

Abstract
BRCA1, with its binding partner BARD1, regulates the cellular response to DNA damage in multiple tissues, yet inherited mutations within BRCA1 result specifically in breast and ovarian cancers. This observation, along with several other lines of evidence, suggests a functional relationship may exist between hormone signaling and BRCA1 function. Our data demonstrates that AKT activation promotes the expression of BRCA1 in response to estrogen and IGF-1 receptor signaling. Further, we have identified a novel AKT phosphorylation site in BRCA1 at S694 which is responsive to activation of these signaling pathways. This rapid increase in BRCA1 protein levels appears to occur independently of new protein synthesis and treatment with the clinically utilized proteasome inhibitor bortezomib similarly leads to a rapid increase in BRCA1 protein levels. Together, these data suggest that AKT phosphorylation of BRCA1 increases total protein expression by preventing proteasomal degradation. AKT activation also appears to support nuclear localization of BRCA1, and co-expression of activated AKT with BRCA1 decreases radiation sensitivity, suggesting this interaction has functional consequences for BRCA1's role in DNA repair. We conclude that AKT regulates BRCA1 protein stability and function through direct phosphorylation of BRCA1. Further, the responsiveness of the AKT-BRCA1 regulatory pathway to hormone signaling may, in part, underlie the tissue specificity of BRCA1 mutant cancers. Pharmacological targets within this pathway could provide strategies for modulation of BRCA1 protein, which may prove therapeutically beneficial for the treatment of breast and ovarian cancers.
Keywords: BRCA1, Estrogen, Breast Cancer, AKT, Proteasomal degradation, Phosphorylation
Introduction
Germline mutations of BRCA1 and subsequent loss of heterozygosity are an important cause of familial breast and ovarian cancer syndromes (1, 2). Although mutations of BRCA1 are rare in sporadic cancer, a percentage of these cases exhibit decreased BRCA1 mRNA expression (3) suggesting that its loss may contribute to tumorigenesis in a proportion of non-hereditary cancers as well. BRCA1 is implicated in the regulation of a number of cellular processes including: DNA repair (4-7), cell cycle checkpoints (8, 9), and transcription (10-12). The function of BRCA1 is, in part, dependent on a direct interaction with BARD1. Both proteins possess N-terminal RING domains and C-terminal BRCT domains (13, 14), and the heterodimerization of the BRCA1 and BARD1 RING domains produces an E3 ubiquitin ligase activity (15, 16). Binding between these proteins may also serve to mutually regulate their nuclear localization (17) and stability (18). Importantly, this interaction is disrupted by several common mutations that occur in cancer patients (19).
Evidence from clinical epidemiology as well as laboratory animal models strongly suggests that tumorigenesis in BRCA1 mutation carriers is hormone dependent. First, oopherectomy in both humans (20) and mice (21) significantly decreases the incidence of cancers initiated by BRCA1 mutation. Second, tamoxifen decreases the risk of developing contralateral breast cancer in patients carrying BRCA1 mutations (20). Third, anti-progesterone therapy prevents tumorigenesis in Brca1 deficient mice (22). However, the majority of BRCA1 mutant tumors are hormone receptor negative (23). Furthermore, BRCA1 transcription is not directly responsive to activation of estrogen receptor (ER), as increases of BRCA1 mRNA are observed only in a delayed and indirect fashion related to proliferation (24, 25).
While ligand bound steroid receptors classically act as nuclear transcription factors, rapid activation of extranuclear cell signaling cascades by both estrogen and progesterone receptors have been described (26). Specifically, ER interacts with and activates PI3-kinase (27, 28), to result in activation of the serine/threonine kinase AKT. Furthermore, insulin-like growth factor receptor (IGFR) signaling, which potently activates AKT, has been implicated in cross-regulation of the ER signaling pathway (29). ER signaling appears to activate the IGFR signaling pathway (30), and conversely IGFR signaling also stimulates ER activity (31). Therefore, in response to hormone receptor signaling, activation of AKT is likely amplified by the convergence of the ER and IGFR signaling pathways.
Previous work has demonstrated that AKT phosphorylates BRCA1 on threonine 509 (32). We therefore chose to investigate whether rapid activation of AKT kinase activity by hormone stimulation could impact the expression and function of BRCA1. In addition to confirming threonine 509 as an AKT phosphorylation site, our findings suggest that AKT phosphorylates BRCA1 at a novel site at serine 694 following estrogen or IGF-1 stimulation. This phosphorylation of BRCA1 by AKT appears to correlate with a rapid stabilization of BRCA1 protein levels and enhanced cell survival following DNA damage.
Results
BRCA1 Protein Expression is Dependent on the PI3K/AKT Signaling Pathway
BRCA1 protein expression and phosphorylation are regulated in a cell cycle specific manner (33, 34). Consistent with these observations, we noted that BRCA1 protein levels were markedly decreased in hormone depleted culture conditions (Figure 1a) in the human breast carcinoma cell lines MCF7 and MDA-MB-231. The upper immunoreactive band in the MCF7 BRCA1 panel represents full length BRCA1 (∼220 kD). The lower band (∼150-180 kD) may represent one of the numerous splice variants of BRCA1 (35) or possibly the BRCA1-IRIS protein (36). These alternative protein products of the BRCA1 locus are independently regulated from the full length p220 protein, and this smaller protein was not consistently expressed in a manner similar to full-length BRCA1 in the experiments presented in this study. We further observed that the levels of activated AKT as assessed by immunoblot for phosphorylated AKT at serine 473 (pS473) consistently correlated with the levels of BRCA1 protein expression while total AKT was unchanged. Therefore we hypothesized that AKT signaling might play an important role in regulating BRCA1 protein expression. To test this, we cultured both MCF7 and MDA-MB-231 breast cell lines as well the human ovarian cancer cell line BG-1 in full serum conditions and treated the cells with the PI3-kinase inhibitor LY294002 at various doses for 18 hours. No changes in cell morphology or culture density were noted in any treatment group at the time of harvest. Furthermore, no Caspase-3 or PARP cleavage was noted in groups treated with the highest dose of LY294002, suggesting that this treatment did not induce apoptosis (Supplemental Figure 1). In all three cell lines, BRCA1 protein levels decreased in a dose-dependent manner with LY294002 treatment (Figure 1b). Also, the BRCA1 protein binding partner BARD1 demonstrated a similar decrease. Immunoblot for pS473-AKT indicated that inhibition of PI3K in these cells resulted in a predictable decrease in AKT activation, which correlated with the decreased BRCA1 protein expression. Since inhibition of PI3K potentially affects pathways other than AKT, we also utilized a direct inhibitor of AKT which prevents its activation in a PH-domain dependent manner (Calbiochem, AKT inhibitor VIII). Treatment of all three cell lines for 18 hours in full serum conditions led to a significant decrease in BRCA1 and BARD1 protein expression, which again correlated with decreased activation of AKT (Figure 1c).
Figure 1. Total BRCA1 protein expression is correlated with AKT activity.
