectionb | Probability of Reinfection by All Shared Variantsc | Classificationd |
|---|---|---|---|---|---|---|---|
| Homologous pairs | |||||||
| 81 --> 81R | 35 | 2 | CAM.01 | 0.061 | 0.118 | 0.024 | Relapse |
| CAM.00 | 0.105 | 0.200 | |||||
| 81R --> 81RR | 34 | 2 | CAM.01 | 0.061 | 0.118 | 0.024 | Relapse |
| CAM.00 | 0.105 | 0.200 | |||||
| 81RR --> 81RRR | 31 | 3 | CAM.00 | 0.105 | 0.284 | 0.049 | Relapse |
| CAM.01 | 0.061 | 0.172 | |||||
| 82 --> 82R | 56 | 4 | CAM.03 | 0.046 | 0.172 | 0.062 | Relapse |
| CAM.00 | 0.105 | 0.360 | |||||
| 82R --> 82RR | 48 | 4 | CAM.03 | 0.046 | 0.172 | 0.0002 | Relapse |
| CAM.01 | 0.061 | 0.223 | |||||
| CAM.00 | 0.105 | 0.360 | |||||
| CAM.46 | 0.003 | 0.013 | |||||
| 96 --> 96R | 71 | 2 | CAM.00 | 0.105 | 0.200 | 0.200 | Indeterminate |
| 123 --> 123R | 68 | 2 | CAM.00 | 0.105 | 0.199 | 0.199 | Indeterminate |
| 126 --> 126R | 85 | 3 | CAM.07 | 0.035 | 0.100 | 0.017 | Relapse |
| CAM.01 | 0.061 | 0.172 | |||||
| 130 --> 130R | 68 | 3 | CAM.12 | 0.013 | 0.284 | 0.002 | Relapse |
| CAM.04 | 0.049 | 0.141 | |||||
| CAM.00 | 0.105 | 0.039 | |||||
| 151 --> 151R | 126 | 4 | CAM.08 | 0.026 | 0.101 | 0.101 | Indeterminate |
| 152 --> 152R | 94 | 4 | CAM.00 | 0.105 | 0.360 | 0.080 | Relapse |
| CAM.01 | 0.061 | 0.223 | |||||
| Heterologous pairs with shared and novel variants | |||||||
| Minority variant expansion | |||||||
| 10 --> 10R | 84 | 3 | CAM.00 | 0.105 | 0.284 | 0.010 | Relapse |
| CAM.11 | 0.012 | 0.034 | |||||
| 68 --> 68R | 99 | 1 | CAM.10 | 0.013 | 0.013 | 0.013 | Relapse |
| 80R --> 80RR | 42 | 7 | CAM.08 | 0.026 | 0.171 | 0.003 | Relapse |
| CAM.12 | 0.013 | 0.089 | |||||
| CAM.00 | 0.105 | 0.541 | |||||
| CAM.02 | 0.066 | 0.379 | |||||
| 87 --> 87R | 81 | 4 | CAM.00 | 0.105 | 0.360 | 0.036 | Relapse |
| CAM.08 | 0.026 | 0.101 | |||||
| 112 --> 112R | 67 | 3 | CAM.00 | 0.105 | 0.284 | 0.009 | Relapse |
| CAM.02 | 0.066 | 0.185 | |||||
| CAM.01 | 0.061 | 0.172 | |||||
| 130R --> 130RR | 43 | 2 | CAM.00 | 0.105 | 0.199 | 0.199 | Indeterminate |
| Shared variant(s) | |||||||
| 36 --> 36R | 99 | 4 | CAM.07 | 0.035 | 0.131 | 0.007 | Relapse |
| CAM.02 | 0.066 | 0.239 | |||||
| CAM.01 | 0.061 | 0.223 | |||||
| 80 --> 80R | 56 | 10 | CAM.00 | 0.105 | 0.672 | 0.050 | Relapse |
| CAM.08 | 0.026 | 0.232 | |||||
| CAM.05 | 0.038 | 0.321 | |||||
| 125 --> 125R | 82 | 9 | CAM.02 | 0.066 | 0.459 | 0.459 | Indeterminate |
| 154 --> 154R | 64 | 3 | CAM.06 | 0.035 | 0.100 | 0.100 | Relapse |
a Listed in descending order of in-host frequency; dominant clone is bolded.
b Calculated as 1-(1-y)x for the recurrent patient with x variants and sharing a single variant of prevalence y.
c Calculated as the product of the reinfection probabilities for all shared variants.
d Recurrent genotypes with ≤10% chance of reinfection with the observed shared variants are classified as relapse.
