

Abstract
The development of gastrointestinal stromal tumors (GISTs) is largely driven by mutations in the KIT and PDGFRα genes. Imatinib mesylate is an oral small molecular tyrosine kinase inhibitor that mainly targets abl, c-KIT, and PDGFRα. Imatinib achieves disease control in approximately 70%–85% of patients with advanced GIST, and the median progression-free survival is 20–24 months. The efficacy of imatinib correlates with tumor kinase mutational status (exon 11 mutations mainly), and some mutations are known to be responsible for primary and secondary imatinib resistance. Beyond these, there are many other mutations that are considered rare and are associated with unknown clinical behavior. In the literature, there are poor and inconsistent data about the inhibitor sensitivity of mutations occurring in the activation-loop domain encoded by exon 17. In this article, we focus on a case of a patient suffering from GIST, harboring an extremely rare KIT activation-loop domain mutation (exon 17 mutation pN822K) treated with imatinib. A review of the literature is also presented.
Keywords: GIST, KIT activation-loop domain mutation, drug resistance, imatinib
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal tract.1 At diagnosis, majority of the tumors are resectable, but approximately 50% of these cases recur.2 The most common sites of primary disease are the stomach (60%) and small intestine (25%).3 Histologically, GISTs have a wide range of morphologies (from spindle cell to epithelioid), but the diagnosis requires an immunopositivity for KIT (CD117)4 or DOG1.5 Most GISTs, but not all, harbor mutations of KIT (approximately 75% of cases),6 or PDGFRα (approximately 5%–8%).7
Imatinib mesylate is an oral small molecular inhibitor of tyrosine kinases; it mainly targets abl, c-KIT, and PDGFRα.8 Imatinib achieves disease control in approximately 70%–85% of patients with advanced GIST, and the median progression-free survival is 20–24 months with a median survival of 5 years.9,10 The spectrum of activity of imatinib is strongly affected by the driver mutations that the tumor harbors and, in particular, the juxtamembrane domain mutations (encoded by exon 11) are the most sensitive, while the extracellular domain mutations (exon 9) require a higher dose of imatinib to achieve good clinical control.10 A subgroup analysis from the ACOSOG Z9001 Trial11 (1 year of adjuvant imatinib in a placebo-controlled trial in 645 patients) did not demonstrate that tumor mutation status independently affects relapse-free survival in either the placebo or imatinib arm. In detail, there was a clear benefit of adjuvant imatinib in patients with KIT exon 11 deletions (not KIT exon 11 insertions or point mutations).
However, drug resistance caused by secondary KIT or PDGFRA mutations eventually develops in 90% of cases.12 In addition to these mutations, there are many others that are considered rare and are associated with unknown clinical behavior. In this article, we report a case of a patient suffering from GIST harboring a rare KIT activation-loop domain mutation (exon 17 mutation pN822K) treated with imatinib.
Case report
In November 2009, a 48-year-old Philippine man without relevant comorbidities was admitted to an Asian hospital to undergo surgical resection of a high-risk ileal 10 cm GIST.
After surgery, he moved to Italy and came to our institute for a second opinion. Tumor tissues for the molecular analysis to sequence c-KIT were not available, so he was treated with a 1-year adjuvant treatment with imatinib at a daily dose of 400 mg daily, according to 2009 international guidelines.
On October 2012, a follow-up abdominal computed tomography (CT) scan detected a 45×32 mm unique local relapse, and treatment with 400 mg a day imatinib was administered again. A tumor biopsy was not performed due to the patient’s refusal.
A CT scan performed after 6 weeks from imatinib onset showed that the lesion increased in size, with no areas of reduced contrast enhancement. Imatinib dosage was then increased to 800 mg a day, but a subsequent CT scan performed after 6 weeks showed no signals of treatment response. The lesion was unique at CT scan and was amenable to radical surgery; on February 27, 2013, the patient underwent surgical disease excision. A large implant of recurrent GIST was visible on the peritoneal surface of the abdominal wall, 8.5 cm in longitudinal diameter. It was apparently increased with respect to the previous CT scan, despite the fact that the patient had not interrupted imatinib administration. An enlarged epiploic appendix of the sigmoid colon was removed for histology, and a peritoneal washing was performed for cytology. The recurrent lesion was eventually radically removed, together with the adherent omentum, taking care not to open the lining capsule surrounding it.