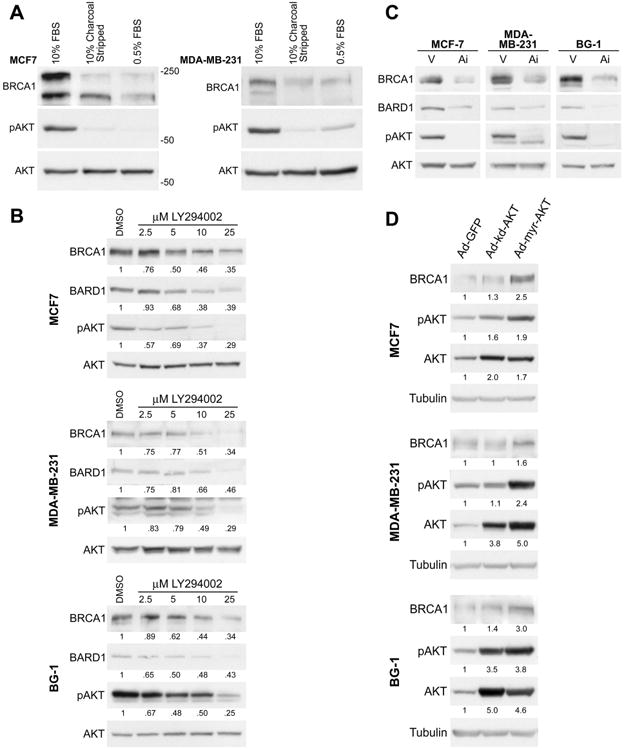
(A) MCF7 and MDA-MB-231 cells were cultured for either 48 hours in 10% charcoal stripped serum (CSS) or 24 hours in serum starved (0.5%) conditions. Control cells were cultured in normal growth medium (10% fetal bovine serum). Cell lysates (100 μg) were immunoblotted for BRCA1, pS473 AKT, and pan AKT. pS473 AKT indicates activation of AKT; pan AKT serves as a loading control. Molecular weights (kD) are indicated. The upper band in the BRCA1 panel for MCF7 cells is the full length (p220) BRCA1 protein. The lower band may represent a splice variant that is not consistently observed across experimental replicates or conditions.
(B) MCF7, MDA-MB-231, and BG-1 cells were cultured in normal growth medium and treated for 18 hours with the PI3-Kinase inhibitor LY294002 at the indicated doses or with vehicle (DMSO). Cell lysates were immunoblotted for BRCA1 and BARD1 proteins, which both decreased in a dose dependent manner in all three cell lines. pS473 AKT was detected to monitor inhibition of the PI3K-AKT signaling pathway; pan AKT levels indicate equal loading. Numbers below each panel represent relative densitometry values compared to the vehicle control.
(C) MCF7, MDA-MB-231, and BG-1 cells were cultured in normal growth medium, then treated for 18 hours with a direct AKT inhibitor (Ai) (5 μM, AKT inhibitor VIII, Calbiochem) or with methanol vehicle (V). Immunoblot for BRCA1 and BARD1 indicates that levels of both proteins decrease with treatment. The inhibitor directly prevents AKT phosphorylation (pS473 AKT immunoblot); pan AKT immunoblot demonstrates equal loading.
(D) MCF7, MDA-MB-231, and BG-1 cells were transduced with the indicated adenoviral vectors at MOI=50 and cultured in 10% CSS medium for 48 hours prior to harvest for immunoblot analysis with the indicated antibodies. Tubulin serves as a loading control. Numbers below each panel represent relative densitometry values compared to the Ad-GFP control.
Since both upstream and direct inhibition of AKT activation correlated with decreased levels of BRCA1 protein expression, we wished to more directly investigate whether AKT effects BRCA1 protein expression. Therefore we asked whether a constitutively activated form of AKT could increase BRCA1 protein expression. We utilized recombinant human adenovirus expressing either a myristolated form of AKT which is constitutively active (Ad-myr-AKT) or a kinase dead mutant of AKT (Ad-kd-AKT) (37). While the kd-AKT contains a K179M point mutation that disrupts the catalytic domain, phosphorylation of kd-AKT S473 can occur normally (38). Cells were cultured in hormone depleted medium to minimize the activation of endogenous AKT, but some phosphorylation of AKT S473 was observed in all groups, perhaps secondary to stimulation of PI3-K activity by adenoviral infection (39). Nonetheless, cells expressing the constitutively active myr-AKT were found to have increased BRCA1 protein levels in comparison to cells expressing either the kinase dead AKT or control GFP protein, suggesting that constitutively active AKT is directly correlated with relatively higher levels of BRCA1 protein expression (Figure 1d). Immunoblot for total AKT levels showed that both AKT transgenes were expressed at approximately equal levels in all three cell lines, and the transgene expression was approximately two to five-fold higher than expression of endogenous AKT, which migrates at the same molecular weight. The observation that constitutively active AKT can increase BRCA1 protein expression while kinase dead AKT cannot strongly suggested that the kinase activity of AKT is directly involved. In total, the results presented in Figure 1 indicate that activation of the PI3K-AKT signaling pathway is associated with increased levels of BRCA1 protein expression in both human breast and ovarian cells.
AKT Appears to Phosphorylate BRCA1 Protein in Cell Culture
As constitutively activated AKT was sufficient to increase levels of BRCA1 protein expression in hormone depleted culture of these cell lines, we next investigated whether AKT was directly phosphorylating BRCA1. To address this question, we utilized a commercially available polyclonal antibody to the phosphorylated form of the AKT substrate consensus sequence R-X-R-X-X-(pT/pS) (PAS antibody, Cell Signaling). Whole cell lysates were prepared from MCF7 cells cultured in medium containing full serum and varying doses of LY294002, similar to the experiments presented in Figure 1b. Immunoblot analysis using the PAS antibody demonstrated a strong immunoreactive potential substrate at approximately 220 kd which decreased in a dose dependent manner with LY294002 treatment (Figure 2a). Reprobing the membrane with a monoclonal antibody to BRCA1 resulted in detection of a substrate migrating at the same molecular weight, which similarly decreased in a LY294002 dose dependent manner. This finding suggested that the identity of the protein recognized by the PAS antibody at this molecular weight could be BRCA1. The immunoblot for pAKT confirmed that AKT activation was inhibited in this experiment.
Figure 2. AKT appears to phosphorylate both endogenous and exogenous BRCA1 protein in cell culture.