Overall, based primarily on deep sequencing data, but using microsatellites to reclassify 2 indeterminate pairs, we concluded that over half (18/29) of the recurrent infections were caused by relapse (Supplementary Figure 3). There was no difference in time to recurrence in those recurrences classified as relapse versus not (median days to recurrence, 68 [range, 31–126] vs 82 [range, 17–115], respectively; P = .8).
DISCUSSION
The lack of genotyping tools to distinguish P. vivax relapses from reinfections in endemic settings has hindered efforts to evaluate antirelapse drugs like primaquine, and to evaluate the epidemiologic burden of infection due to relapses. While multiple studies have previously aimed to characterize relapses using highly discriminatory molecular markers, they invariably reach the conclusion that relapses frequently display novel genotypes compared to those detected initially, making it impossible to distinguish them from novel genotypes arising from new mosquito bites [12–15, 30, 31].
The appearance of novel genotypes even among known relapses has at least 3 plausible contributing explanations. First, persons living in endemic areas who have been exposed to multiple vivax infections over a lifetime likely harbor a diverse collection of hypnozoites within their liver [9, 32]. Reactivation of latent hypnozoites from this library of past infections can lead to emergence of variants not present in the most recently observed episode. Second, as demonstrated by the examples of minority variant expansion found within our cohort by deep sequencing, apparently novel clones at relapse may in fact have existed originally as minority variants below the limit of detection. In our patient with 3 recurrences, deep sequencing suggested successive relapses with a minority clone increasing in proportion at each relapse. However, the microsatellite picture was more confusing—only by examining minor alleles not originally counted could the same variants be appreciated through successive episodes, highlighting the loss of useful information when minority variants are missed (Figure 4). Finally, infections inoculated by a single mosquito bite have been shown to contain different but closely related genotypes, representing sporozoites that arose from meiotic recombination within the mosquito [33–36]. The detectability of these “sibling” genotypes likely varies at different samplings.
To address the above, we propose a relapse classification scheme that assumes that consecutive vivax malaria episodes arising from relapse will often contain heterologous hypnozoites, but should also be composed of recurring variants arising from the same latent hypnozoite reservoir within an individual. Our previous work using heteroduplex tracking assays suggested that variant overlap is common among relapses when minority variants are detected [16, 37, 38]. Similarly, in a comprehensive microsatellite genotyping study of Brazilians who developed relapse without re-exposure to a malaria-endemic setting, more homology was noted between consecutive relapse episodes when nondominant microsatellite alleles were taken into consideration [15].
Here, we build on these previous findings by using amplicon deep sequencing to highlight patterns of multivariant overlap that are unlikely to happen by chance. To detect this overlap, we targeted pvmsp1, an antigenic marker with great nucleotide diversity, to sensitively detect minority parasite clones. By evaluating pvmsp1 variant overlap, we identified over half of recurrence pairs as representing probable relapse. This is biologically plausible in a region where transmission is relatively low (on average, persons are exposed to <1 infected mosquito bite per year) but where local vivax strains are known to cause frequent relapse [9].
One could argue that microsatellites were found to be more discriminatory in our cohort and are also neutral to immune selection, and that these advantages outweigh the problem of less sensitive and specific detection of minority alleles. However, the very hypervariability of microsatellite repeats may render them less useful for comparing individual genotypes longitudinally within persons to look for recurring variants. We propose that different microsatellite alleles found at relapse may often represent closely related sibling parasite subpopulations that arose from the same mosquito inoculation [33]. Genotyping may variably detect microsatellite subclones that are different sizes, but nonetheless reactivate on a clonal basis and demonstrate the same antigenic clone (ie, pvmsp1). In other words, if microsatellite siblings are branches of the same tree, differential hypnozoite reactivation likely occurs at the tree level within a forest, instead of at the branch level. Such nuances can likely only be explored with whole-genome analysis of single clones within polyclonal relapsing infections [35, 39]. Direct comparison of such data with microsatellite and deep sequencing results would help clarify what level of clone differentiation is important for describing relapse phenotypes.