Macroscopically, the tumor was roundish and with a hard consistency; the maximum diameter was 8.5 cm. The cut surface was grayish and dishomogeneous for the presence of hemorrhagic areas. Histologically, the tumor was composed of bland spindle cells (Figures 1 and 2A). There were no areas of tumor necrosis. Many dilated and thrombosed vessels resembling similar findings seen in neurogenic tumors were intermingled within the tumor cells. Immunocytochemical stains revealed strong cytoplasmic expression of CD117, DOG1, and CD34 (Figure 2B and C). No expression was detected for desmin and S100 protein. c-KIT (exons 9, 11, 13, and 17) and PDGFRα (exons 12, 14, and 18) mutational analyses were performed by bidirectional Sanger sequencing, using BigDye Terminator chemistry, on a 3500 Dx Genetic Analyzer. The test results showed a single mutation in exon 17 of the c-KIT gene (pN822K; Figure 3), confirmed in two independent amplifications, while the PDGFRα mutational status was wild type. Based on the evidence of prior response to imatinib (no tumor shrinkage at instrumental evaluation and no pathological response at the histological report) and the evidence of this rare mutation, treatment with imatinib was not restarted and we decided to begin a clinical–instrumental follow-up every 3–4 months.
Figure 1.
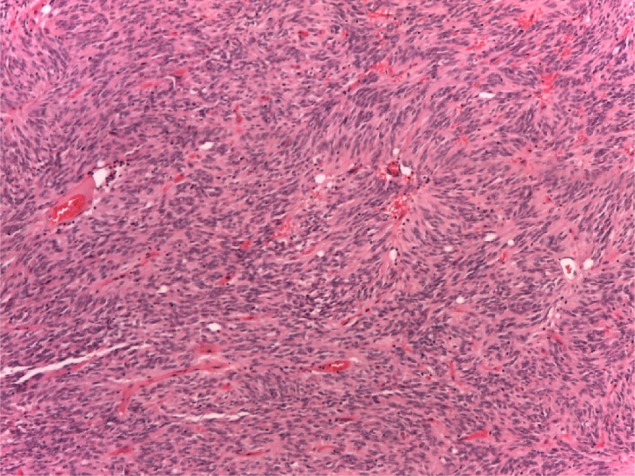
The tumor was composed of a monotonous spindle-cell proliferation.
Notes: Hematoxylin and eosin stain; original magnification: 10×.
Figure 2.
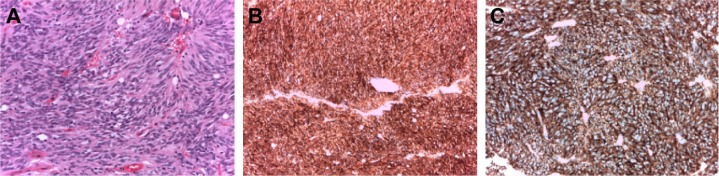
Gastrointestinal stromal tumor cell histology.
Notes: (A) Tumor cells were cytologically bland. No areas of necrosis or atypical mitoses were detected (hematoxylin and eosin stain; original magnification: 10×). (B) Tumor cells expressed strong and diffuse positivity for CD117 (KIT) (original magnification: 20×). (C) Tumor cells expressed strong and diffuse positivity for DOG-1 (original magnification: 20×).
Figure 3.

Electropherogram obtained by bidirectional Sanger sequencing of two independent amplifications, both showing a pN822K mutation.
At the time of this report, 18 months after surgical resection of the relapsed disease, the patient is still in complete remission.
Discussion
In this article, we have described the case of a patient affected by a GIST harboring an extremely rare KIT exon 17 mutation, pN822K, treated with imatinib. The description of this case has significant caveats because we cannot classify this mutation as intrinsic or acquired resistance, since the sample from the first surgery was not available to perform gene sequencing analyses. In our case, the mutation was associated with resistance to imatinib.
The development of GISTs is largely driven by mutations in the KIT and PDGFRα genes. KIT and PDGFRα secondary mutations, or nonsensitive primary mutations, represent the principal cause of resistance to imatinib; other mechanisms can involve the activation of different pathways (eg, k-ras and BRAF).6,13–17
Regarding the epidemiology, mutations affecting the kinase domain in untreated primary resected GISTs are very rare (each <1%), mainly occurring at codon 642 in exon 13 and at codons 816, 820, and 823 in exon 17.18 Table 1 depicts the frequency (0.4%–4.5%) of c-KIT exon 17 mutations, as reported by several retrospective series of imatinib-naïve GIST patients.19–27
Table 1.