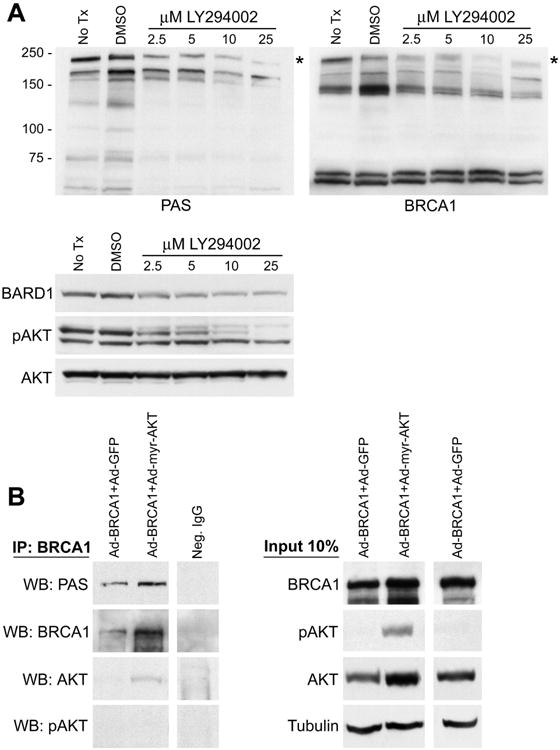
(A) MCF7 cells cultured in normal growth medium were treated with the indicated doses of LY294002 for 18 hours and then harvested for immunoblot analysis. Controls include both untreated cells and DMSO vehicle treated cells. Molecular weights (kD) are indicated. The immunoblot was probed with rabbit polyclonal phospho-AKT-substrate antibody (PAS), then stripped and reprobed with a mouse monoclonal BRCA1 antibody. An immunoreactive band was observed (*) of the same molecular weight with both antibodies which decreased in a dose-dependent manner upon LY294002 treatment. Immunoblots for BARD1, pS473 AKT, and pan AKT are presented in the third panel. The pS473 AKT antibody bound to a cross-reactive band of lower molecular weight in this experiment which was not recognized by the pan AKT antibody.
(B) MCF7 cells were transduced with the indicated combinations of adenovirus vectors. MOI=10 for Ad-BRCA1; MOI=50 for Ad-myr-AKT and control Ad-GFP. Following transduction the cells were cultured in CSS medium for 48 hours prior to harvest. Immunoprecipitation of BRCA1 was performed with two monoclonal anti-BRCA1 antibodies and then samples were immunoblotted with the indicated antibodies. Control immunoprecipitation was performed using non-specific IgG with lysate from cells transduced with Ad-BRCA1+Ad-GFP. 10% of the lysate used for immunoprecipitation was analyzed by immunoblot as an input control. The myr-AKT protein runs at a similar molecular weight as the endogenous protein. Tubulin indicates equal loading.
To further investigate whether the target protein at 220 kd was indeed BRCA1 we expressed exogenous BRCA1 with either myr-AKT or a GFP control protein using adenoviral vectors in MCF7 cells cultured in hormone depleted medium. Immunoprecipitation using pooled BRCA1 monoclonal antibodies followed by immunoblot with the PAS antibody demonstrated an immunoreactive band at ∼220 kD, suggesting the identity of this substrate was BRCA1 (Figure 2b). Reprobing the membrane with BRCA1 antibodies further suggested the identity of this band was BRCA1. Small amounts of AKT also appeared to co-immunoprecipitate with BRCA1, demonstrating that there may be an interaction between these two proteins. A control immunoprecipitation was performed with non-specific IgG utilizing protein lysate from the Ad-BRCA1+Ad-GFP group. Over-expression of exogenous BRCA1 was utilized in this experiment because the immunoprecipitation conditions were optimized for specificity at the expense of efficiency in the purification of BRCA1 from the sample. It should be noted that exogenous BRCA1 expressed from the RGD-modified adenoviral vector reaches much higher total protein levels than endogenous BRCA1 (40). Therefore, co-expression of myr-AKT does not appear to significantly enhance total levels of exogenous BRCA1 compared to control (Figure 2b, input lanes), in contrast to the ability of myr-AKT to significantly enhance endogenous BRCA1 expression (Figure 1d). Furthermore, transient activation of endogenous AKT by adenoviral infection leads to some phosphorylation of BRCA1 protein in control Ad-BRCA1+Ad-GFP samples. Therefore, to confirm that the epitope on BRCA1 recognized by the PAS antibody truly is phosphorylated, we treated samples from these experimental groups with λ-phosphatase prior to immunoblot analysis. This treatment abolished the signal produced by the PAS antibody without affecting the signal from the total BRCA1 antibody (Supplemental Figure 2), indicating that recognition of the epitope by the PAS antibody is indeed phospho-specific. Overall, these results strongly suggest that AKT phosphorylates BRCA1 in vivo and this phosphorylation appears to contribute to AKT regulation of BRCA1 protein levels.
AKT Directly Phosphorylates BRCA1 in vitro at both S694 and T509
We wished to further examine phosphorylation of BRCA1 by AKT. Therefore, we performed in vitro kinase reactions using purified full length BRCA1 and purified active AKT1. Figure 3a demonstrates that full length BRCA1 is phosphorylated by AKT in vitro. This finding is consistent with a previous report that suggested full length BRCA1 is phosphorylated by AKT and identified T509 as the target residue using GST fusion proteins (32). We were interested whether additional sites of phosphorylation existed in BRCA1 and therefore analyzed human and mouse BRCA1 protein sequences using Scansite (http://scansite.mit.edu) for other possible AKT consensus sites that appeared to be conserved. The three AKT consensus sites of greatest probability identified were: T509, T696, and T1246. We noted that T696 was not conserved in the aligned mouse sequence, but that nearby S694 of human BRCA1 was conserved and was also identified by Scansite as a potential AKT phosphorylation site. Therefore, we constructed GST-fusion proteins containing approximately 200 amino acids of the human BRCA1 protein sequence spanning T509, S694/T696, or T1246 and utilized these purified fusion proteins in an in vitro AKT kinase assay. Results of this experiment showed that both the T509 and S694/T696 constructs were strongly phosphorylated, while T1246 was not (Figure 3b). As the T509 site had been previously identified by others, we produced a fusion protein containing a T509A point mutation which was not phosphorylated in vitro by AKT, confirming that T509 is the site of phosphorylation in this construct. Due to the proximity of the S694 and T696 sites we chose not to produce point mutations of these residues, hypothesizing that a point mutation within the AKT consensus sequence might disrupt phosphorylation of the other possible site as well. Therefore, we instead analyzed the S694/T696 fusion protein by electrospray mass spectrometry and identified the conserved S694 residue as a site of phosphorylation by AKT (Figure 3c).
Figure 3. AKT phosphorylates BRCA1 at both S694 and T509.
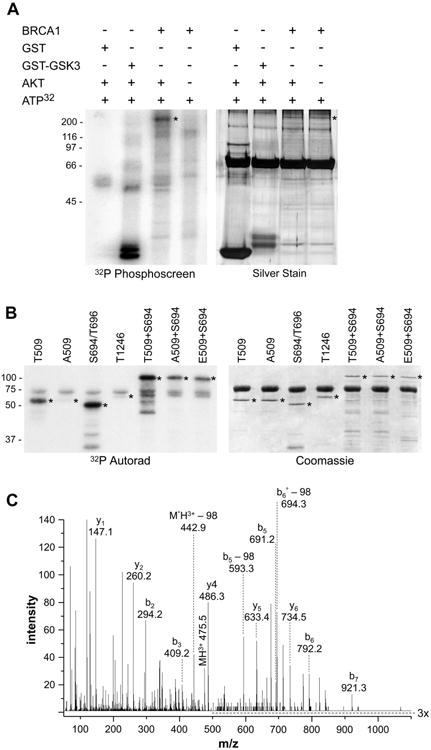
(A) Full length, purified BRCA1 protein (250 ng/lane) was added to an in vitro kinase assay plus or minus active AKT1 kinase. GST and GST-GSK3 were included as negative and positive controls respectively. Molecular weights (kD) are indicated. Reaction products were separated by SDS-PAGE and 32P-labelled products were detected by exposure to a phosphor screen. Silver stain demonstrated equal loading of BRCA1 (*) in the experimental lanes. The predominant band in all lanes is BSA (a component of the enzyme dilution buffer).