The major obvious limitation to our analyses is our reliance on samples from an endemic cohort, and therefore uncertainty arises regarding which of the recurrent infections were actually caused by relapse arising from hypnozoites, as opposed to reinfection from a mosquito bite or recrudescence of blood-stage parasites due to drug resistance. We feel that recurrence due to recrudescence was unlikely, as all subjects were treated with directly observed artemisinin-based combination therapy known to be highly effective against P. vivax and cleared their parasites rapidly [19]. On the other hand, reinfections from mosquito bites likely did occur. However, in an area of relatively low transmission, we expect that the majority of recurrences were due to relapse.
The lack of a gold standard to determine the source of recurrences is a great challenge to vivax genotyping studies in endemic areas. Our strategy of using local population diversity as a context for determining when recurring variants were not likely to occur by chance can be applied to settings where a significant amount of genetic diversity exists. It will underestimate the proportion of relapses, as a monoclonal relapse of a variant that may have been missed originally based on sampling variability, or because it arose from a historical infection, would not be classified as a probable relapse [13, 40]. However, the scheme can be refined based on a greater insight into the detectability of variants over time, the use of more than 1 sequenced marker, and a greater understanding of what level of variant differentiation correlates with relapse. While the outcomes of individual recurrences cannot be distinguished with absolute confidence, our method provides a framework for estimating the proportion of recurrences likely due to relapse. Otherwise, complicated design strategies are needed for the same purpose [41].
Future studies should employ deep sequencing at multiple loci, continue to explore how to integrate results from microsatellite markers, and apply these and whole-genome-based analyses to isolates collected in antirelapse trials where exposure is limited and relapse outcomes are more definitively known [31, 42, 43]. Ultimately, well-characterized relapsing parasites collected in field studies could be used in genome-wide association studies to understand any genetic determinants of relapse. Such an understanding would accelerate the development of effective antirelapse therapies and inform strategies that are needed to successfully eliminate vivax malaria. Our study, by characterizing genetic signatures of relapse, represents a first step toward this important goal.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We are indebted to the study volunteers, as well as the dedicated field staff at the Armed Forces Research Institute of Medical Sciences (AFRIMS) and the Cambodia National Malaria Program who carried out the clinical study. We thank Steve Meshnick for his review of the manuscript. The views expressed in this paper are those of the authors and do not represent the official position of the US Department of Defense.
Financial support. This work was supported by the National Institutes of Health (grants K08 AI110651 to J. T. L., R01 AI089819 to J. J. J., R01 AI099473-03 to J. A. B.) and the US Army Medical Materiel Development Activity, Fort Detrick, MD. J. T. L. was also supported by an American Society of Tropical Medicine and Hygiene (ASTMH)/Burroughs Wellcome Postdoctoral Fellowship in Tropical Infectious Disease.
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Mueller I, Galinski MR, Baird JK, et al. Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect Dis 2009; 9:555–66. [DOI] [PubMed] [Google Scholar]
- 2.Arnott A, Barry AE, Reeder JC. Understanding the population genetics of Plasmodium vivax is essential for malaria control and elimination. Malar J 2012; 11:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cotter C, Sturrock HJW, Hsiang MS, et al. The changing epidemiology of malaria elimination: new strategies for new challenges. Lancet 2013; 382:900–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wells TNC, Burrows JN, Baird JK. Targeting the hypnozoite reservoir of Plasmodium vivax: the hidden obstacle to malaria elimination. Trends Parasitol 2010; 26:145–51. [DOI] [PubMed] [Google Scholar]