Frequency of KIT exon 17 mutations in untreated patients with GIST from retrospective studies
| References | Patient (N) | Exon 17 mutations (N) | Type(s) of mutation | Primitive site |
|---|---|---|---|---|
| Subramanian et al19 | 26 | 1 (3.8%) | N822K | Gastric |
| Wasag et al20 | 28 | 1 (3.5%) | N822K | Duodenum |
| Rossi et al21 | 167 | 1 (0.6%) | Not reported | Not reported |
| Arne et al22 | 204 | 1 (0.5%) | Not reported | Not reported |
| Kern et al23 | 95 | 2 (2.1%) | N822K; G826E | Gastric |
| Calabuig-Fariñas et al24 | 154 | 3 (1.9%) | D816V; N822K; V823D | Not reported |
| Wozniak et al25 | 427 | 2 (0.4%) | N822K; N822H | Not reported |
| Kang et al26 | 22 | 1 (4.5%) | Not reported | Not reported |
| Joensuu et al27 | 1,505 | 10 (0.6%) | Not reported | 6/10 nongastric |
Abbreviations: GIST, gastrointestinal stromal tumor; N, number.
Very little data are available regarding imatinib sensitivity in GISTs harboring kinase-domain mutations. In a smaller study by Bozzi et al,28 it was found that two out 18 imatinib-treated patients had secondary exon 17 KIT mutations (N822K, D816H) associated with exon 11 KIT mutations (dup W577-R586, Ex11:V559A). In a Korean study of 290 patients,29 two patients were enrolled with primary exon 17 mutations (A794T and G812D), and both attained a partial response. However, the paper did not report the long-term outcomes of these patients. Conversely, the development of subsequent resistance to imatinib is more frequent, as was demonstrated by a meta-analysis of ten trials (>1,000 patients).30 It was shown that the prevalence of second-site KIT or PDGFRA mutations was 61.3%; specifically, the prevalence of exon 17 mutation was the most common, at 54.5%.
In a Phase II study of imatinib conducted with 147 patients with GIST,31 molecular analyses were performed using samples from ten patients with primary resistance and 33 patients with secondary resistance. Uncommon kinase mutations were most common in GISTs with secondary resistance when compared to those with primary resistance tumors (67% vs 10%, respectively; P=0.002).
Recently, a retrospective analysis of 93 patients treated with neoadjuvant imatinib32 showed that exons 9, 13, and 17 mutations were rare, and that they seem to confer a major risk of recurrence.
Two preclinical studies have attempted to predict the association between exon 17 mutations and inhibitor sensitivity, yielding contradictory results.33,34 The first study with a xenograft model of GIST harboring an exon 17 D816H mutation has shown a response to intermittent or continuous imatinib treatment;33 the second study explored the response of an exon 17 (A818T) knock-in mouse to imatinib, showing that it was refractory.34
In the literature, there are a few clinical reports about GISTs harboring exon 17 mutations showing a worse prognosis when treated with imatinib or sunitinib.35–38 In 2006, Loughrey et al35 described a case of a female patient affected by small-intestine GIST who progressed after 1 year of treatment of imatinib, and the mutational analysis performed over a peritoneal disease sample revealed a KIT exon 11, 21 amino acid deletion (identical to that found in the primary tumor) in association with an exon 17 point mutation resulting in an Asn822Lys substitution in the kinase domain. In 2009, a retrospective analysis showed that four out of ten patients refractory to imatinib harbored a mutation in exon 17 (T2467G).36 Subsequently, in another retrospective series, a different mutation in exon 17 (Y823D) was found in nine out 18 refractory patients.37
In 2010, a case was described of a patient with advanced gastric GIST harboring an exon 11 KIT mutation who, after a 3-year imatinib treatment, developed two new liver lesions harboring two different KIT exon 14 (c.2096C[T]) and KIT exon 17 (c.2554T[G]) mutations with dissociated clinical behavior (both failed to benefit from increasing imatinib dose, while the exon 14 lesion benefited from a sunitinib switch).38
Regarding the focus of our report, the exon 17 N822K mutation is extremely rare and, so far, it is associated with unpredictable clinical behavior. Table 2 summarizes the description of all case reports about GIST harboring KIT exon 17 mutation N822K. In 2009, Nishida et al39 reported that four patients with GIST harboring secondary/tertiary exon 17 mutations (D816H, W823D, D822K, plus N822K) did not benefit from sunitinib treatment. In 2011, Hanson et al40 described a complex case of a female patient affected from a small-intestine rhabdoid GIST harboring KIT exon 11 579–580 insertion. At the excision of the mass, peritoneal metastases were noted, and she was subsequently treated with 1 year of imatinib. Then, 3 years later, there was an abdominal recurrence harboring an exon 17 N822K mutation, together with an exon 11 mutation. After the excision of the mass, she was treated with 2 years of sunitinib in the absence of a macroscopic evidence of the disease. Later, a third recurrence was excised (sunitinib was given in absence of the macroscopic disease so it cannot be speculated if the disease was responsive to sunitinib) and this did not harbor an exon 17 mutation. The authors speculated that there was a preexisting clone harboring an exon 17 mutation that was selected following imatinib treatment. In 2013, a retrospective analysis of 38 patients who developed imatinib resistance were documented 12 cases with exon 17 mutations (including also N822K mutations), and all the patients with these secondary mutations failed to benefit from switching treatment to sunitinib.41
Table 2.