(B) GST-BRCA1 fusion proteins (1 μg) which encompass the indicated potential AKT phosphorylation sites were added to an AKT1 kinase assay and phospho-proteins were detected by 32P-autoradiography (*). Coomassie staining demonstrated equal amounts of all fusion proteins in the kinase reactions (*). The predominant band in all lanes is BSA. Molecular weights (kD) are indicated.
(C) Tandem mass spectra used to identify phosphopeptide RHD(pS)DTFPELK from GST-BRCA1-S694/T696. Eleven unique peptides were used to identify the protein with an expect value of 2.9*10-9 (MS rms error of 44 ppm). The precursor, b and y ions are labeled above. Ions b5, b5-98, and y6 indicate serine phosphorylation. ° symbolizes loss of water. The m/z range of 500 to 1100 is magnified 3 times for better visualization.
We also wished to test whether phosporylation at the T509 site affected the efficiency of phosphorylation at the novel S694 site by AKT in vitro. We created GST fusion proteins containing 400 amino acid segments of human BRCA1 which encompassed the S694 site with a wild-type T509 site, a T509A point mutant which cannot be phosphorylated, or a T509E point mutant which mimics the charge created by phosphorylation at the 509 position. The A509+S694 and E509+S694 constructs were both phosphorylated by AKT equally, suggesting that neither mutation affected the ability of S694 to be phosphorylated (Figure 3b). By densitometry analysis, the intensity of these signals was approximately equal to one half of the signal obtained with the wild-type T509+S694 construct, which is the expected result as the mutant constructs can only incorporate one half the amount of 32P as the wild-type construct. This result suggests that prior phosphorylation at T509 is not required for efficient phosphorylation of the S694 site by AKT in vitro.
AKT Phosphorylates Endogenous BRCA1 at S694 in Response to Hormone Stimulation
After identifying a potentially novel AKT phosphorylation site within BRCA1 we then generated a rabbit polyclonal antibody specific to the phosphorylated epitope at S694. Initial characterization of this antibody by immunoblot analysis of MCF7 whole cell lysates revealed immunoreactivity with a band of approximately 220 kD, which was parallel to the band recognized by a well-characterized, commercially available anti-BRCA1 antibody (Figure 4a, lanes 1-3). Further, pre-incubation of the pS694 antibody with the phosphorylated peptide antigen prior to incubation with the PVDF membrane largely blocked the signal from this band (Figure 4a, lanes 4-5). Immunoprecipitation using pooled monoclonal antibodies towards BRCA1 followed by immunoblot with the pS694 antibody demonstrated an immunoreactive band at the correct molecular weight (Figure 4b), and reprobing this membrane with a pan-BRCA1 antibody recognized the same band. Finally, λ-phosphatase treatment of lysates prior to SDS-PAGE abrogated the pS694 antibody signal while total BRCA1 levels were equal between treated and untreated lysates (Figure 4c). Together, this data indicates that the pS694-BRCA1 antibody exhibits reasonable phospho-specificity for BRCA1.
Figure 4. Characterization of pS694 BRCA1 polyclonal antibody.
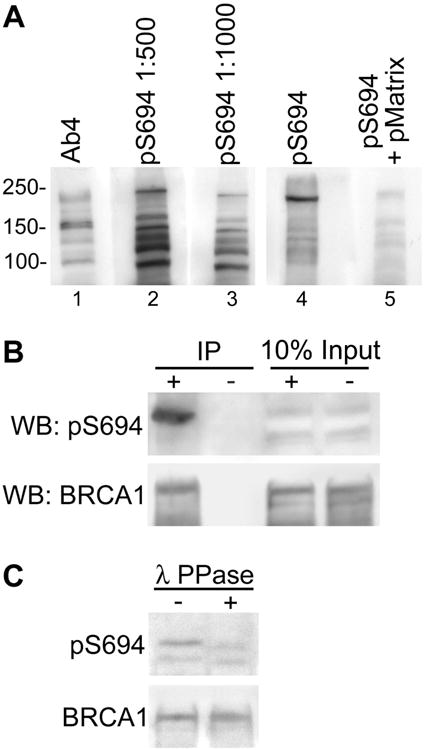
(A) Whole cell extracts were prepared from MCF7 cells and separated by SDS-PAGE. After transfer, the PVDF membrane was split longitudinally and each lane was incubated separately with the indicated antibodies: mouse monoclonal anti-BRCA1 (Ab4) and rabbit polyclonal anti-pS694 BRCA1 (at dilutions of 1:500 and 1:1000). Additionally, pS694 BRCA1 antibody at a dilution of 1:500 was incubated with the membrane either without or with pre-incubation with the specific phosphorylated antigen matrix (lanes four and five respectively) as a control for specificity of the antibody. Separately incubated lanes (1-3 and 4-5) were reassembled and exposed to the same piece of film for chemiluminescent detection. Molecular weights (kD) are indicated.
(B) MCF7 whole cell lysates were prepared for immunoprecipitation with two monoclonal BRCA1 antibodies (+) or with non-specific IgG control (-). Immunoblot was performed first with pS694 BRCA1 antibody and then the membrane was stripped and reprobed with pan BRCA1 antibody. Ten percent of the total lysate used for immunoprecipitation was run as a loading control (input).
(C) MCF7 whole cell lysates were prepared in lysis buffer without phosphatase inhibitors. Lysates were treated with λ-phosphatase (+) or not (-) and then immunoblotted with pS694 BRCA1 antibody. The membrane was stripped and reprobed with pan BRCA1 antibody.
We hypothesized that stimulation of breast and ovarian cells with hormones that regulate normal physiology and proliferation might lead to activation of AKT and downstream phosphorylation of BRCA1 at S694. To test this we stimulated MCF7, MDA-MB-231, and BG-1 cells with either estradiol (E2) or insulin-like growth factor 1 (IGF-1) over a brief time course (0.5 to 4 hours) before collecting cell lysates for immunoblot. Figure 5a demonstrates that E2 stimulation of the ER-positive MCF7 cell line leads to a rapid increase in total BRCA1 and BARD1 protein levels. We also observed similar increases in the levels of phosphorylation at S694 of BRCA1 which correlated with the total levels of BRCA1 protein. Again, the increase of BRCA1 protein occurred concurrently with activation of AKT, as evidenced by increased pAKT levels.
Figure 5. Hormone stimulation results in rapid phosphorylation and increased BRCA1 protein levels.