- 5.White MT, Karl S, Battle KE, Hay SI, Mueller I, Ghani AC. Modelling the contribution of the hypnozoite reservoir to Plasmodium vivax transmission. Elife 2014; 3:e04692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baird JK, Leksana B, Masbar S, et al. Diagnosis of resistance to chloroquine by Plasmodium vivax: timing of recurrence and whole blood chloroquine levels. Am J Trop Med Hyg 1997; 56:621–6. [DOI] [PubMed] [Google Scholar]
- 7.Pukrittayakamee S, Clemens R, Chantra A, et al. Therapeutic responses to antibacterial drugs in vivax malaria. Trans R Soc Trop Med Hyg 2001; 95:524–8. [DOI] [PubMed] [Google Scholar]
- 8.Mayxay M, Pukrittayakamee S, Newton PN, White NJ. Mixed-species malaria infections in humans. Trends Parasitol 2004; 20:233–40. [DOI] [PubMed] [Google Scholar]
- 9.White NJ. Determinants of relapse periodicity in Plasmodium vivax malaria. Malar J 2011; 10:297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Douglas NM, Nosten F, Ashley EA, et al. Plasmodium vivax recurrence following falciparum and mixed species malaria: risk factors and effect of antimalarial kinetics. Clin Infect Dis 2011; 52:612–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smithuis F, Kyaw MK, Phe O, et al. Effectiveness of five artemisinin combination regimens with or without primaquine in uncomplicated falciparum malaria: an open-label randomised trial. Lancet Infect Dis 2010; 10:673–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Imwong M, Snounou G, Pukrittayakamee S, et al. Relapses of Plasmodium vivax infection usually result from activation of heterologous hypnozoites. J Infect Dis 2007; 195:927–33. [DOI] [PubMed] [Google Scholar]
- 13.Chen N, Auliff A, Rieckmann K, Gatton M, Cheng Q. Relapses of Plasmodium vivax infection result from clonal hypnozoites activated at predetermined intervals. J Infect Dis 2007; 195:934–41. [DOI] [PubMed] [Google Scholar]
- 14.Restrepo E, Imwong M, Rojas W, Carmona-Fonseca J, Maestre A. High genetic polymorphism of relapsing P. vivax isolates in northwest Colombia. Acta Trop 2011; 119:23–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Araujo FCF, de Rezende AM, Fontes CJF, Carvalho LH, Alves de Brito CF. Multiple-clone activation of hypnozoites is the leading cause of relapse in Plasmodium vivax infection. PLOS ONE 2012; 7:e49871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin JT, Patel JC, Kharabora O, et al. Plasmodium vivax isolates from Cambodia and Thailand show high genetic complexity and distinct patterns of P. vivax multidrug resistance gene 1 (pvmdr1) polymorphisms. Am J Trop Med Hyg 2013; 88:1116–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orjuela-Sánchez P, Sá JM, Brandi MCC, et al. Higher microsatellite diversity in Plasmodium vivax than in sympatric Plasmodium falciparum populations in Pursat, Western Cambodia. Exp Parasitol 2013; 134:318–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thanapongpichat S, McGready R, Luxemburger C, et al. Microsatellite genotyping of Plasmodium vivax infections and their relapses in pregnant and non-pregnant patients on the Thai-Myanmar border. Malar J 2013; 12:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lon C, Manning JE, Vanachayangkul P, et al. Efficacy of two versus three-day regimens of dihydroartemisinin-piperaquine for uncomplicated malaria in military personnel in northern Cambodia: an open-label randomized trial. PLOS ONE 2014; 9:e93138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parobek CM, Bailey JA, Hathaway NJ, Socheat D, Rogers WO, Juliano JJ. Differing patterns of selection and geospatial genetic diversity within two leading Plasmodium vivax candidate vaccine antigens. PLOS Negl Trop Dis 2014; 8:e2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altshuler D, Pollara VJ, Cowles CR, et al. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature 2000; 407:513–6. [DOI] [PubMed] [Google Scholar]
- 22.Rezende AM, Tarazona-Santos E, Fontes CJF, et al. Microsatellite loci: determining the genetic variability of Plasmodium vivax. Trop Med Int Health 2010; 15:718–26. [DOI] [PubMed] [Google Scholar]
- 23.Karunaweera ND, Ferreira MU, Hartl DL, Wirth DF. Fourteen polymorphic microsatellite DNA markers for the human malaria parasite Plasmodium vivax. Molecular Ecology Notes 2007; 7:172–5. [Google Scholar]