Summary of case reports focusing on KIT exon 17 pN822K mutation
| References | Patient (N) | Primitive site(s) | Type(s) of mutation | Refractory to |
|---|---|---|---|---|
| Nishida et al39 | 4 | Three small intestine; stomach | D816H; W823D; D822K; N822K | Imatinib; sunitinib |
| Hanson et al40 | 1 | Small intestine | N822K | Imatinib |
| Gao et al41 | 4 | Not reported | N822K | Imatinib; sunitinib |
| Singeltary et al42 | 1 | Rectum | N822K | Imatinib; sunitinib |
| Our case | 1 | Small intestine | N822K | Imatinib |
Abbreviation: N, number.
Interestingly, a second case of a 54-year-old white male with a rectal GIST harboring an N822K mutation at c-KIT exon 17 was described.42 The patient did not respond to imatinib (neither when administered at 400 mg daily nor at 800 mg daily), nor to sunitinib. After two lines of treatment, a third line was attempted with sorafenib, which resulted in a short-lasting partial response (progression with new lesions at 6-month treatment); so far, it was decided that imatinib be added to sorafenib, attaining 2-year disease stabilization.
This case report documented imatinib resistance in a patient with a rectal GIST harboring a c-KIT N8222K mutation, which was proven both by radiological disease progression and with the absence of pathological features of imatinib response.
Conclusion
In conclusion, this report suggests that patients with GIST harboring this rare mutation can be resistant to imatinib. Nowadays, sunitinib remains the standard of care for imatinib-refractory GISTs, regardless the status of their secondary KIT mutation. However, genotype analysis showed that patients with a secondary KIT mutation involving the activation-loop domain have poor progression-free survival and overall survival.43,44 Novel tyrosine kinase inhibitors (nilotinib, regorafenib, sorafenib) have been tested in preclinical and clinical settings.45–48 Two Phase III randomized trials (nilotinib, regorafenib)45,46 and two Phase II trials (sorafenib)47,48 have shown a modest benefit of these drugs in patients with imatinib/sunitinib-refractory GIST. Subgroup analyses of secondary mutations of KIT were not performed to check if these drugs could be active in the presence of activation-loop domain mutations. A rebiopsy should be encouraged to better define the mechanisms of acquired resistance, and an effort should be made to retrospectively and prospectively analyze the outcomes of patients with GIST harboring rare mutations treated in clinical trials.
Preclinical studies tested the sensitivity of imatinib-resistant GIST cells to PKC412 and found that KIT-V654A and PDGFRA-D842V mutants were sensitive to PKC412.49 Indeed, preclinical studies could help to select drugs active for loop domain mutations: a molecular modeling analysis showed that the fragment deletion of exon 11 and the point mutation on exon 17 would lead to a shift of KIT conformational equilibrium toward an active form, for which nilotinib and sorafenib bind with higher affinity than imatinib and sunitinib; thus far, they could be tested in these settings.50
Acknowledgments
The authors thank Mr William Russell-Edu for help with English editing. The authors’ local Ethical committee approved this retrospective study, and patients provided informed consent.