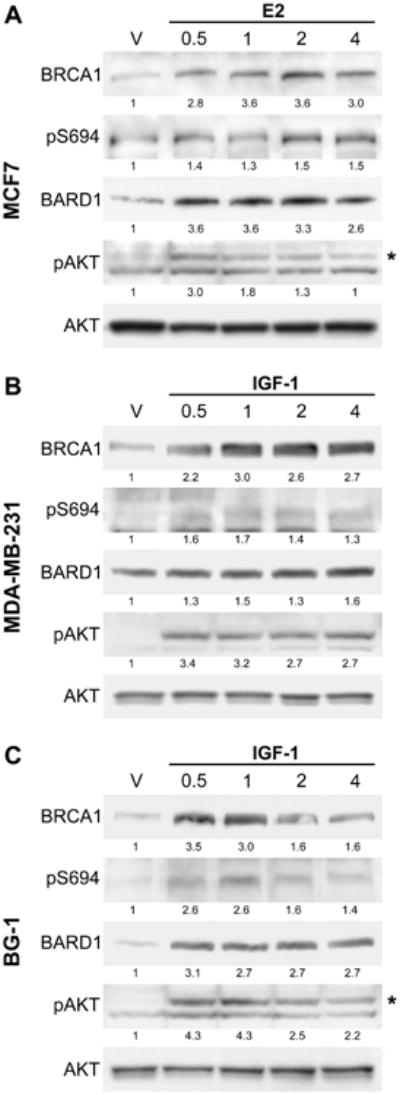
(A) Time course experiment of MCF7 cells incubated for 48 hours in 10% CSS medium and then stimulated with 10 nM of estradiol (E2) for the indicated times (in hours) prior to harvest. Control ethanol vehicle (V) treatment was performed for 4 hours. Whole cell lysates were immunoblotted with the indicated antibodies. The upper band (*) seen in the pS473 AKT antibody panel is specifically recognized by the pan AKT antibody; the lower band is a cross-reaction. Numbers below each panel represent relative densitometry values compared to the vehicle control.
(B,C) Time course experiment of MDA-MB-231 cells (B) or BG-1 cells (C) incubated for 24 hours in serum starved medium and then stimulated with 100 ng/mL of recombinant IGF-1 for the indicated times (in hours). Control BSA vehicle (V) treatment was performed for 4 hours. Whole cell lysates were immunoblotted with the indicated antibodies. The upper band (*) seen in the pS473 AKT antibody panel for BG-1 cells is specifically recognized by the pan AKT antibody; the lower band is a cross-reaction.
Stimulation of ER-negative MDA-MB-231 and ER-positive BG-1 cells with IGF-1 similarly resulted in a rapid increase of BRCA1 and BARD1 protein levels, which coordinated with increased pS694 signal and AKT activation (Figure 5b, 5c). We also attempted to stimulate the ER-positive BG-1 ovarian cell line with E2. However, this treatment did not activate AKT and subsequently did not increase BRCA1 protein levels (data not shown), suggesting that the ability of estrogen receptor to stimulate the AKT pathway may be cell line and perhaps tissue specific. Overall, these results indicate that hormone stimulation of both breast and ovarian derived cell lines leads to rapid activation of AKT and phosphorylation of endogenous BRCA1 at S694, concurrent with a rapid increase of total BRCA1 protein levels.
Rapid Estrogen Stimulation of Increased BRCA1 and BARD1 Protein Levels Occurs Through Post-translational Mechanisms Requiring AKT
We then chose to further investigate the mechanism of the rapid increase in BRCA1 and BARD1 protein levels in the MCF7 cell line following E2 treatment. To account for changes in BRCA1 protein levels that might be associated with proliferation (33) and indirect transcriptional upregulation of BRCA1 mRNA by estrogen signaling (24, 41) we modified our time course, adding an extended 24 hour time point to the early (0.5 h) and intermediate (4 h) time points. Estradiol treatment consistently induced a two to four-fold increase of BRCA1 and BARD1 protein levels by one half hour (Figure 6a, 6b). In additional experiments, we demonstrated that E2 stimulation leads to appreciable increases in phospho-AKT within 5 minutes, with peak activation routinely observed at 30 minutes, and with a return to baseline levels variably occurring between one and two hours (Supplemental Figure 3). To demonstrate that the E2 effect on BRCA1 protein levels was mediated by the AKT pathway, we pretreated cells with LY294002 for one hour prior to stimulation with E2. This treatment blocked the rapid activation of AKT observed at the 0.5 hour time point, and resulted in a complete lack of BRCA1 and BARD1 protein increase (Figure 6a). Conversely, pretreatment with inhibitors of MAP-Kinase pathways had no effect on the rapid accumulation of BRCA1 and BARD1 protein following estrogen treatment (Supplemental Figure 4), suggesting that this effect specifically requires the AKT pathway and not other mitogen stimulated pathways.
Figure 6. Rapid estrogen stimulation of increased BRCA1 and BARD1 protein levels requires AKT activation but not new protein synthesis.
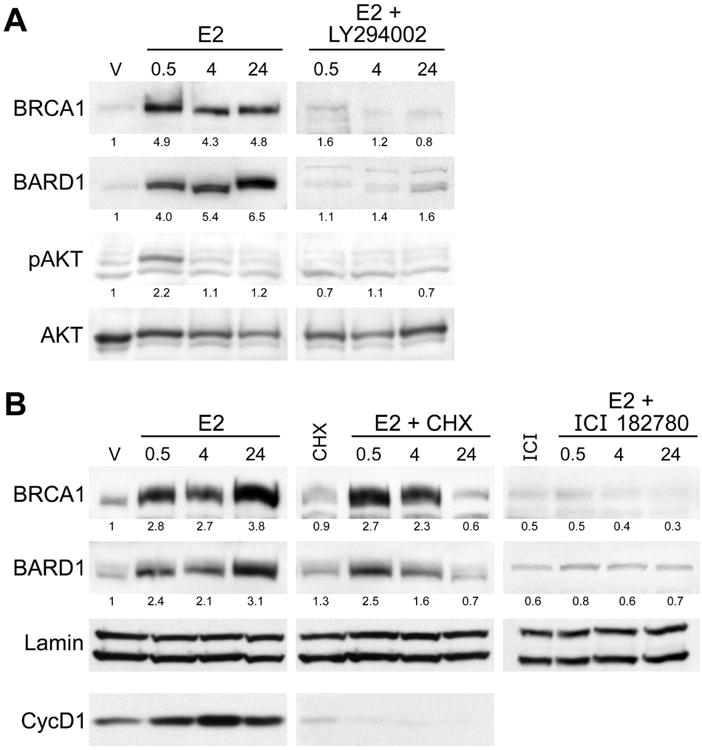
(A) MCF7 cells cultured in 10% CSS medium were stimulated with 10 nM E2 or pretreated with 10 μM LY294002 for 1 hour before stimulation with E2 for indicated amount of time (in hours). Immunoblot for BRCA1 and BARD1 proteins demonstrates that pre-incubation with LY294002 blocks the increase in total protein levels observed with E2 treatment alone. Samples treated with LY294002 alone showed similar levels of BRCA1 and BARD1 expression as the E2 + LY294002 groups (data not shown). pS473-AKT was detected to monitor AKT activation. Numbers below each panel represent relative densitometry values compared to the vehicle control.