- 24.Sutton PL. A call to arms: on refining Plasmodium vivax microsatellite marker panels for comparing global diversity. Malar J 2013; 12:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Havryliuk T, Ferreira MU. A closer look at multiple-clone Plasmodium vivax infections: detection methods, prevalence and consequences. Mem Inst Oswaldo Cruz 2009; 104:67–73. [DOI] [PubMed] [Google Scholar]
- 26.Mideo N, Kennedy DA, Carlton JM, Bailey JA, Juliano JJ, Read AF. Ahead of the curve: next generation estimators of drug resistance in malaria infections. Trends Parasitol 2013; 29:321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Juliano JJ, Kwiek JJ, Cappell K, Mwapasa V, Meshnick SR. Minority-variant pfcrt K76T mutations and chloroquine resistance, Malawi. Emerging Infect Dis 2007; 13:872–7. [DOI] [PubMed] [Google Scholar]
- 28.Kwiek JJ, Alker AP, Wenink EC, Chaponda M, Kalilani LV, Meshnick SR. Estimating true antimalarial efficacy by heteroduplex tracking assay in patients with complex Plasmodium falciparum infections. Antimicrob Agents Chemother 2007; 51:521–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Norman GR, Streiner DL. Biostatistics: the bare essentials. Hamilton, Ontario: Bc Decker, 2008. [Google Scholar]
- 30.Orjuela-Sánchez P, da Silva NS, da Silva-Nunes M, Ferreira MU. Recurrent parasitemias and population dynamics of Plasmodium vivax polymorphisms in rural Amazonia. Am J Trop Med Hyg 2009; 81:961–8. [DOI] [PubMed] [Google Scholar]
- 31.Kim J-R, Nandy A, Maji AK, et al. Genotyping of Plasmodium vivax reveals both short and long latency relapse patterns in Kolkata. PLOS ONE 2012; 7:e39645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imwong M, Boel ME, Pagornrat W, et al. The first Plasmodium vivax relapses of life are usually genetically homologous. J Infect Dis 2012; 205:680–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nkhoma SC, Nair S, Cheeseman IH, et al. Close kinship within multiple-genotype malaria parasite infections. Proc Biol Sci 2012; 279:2589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conway DJ, Greenwood BM, McBride JS. The epidemiology of multiple-clone Plasmodium falciparum infections in Gambian patients. Parasitology 1991; 103(Pt 1):1–6. [DOI] [PubMed] [Google Scholar]
- 35.Bright AT, Manary MJ, Tewhey R, et al. A high resolution case study of a patient with recurrent Plasmodium vivax infections shows that relapses were caused by meiotic siblings. PLOS Negl Trop Dis 2014; 8:e2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sutton PL, Neyra V, Hernandez JN, Branch OH. Plasmodium falciparum and Plasmodium vivax infections in the Peruvian Amazon: propagation of complex, multiple allele-type infections without super-infection. Am J Trop Med Hyg 2009; 81:950–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Givens MB, Lin JT, Lon C, et al. Development of a capillary electrophoresis-based heteroduplex tracking assay to measure in-host genetic diversity of initial and recurrent Plasmodium vivax infections in Cambodia. J Clin Microbiol 2014; 52:298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andrianaranjaka V, Lin JT, Golden C, Juliano JJ, Randrianarivelojosia M. Activation of minority-variant Plasmodium vivax hypnozoites following artesunate+amodiaquine treatment in a 23-year old man with relapsing malaria in Antananarivo, Madagascar. Malar J 2013; 12:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nair S, Nkhoma SC, Serre D, et al. Single-cell genomics for dissection of complex malaria infections. Genome Res 2014; 24:1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koepfli C, Schoepflin S, Bretscher M, et al. How much remains undetected? Probability of molecular detection of human Plasmodia in the field. PLOS ONE 2011; 6:e19010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Betuela I, Rosanas-Urgell A, Kiniboro B, et al. Relapses contribute significantly to the risk of Plasmodium vivax infection and disease in Papua New Guinean children 1–5 years of age. J Infect Dis 2012; 206:1771–80. [DOI] [PubMed] [Google Scholar]
- 42.Sutanto I, Tjahjono B, Basri H, et al. Randomized, open-label trial of primaquine against vivax malaria relapse in Indonesia. Antimicrob Agents Chemother 2013; 57:1128–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Llanos-Cuentas A, Lacerda MV, Rueangweerayut R, et al. Tafenoquine plus chloroquine for the treatment and relapse prevention of Plasmodium vivax malaria (DETECTIVE): a multicentre, double-blind, randomised, phase 2b dose-selection study. Lancet 2014; 383:1049–58. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.