Footnotes
Disclosure
None of the authors, nor the European Institute of Oncology, received any payment or support in kind for any aspect of the submitted work. The authors report no conflicts of interest in this work.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
References
- 1.Nilsson B, Bümming P, Meis-Kindblom JM, et al. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era – a population-based study in western Sweden. Cancer. 2005;103(4):821–829. doi: 10.1002/cncr.20862. [DOI] [PubMed] [Google Scholar]
- 2.Corless CL. Gastrointestinal stromal tumors: what do we know now? Mod Pathol. 2014;27(Suppl 1):S1–S16. doi: 10.1038/modpathol.2013.173. [DOI] [PubMed] [Google Scholar]
- 3.Miettinen M, Lasota J. Gastrointestinal stromal tumors (GISTs): definition, occurrence, pathology, differential diagnosis and molecular genetics. Pol J Pathol. 2003;54(1):3–24. [PubMed] [Google Scholar]
- 4.Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998;152(5):1259–1269. [PMC free article] [PubMed] [Google Scholar]
- 5.West RB, Corless CL, Chen X, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol. 2004;165(1):107–113. doi: 10.1016/S0002-9440(10)63279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Debiec-Rychter M, Sciot R, Le Cesne A, et al. EORTC Soft Tissue and Bone Sarcoma Group. Italian Sarcoma Group. Australasian GastroIntestinal Trials Group KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42(8):1093–1103. doi: 10.1016/j.ejca.2006.01.030. [DOI] [PubMed] [Google Scholar]
- 7.Corless CL, Schroeder A, Griffith D, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23(23):5357–5364. doi: 10.1200/JCO.2005.14.068. [DOI] [PubMed] [Google Scholar]
- 8.Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 2000;96(3):925–932. [PubMed] [Google Scholar]
- 9.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 10.von Mehren M, Heinrich MC, Joensuu H, Blanke CD, Wehrle E, Demetri D. Follow-up results after 9 years (yrs) of the ongoing, phase II B2222 trial of imatinib mesylate (IM) in patients (pts) with metastatic or unresectable KIT+ gastrointestinal stromal tumors (GIST) [abstract] J Clin Oncol. 2011;29(Suppl) abstr 10016. [Google Scholar]
- 11.Corless CL, Ballman KV, Antonescu CR, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32(15):1563–1570. doi: 10.1200/JCO.2013.51.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127–1134. doi: 10.1016/S0140-6736(04)17098-0. [DOI] [PubMed] [Google Scholar]
- 13.Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342–4349. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 14.Debiec-Rychter M, Wasag B, Stul M, et al. Gastrointestinal stromal tumours (GISTs) negative for KIT (CD117 antigen) immunoreactivity. J Pathol. 2004;202(4):430–438. doi: 10.1002/path.1546. [DOI] [PubMed] [Google Scholar]
- 15.Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST) Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28(7):1247–1253. doi: 10.1200/JCO.2009.24.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heinrich MC, Owzar K, Corless CL, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26(33):5360–5367. doi: 10.1200/JCO.2008.17.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miranda C, Nucifora M, Molinari F, et al. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(6):1769–1776. doi: 10.1158/1078-0432.CCR-11-2230. [DOI] [PubMed] [Google Scholar]
- 18.Lasota J, Corless CL, Heinrich MC, et al. Clinicopathologic profile of gastrointestinal stromal tumors (GISTs) with primary KIT exon 13 or exon 17 mutations: a multicenter study on 54 cases. Mod Pathol. 2008;21(4):476–484. doi: 10.1038/modpathol.2008.2. [DOI] [PubMed] [Google Scholar]
- 19.Subramanian S, West RB, Corless CL, et al. Gastrointestinal stromal tumors (GISTs) with KIT and PDGFRA mutations have distinct gene expression profiles. Oncogene. 2004;23(47):7780–7790. doi: 10.1038/sj.onc.1208056. [DOI] [PubMed] [Google Scholar]