(B) Immunoblots for indicated proteins from MCF7 cells cultured for 48 hours in CSS medium and then stimulated with 10 nM estrogen (E2) for the indicated amount of time (in hours) or with ethanol as vehicle control (V). Cells were pretreated with either 25 μM cycloheximide (CHX) or 1 μM ICI 182780 for 1 hour prior to addition of E2. Control groups were treated with inhibitor alone and matched to the 0.5 hour time point. Cyclin D1 was analyzed only in the E2 and E2+CHX treated samples as a control for the effectiveness of CHX treatment. Lamin immunoblot indicates equal loading. Numbers below each panel represent relative densitometry values compared to vehicle control.
The rapid increase of BRCA1 and BARD1, which requires AKT activation, and the association of this increase with AKT-dependent phosphorylation of pS694 on BRCA1 all suggest that the mechanism involves primarily post-translational regulation. To test this hypothesis and to rule out new protein synthesis as the mechanism of BRCA1 and BARD1 protein increase, we performed a similar pretreatment experiment with the protein synthesis inhibitor cycloheximide (CHX) prior to E2 stimulation. Interestingly, this treatment did not block the rapid accumulation of BRCA1 and BARD1 proteins at 0.5 and 4 hours, but did abrogate expression at 24 hours of stimulation demonstrating that the rapid changes in BRCA1/BARD1 protein levels following E2 treatment occur independently of new protein synthesis (Figure 6b). We also pretreated cells with the pure ER antagonist ICI 182780 (fulvestrant) and blocked the accumulation of BRCA1 and BARD1 at all time points, establishing that the mechanism involves E2 activation of the estrogen receptor. Finally, qPCR analysis indicated that E2 treatment did not increase BRCA1 or BARD1 mRNA at early time points (Supplemental Figure 5) and flow cytometry showed that cell cycle progression was not noted until after 8 hours of E2 treatment (Supplemental Figure 6). These results indicate that BRCA1 accumulation at early time points is not due to ER-mediated transcription or cell cycle progression. In total, this data supports the conclusion that estrogen receptor signaling in MCF7 cells leads to a rapid activation of the AKT signaling pathway, leading to phosphorylation of BRCA1 and subsequently increased protein stability resulting in an increased level of total protein. This mechanism is consistent with the non-genomic activation of several cell signaling pathways by ligand-bound estrogen receptor (26, 28, 42). These observations suggest that the half-life of BRCA1 in hormone depleted conditions is very short, and that E2 signaling leads to a rapid and dramatic increase in protein stability. In support of this, we found that the half-life of BRCA1 protein in MCF7 cells following E2 stimulation is approximately 6 hours (Supplemental Figure 7).
Proteasome Inhibition Results in Rapid Accumulation of BRCA1 Protein
We next sought to determine if constitutive degradation of BRCA1 and BARD1 by the 26S proteasome was responsible for the constitutively low levels of these proteins in cells cultured in hormone depleted medium. MCF7 cells cultured in hormone depleted medium were treated with the proteasome inhibitor MG132 for either 0.5, 4, or 8 hours. This treatment resulted in accumulation of both BRCA1 and BARD1 proteins beginning within a half hour after the addition of MG132 (Figure 7a), similar to the effect seen after estrogen treatment. We noted in preliminary experiments that MG132 treatment led to the dramatic activation of AKT, confounding whether the accumulation of BRCA1 and BARD1 was due to proteasome inhibition or to activation of AKT. Therefore, we co-treated cells with LY294002 and MG132 to prevent AKT activation, and found that BRCA1 and BARD1 protein levels increased rapidly under these conditions as well. This result indicates that prevention of proteasome-mediated degradation of BRCA1 and BARD1 appears to be downstream of AKT activity. We also tested whether a proteasome inhibitor used clinically, bortezomib (Velcade), could restore BRCA1 and BARD1 protein in a similar experiment. Both doses of bortezomib tested rapidly increased BRCA1 and BARD1 expression, and only minor activation of AKT was observed by 8 hours (Figure 7b). These results demonstrate that treatment with two individual proteasome inhibitors rapidly increases BRCA1 and BARD1 protein levels. The timing of this increase in protein levels is similar to results observed after estrogen treatment leading to activation of AKT. The fact that proteasome inhibition can similarly lead to increased BRCA1 protein levels regardless of the activation of AKT strongly suggests that this mechanism functions downstream of AKT signaling to regulate BRCA1 protein expression.
Figure 7. Proteasome inhibition rapidly increases BRCA1 protein levels.
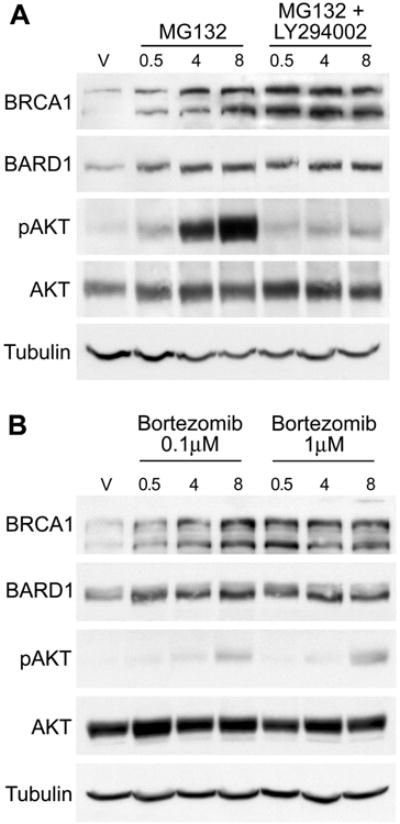
(A) MCF7 cells were cultured in CSS medium for 48 hours prior to treatment with 20 μM MG132 alone or in combination with 10 μM LY294002 for the indicated amount of time (in hours). Control cells were treated with EtOH vehicle (V) alone. Cell lysates were immunoblotted with the indicated antibodies. Tubulin is included as an additional loading control.
(B) Similarly cultured MCF7 cells were treated with the clinically utilized proteasome inhibitor bortezomib at either 0.1 μM or 1 μM for the indicated times prior to harvest for immunoblot analysis.
AKT Activity Appears to Support Nuclear Accumulation of BRCA1
BRCA1 is a nuclear localized protein that forms discrete foci during S-phase and following DNA damage (43, 44). Hinton et al. (45) have demonstrated that a BRCA1 T509A mutant protein is inefficiently transported into the nucleus, suggesting that AKT phosphorylation of BRCA1 might regulate its nuclear translocation. To further address whether AKT activity affected BRCA1 subcellular localization, we performed immunofluorescence studies using MCF7 cells cultured in hormone depleted medium that had been transduced with adenoviral vectors expressing activated AKT (Ad-myr-AKT), kinase dead AKT (Ad-kd-AKT), or control vector (Ad-LacZ). We performed the experiment either with or without the co-expression of exogenous wild-type BRCA1 (Ad-BRCA1). The cells were stained with two independent anti-BRCA1 antibodies to confirm specificity of the staining for BRCA1 (46), and within each experiment 200 cells from each transduction group were scored for expression of BRCA1 as nuclear, cytoplasmic, mixed, or absent. Expression of myr-AKT, but not kd-AKT or LacZ, restored significant nuclear expression of endogenous BRCA1 protein (Figure 8a, upper panels), similar to immunoblot results (Figure 1d). Furthermore, co-expression of exogenous BRCA1 with myr-AKT led to an even greater percentage of cells exhibiting nuclear expression of BRCA1 compared to transduction with Ad-myr-AKT alone (Figure 8a, 8b). Interestingly, about 50% of cells co-transduced with Ad-BRCA1 and Ad-kd-AKT demonstrated low levels of BRCA1 staining almost exclusively in the cytoplasm. This observation suggests that the kd-AKT protein can act in a dominant negative fashion to prevent nuclear accumulation of BRCA1. Finally, estrogen treatment of MCF7 cells cultured in hormone depleted medium also restored focal nuclear staining for endogenous BRCA1, which was blocked by pretreatment with LY294002 (Supplemental Figure 8). Together, these data indicate that AKT signaling plays an important role in facilitating nuclear localization of BRCA1.
Figure 8. AKT Promotes Nuclear Localization of BRCA1.
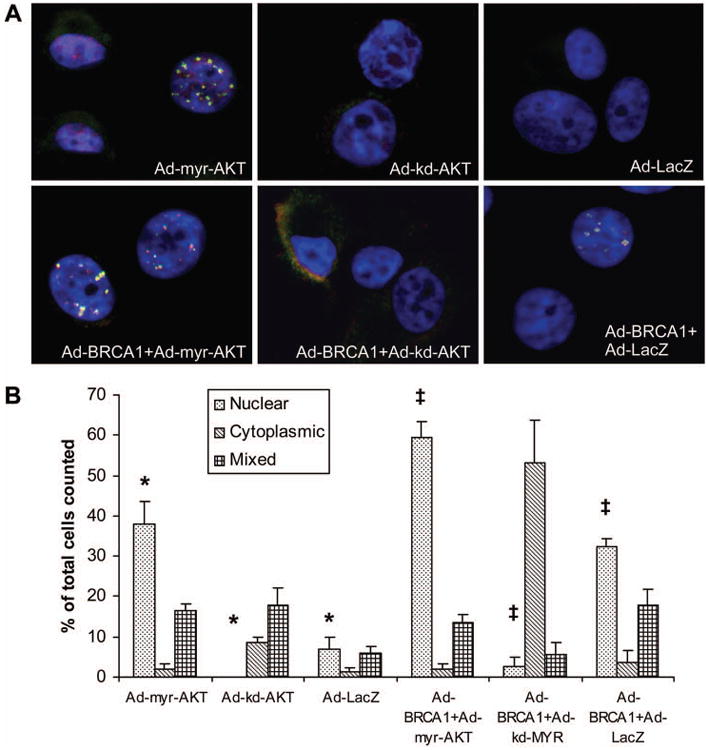
(A) MCF7 cells were transduced with the indicated adenoviral vectors, cultured for 48 hours in CSS medium, then fixed and stained with both an N-terminal mouse monoclonal BRCA1 antibody (red) and an exon 11 directed rabbit polyclonal BRCA1 antibody (green). Yellow indicates overlap of both signals to demonstrate specific staining for BRCA1. Nuclei were counterstained with DAPI. The following MOI's were used: 100 for Ad-myr-AKT, Ad-kd-AKT, and control Ad-LacZ; 25 for Ad-BRCA1.
(B) Quantification of BRCA1 subcellular localization. Two hundred cells were scored for each group in three independent experiments for BRCA1localization as either primarily nuclear, primarily cytoplasmic, or mixed between both cellular compartments. Unstained cells were included in the count but are not represented on the graph. The percentage of cells with nuclear BRCA1 was significantly increased for Ad-myr-AKT groups in comparison to both Ad-kd-AKT and Ad-LacZ (*) and for Ad-BRCA1+Ad-myr-AKT groups in comparison to both Ad-BRCA1+Ad-kd-AKT and Ad-BRCA1+Ad-LacZ (‡), (p≤0.01 for all comparisons by student's t-test, error bars represent SEM, N=3).
Co-expression of Activated AKT and Wild-type BRCA1 Improves Radiation Survival
Loss of the DNA repair function of BRCA1 results in hypersensitivity of cells to ionizing radiation (4, 6). We hypothesized that the low levels of BRCA1 and BARD1 protein expressed in MCF7 cells cultured in hormone depleted conditions would result in increased sensitivity to radiation treatment. Further, we predicted that the interaction of BRCA1 and activated AKT would decrease radiation sensitivity. To test this hypothesis, we transduced cells with either wild-type or a truncated (1853stop) BRCA1 protein in combination with myr-AKT, kd-AKT, or GFP control. This truncated form of BRCA1 is unable to regulate DNA repair (6). We also transduced cells with Ad-myr-AKT, Ad-kd-AKT, or Ad-GFP alone, and included mock-transduced cells as an additional control. All groups were cultured in hormone depleted medium for 24 hours, then exposed to 0, 1, 2, or 4 Gy of ionizing radiation, and returned to culture for another 48 hours following irradiation. Then all groups were changed into normal culture medium for colony outgrowth which was quantitated at three weeks. Only cells co-expressing wild-type BRCA1 and myr-AKT demonstrated significantly improved survival compared to other groups (Figure 9; percentage survival of 95, 67, and 18 for 1, 2, and 4 Gy, respectively). Expression of either exogenous BRCA1 with control GFP or expression of myr-AKT alone trended towards a slightly improved survival compared to the remaining transduction groups, but this change was not significant and was much smaller than that observed for the expression of both proteins together. Furthermore, neither the group expressing truncated BRCA1 (BR1853) with activated AKT (myr-AKT) nor the group expressing mutant AKT (kd-AKT) with wild-type BRCA1 demonstrated any improvement in survival. This combination of results suggests that expression of functional BRCA1 and activated AKT are together contributing to the observed increase in survival. We also performed immunoblots on lysates from parallel cultures harvested at the time of irradiation (Supplemental Figure 9). These demonstrated that the level of exogenous BRCA1 expressed from the adenoviral vector was relatively similar whether myr-AKT or control GFP protein was co-expressed (similar to Figure 2b). Therefore, the improved survival of cells expressing BRCA1 with myr-AKT compared to those expressing BRCA1 with control GFP suggests that activated AKT may positively support the role of BRCA1 in the regulation of DNA repair in addition to stabilizing BRCA1 expression.
Figure 9. Co-expression of Activated AKT and BRCA1 Improves Radiation Survival.