- 20.Wasag B, Debiec-Rychter M, Pauwels P, et al. Differential expression of KIT/PDGFRA mutant isoforms in epithelioid and mixed variants of gastrointestinal stromal tumors depends predominantly on the tumor site. Mod Pathol. 2004;17(8):889–894. doi: 10.1038/modpathol.3800136. [DOI] [PubMed] [Google Scholar]
- 21.Rossi S, Gasparotto D, Toffolatti L, et al. Molecular and clinicopathologic characterization of gastrointestinal stromal tumors (GISTs) of small size. Am J Surg Pathol. 2010;34(10):1480–1491. doi: 10.1097/PAS.0b013e3181ef7431. [DOI] [PubMed] [Google Scholar]
- 22.Arne G, Kristiansson E, Nerman O, et al. Expression profiling of GIST: CD133 is associated with KIT exon 11 mutations, gastric location and poor prognosis. Int J Cancer. 2011;129(5):1149–1161. doi: 10.1002/ijc.25755. [DOI] [PubMed] [Google Scholar]
- 23.Kern A, Görgens H, Dittert DD, et al. Mutational status of KIT and PDGFRA and expression of PDGFRA are not associated with prognosis after curative resection of primary gastrointestinal stromal tumors (GISTs) J Surg Oncol. 2011;104(1):59–65. doi: 10.1002/jso.21905. [DOI] [PubMed] [Google Scholar]
- 24.Calabuig-Fariñas S, López-Guerrero JA, Navarro S, et al. Evaluation of prognostic factors and their capacity to predict biological behavior in gastrointestinal stromal tumors. Int J Surg Pathol. 2011;19(4):448–461. doi: 10.1177/1066896911402327. [DOI] [PubMed] [Google Scholar]
- 25.Wozniak A, Rutkowski P, Piskorz A, et al. Polish Clinical GIST Registry Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST): Polish Clinical GIST Registry experience. Ann Oncol. 2012;23(2):353–360. doi: 10.1093/annonc/mdr127. [DOI] [PubMed] [Google Scholar]
- 26.Kang G, Lee J, Jang KT, et al. Multiplex mutation screening by mass spectrometry in gastrointestinal stromal tumours. Pathology. 2012;44(5):460–464. doi: 10.1097/PAT.0b013e3283559c45. [DOI] [PubMed] [Google Scholar]
- 27.Joensuu H, Rutkowski P, Nishida T, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33(6):634–642. doi: 10.1200/JCO.2014.57.4970. [DOI] [PubMed] [Google Scholar]
- 28.Bozzi F, Conca E, Manenti G, et al. High CD133 expression levels in gastrointestinal stromal tumors. Cytometry B Clin Cytom. 2011;80(4):238–247. doi: 10.1002/cyto.b.20589. [DOI] [PubMed] [Google Scholar]
- 29.Kang HJ, Ryu MH, Kim KM. Imatinib efficacy by tumor genotype in Korean patients with advanced gastrointestinal stromal tumors (GIST): The Korean GIST Study Group (KGSG) study. Acta Oncol. 2012;51(4):528–536. doi: 10.3109/0284186X.2011.636753. [DOI] [PubMed] [Google Scholar]
- 30.Lee JH, Kim Y, Choi JW, Kim YS. Correlation of imatinib resistance with the mutational status of KIT and PDGFRA genes in gastrointestinal stromal tumors: a meta-analysis. J Gastrointestin Liver Dis. 2013;22(4):413–418. [PubMed] [Google Scholar]
- 31.Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24(29):4764–4774. doi: 10.1200/JCO.2006.06.2265. [DOI] [PubMed] [Google Scholar]
- 32.Bednarski BK, Araujo DM, Yi M, et al. Analysis of prognostic factors impacting oncologic outcomes after neoadjuvant tyrosine kinase inhibitor therapy for gastrointestinal stromal tumors. Ann Surg Oncol. 2014;21(8):2499–2505. doi: 10.1245/s10434-014-3632-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Revheim ME, Kristian A, Malinen E, et al. Intermittent and continuous imatinib in a human GIST xenograft model carrying KIT exon 17 resistance mutation D816H. Acta Oncol. 2013;52(4):776–782. doi: 10.3109/0284186X.2013.770920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishikawa T, Nakai N, Liu NN, et al. In vivo effect of imatinib on progression of cecal GIST-like tumors in exon 17-type c-kit knock-in mice. Lab Invest. 2009;89(10):1161–1168. doi: 10.1038/labinvest.2009.78. [DOI] [PubMed] [Google Scholar]
- 35.Loughrey MB, Beshay V, Dobrovic A, Zalcberg J, Waring PM. Pathological response of gastrointestinal stromal tumour to imatinib treatment correlates with tumour KIT mutational status in individual tumour clones. Histopathology. 2006;49(1):99–100. doi: 10.1111/j.1365-2559.2006.02336.x. [DOI] [PubMed] [Google Scholar]
- 36.Wang CM, Fu H, Zhao GF, et al. Secondary resistance to imatinib in patients with gastrointestinal stromal tumors through an acquired KIT exon 17 mutation. Mol Med Rep. 2009;2(3):455–460. doi: 10.3892/mmr_00000121. [DOI] [PubMed] [Google Scholar]
- 37.Wang CM, Huang K, Zhou Y, et al. Molecular mechanisms of secondary imatinib resistance in patients with gastrointestinal stromal tumors. J Cancer Res Clin Oncol. 2010;136(7):1065–1071. doi: 10.1007/s00432-009-0753-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cameron S, Savvoukidis T, Armbrust T, et al. Analysis of a case with disappearance of the primary gastrointestinal stromal tumor and progressive liver metastases under long-term treatment with tyrosine kinase inhibitors. Med Oncol. 2010;27(2):213–218. doi: 10.1007/s12032-009-9193-0. [DOI] [PubMed] [Google Scholar]
- 39.Nishida T, Takahashi T, Nishitani A, et al. Japanese Study Group on GIST Sunitinib-resistant gastrointestinal stromal tumors harbor cis-mutations in the activation loop of the KIT gene. Int J Clin Oncol. 2009;14(2):143–149. doi: 10.1007/s10147-008-0822-y. [DOI] [PubMed] [Google Scholar]
- 40.Hanson JA, Trent JC, Yang D, Cooper K. Small-intestinal rhabdoid gastrointestinal stromal tumor (GIST): mutation analysis and clinical implications of a rare morphological variant. Int J Surg Pathol. 2011;19(5):653–657. doi: 10.1177/1066896911404413. [DOI] [PubMed] [Google Scholar]
- 41.Gao J, Tian Y, Li J, Sun N, Yuan J, Shen L. Secondary mutations of c-KIT contribute to acquired resistance to imatinib and decrease efficacy of sunitinib in Chinese patients with gastrointestinal stromal tumors. Med Oncol. 2013;30(2):522. doi: 10.1007/s12032-013-0522-y. [DOI] [PubMed] [Google Scholar]
- 42.Singeltary B, Ghose A, Sussman J, Choe K, Olowokure O. Durable response with a combination of imatinib and sorafenib in KIT exon 17 mutant gastrointestinal stromal tumor. J Gastrointest Oncol. 2014;5(1):E27–E29. doi: 10.3978/j.issn.2078-6891.2013.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–1338. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 44.Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–5359. doi: 10.1200/JCO.2007.15.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reichardt P, Blay JY, Gelderblom H, et al. Phase III study of nilotinib versus best supportive care with or without a TKI in patients with gastrointestinal stromal tumors resistant to or intolerant of imatinib and sunitinib. Ann Oncol. 2012;23(7):1680–1687. doi: 10.1093/annonc/mdr598. [DOI] [PubMed] [Google Scholar]
- 46.Demetri GD, Reichardt P, Kang YK, et al. GRID study investigators Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295–302. doi: 10.1016/S0140-6736(12)61857-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park SH, Ryu MH, Ryoo BY, et al. Sorafenib in patients with metastatic gastrointestinal stromal tumors who failed two or more prior tyrosine kinase inhibitors: a phase II study of Korean gastrointestinal stromal tumors study group. Invest New Drugs. 2012;30(6):2377–2383. doi: 10.1007/s10637-012-9795-9. [DOI] [PubMed] [Google Scholar]
- 48.Kindler HL, Campbell NP, Wroblewski K, et al. Sorafenib (SOR) in patients (pts) with imatinib (IM) and sunitinib (SU)-resistant (RES) gastrointestinal stromal tumors (GIST): final results of a University of Chicago Phase II Consortium trial [abstract] J Clin Oncol. 2011;29(Suppl) abstr 10009. [Google Scholar]
- 49.Debiec-Rychter M, Cools J, Dumez H, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128(2):270–279. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 50.Hsueh YS, Lin CL, Chiang NJ, et al. Selecting tyrosine kinase inhibitors for gastrointestinal stromal tumor with secondary KIT activation-loop domain mutations. PLoS One. 2013;8(6):e65762. doi: 10.1371/journal.pone.0065762. [DOI] [PMC free article] [PubMed] [Google Scholar]