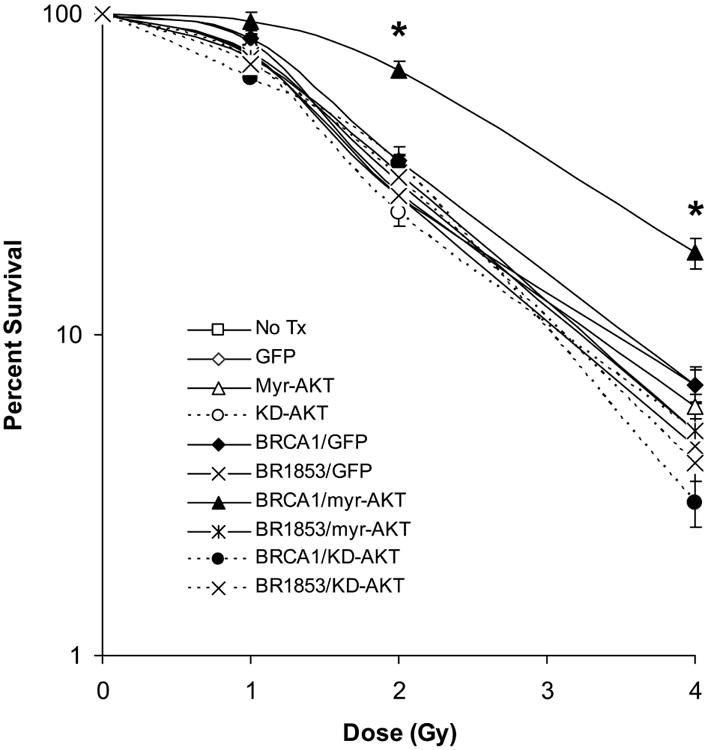
MCF7 cells were transduced with the indicated adenoviral vectors and plated at low density in triplicate in six-well plates. Cells were cultured in CSS medium for 24 hours prior to irradiation with indicated doses and maintained in CSS medium for an additional 48 hours following irradiation. Cells were then switched into normal growth medium to allow for colony outgrowth. Survival is presented on a logarithmic scale as the number of remaining colonies at each dose divided by the average number of colonies in unirradiated wells of the same transduction group. The following MOI's were used: 100 for Ad-myr-AKT, Ad-kd-AKT, and control Ad-GFP; 10 for Ad-BRCA1 and Ad-BR1853. Error bars represent SEM, N=3. A student's t-test was performed for BRCA1/myr-AKT vs. mock, GFP, myr-AKT, BRCA1/GFP, or BR1853/myr-AKT and found to be significant at p≤ 0.01 for both 2 and 4 Gy treatment groups (*) in all cases.
Discussion
BRCA1 is widely expressed in multiple tissues and plays a critical role in development as BRCA1 null mice die at a very early embryonic stage due to a severe proliferative defect (47). Yet maintenance of BRCA1 function in the adult breast and ovary seems to be of utmost importance since women carrying BRCA1 mutations have an extremely high risk for development of cancer specifically in these tissues. The relationship between hormone signaling and BRCA1 function in the breast and ovary is complex. Despite the fact that the majority of BRCA1-associated tumors are estrogen and progesterone receptor negative, antagonism of ER and PR signaling pathways clearly reduces the incidence of tumor formation in both patients with BRCA1 mutation and in Brca1 mutant mouse models (21, 22, 48). Conversely, synthetic constitutive activation of ER signaling accelerates tumorigenesis in Brca1 mutant mice while ultimately resulting in ER negative tumors (49). To explain these observations two primary hypotheses have been put forth: 1) that estrogen and/or progesterone signaling serve to regulate BRCA1 expression and function (24, 50) and 2) that BRCA1 may serve to modulate estrogen receptor and/or progesterone receptor signaling (22, 51, 52). It should be noted that these two models are not mutually exclusive, as a regulatory feedback loop would be possible if both hypotheses are correct. Furthermore, IGF receptor plays important roles in both mammary development (53, 54) and tumorigenesis (55), and cross regulation between ER and IGFR signaling pathways has been demonstrated (30, 56).
In this work, we show that AKT signaling dynamically regulates BRCA1 protein stability in a post-translational manner. Stimulation of both human breast and ovarian cell lines with biologically important hormones rapidly activates AKT and leads to phosphorylation of BRCA1 on the newly identified site, S694. We also confirm the previous work of Altiok et al. (32) by demonstrating AKT phosphorylation of BRCA1 at T509. Our data, as well as that of others (45), indicates that AKT-mediated phosphorylation of BRCA1 may regulate the nuclear localization and function of BRCA1 in addition to stabilizing protein expression. However, this study does not directly address the relative contributions of AKT phosphorylation of BRCA1 at S694 versus T509 to the regulation of BRCA1 protein stability and function. While we demonstrate that S694 is rapidly phosphorylated in cell culture following stimulation with estradiol or IGF-1, we have not addressed if phosphorylation also occurs at T509 under the same conditions. Further investigation in this area should address the individual contributions of S694 and T509 phosphorylation to the biologic regulation of BRCA1 by AKT described in this study.
There is accumulating evidence that BRCA1 is capable of dampening signaling through estrogen receptor (51, 57), IGF-1 receptor (58), and progesterone receptor (52, 59). More recently, loss of BRCA1 expression was associated with increased activity of the ER signaling pathway mediated by AKT (60). As well, BRCA1 has also been reported to negatively regulate AKT activation (61). In this context, our data appears to complete a negative feedback loop in which hormonal activation of proliferation and survival pathways in the mammary gland may serve to immediately stabilize the expression and function of BRCA1 via AKT. This would result in the preparation of DNA repair pathways which may become necessary during proliferation as well as leading to a negative feedback that shuts off the mitogenic signal. Therefore, we propose that this regulatory system may, in part, explain the hormone-dependence of BRCA1 associated tumorigenesis. Nevertheless, further investigation will be required to explain why the loss of additional factors, such as ER, PR, and p53, is required for the transformation to outright malignancy.
Finally, these results indicate that manipulation of BRCA1 protein levels may be feasible in clinical settings. Bortezomib (Velcade) rapidly increases BRCA1 levels in tissue culture cells under conditions where the protein is highly unstable. Most ER/PR negative breast cancers are similar to BRCA1 mutant cancers in regard to molecular and clinical characteristics (62-64). These highly aggressive breast tumors, as well as many ovarian tumors, often have decreased BRCA1 expression despite having two intact BRCA1 alleles (3, 65). Our data suggests that bortezomib treatment could increase BRCA1 expression in these cases and thereby may provide therapeutic benefit. Additionally, inhibition of hormone receptors or the associated downstream effector proteins is the focus of numerous clinical trials. Our data provides a basic rationale for the combination of these inhibitors with bortezomib (or other protease inhibitors). Further investigation of the PI3K-AKT-BRCA1 regulatory pathway and its responsiveness to hormone signaling in vivo should increase our understanding of the biological relationship between hormone receptors and BRCA1 function and could provide additional therapeutic targets.
Materials and Methods
Cell Culture and Chemicals
MCF7 and MDA-MB-231 cells were obtained from the American Type Culture Collection (HTB-22, HTB-26 respectively). BG-1 cells were kindly provided by Dr. Kenneth Korach (NIEHS/NIH). All lines were normally maintained at 37°C with 5% CO2 in DMEM with 1 mM L-glutamine (Gibco) supplemented with 10% Fetalclone serum (HyClone), 1% non-essential amino acids (Gibco), and 1% insulin/transferrin/selenium-A (Gibco). For hormone depleted conditions (denoted as CSS), cells were cultured in phenol-free DMEM with 1mM L-glutamine and 25 mM HEPES supplemented with 10% charcoal/dextran treated fetal bovine serum (Gemini Bioproducts). Serum starvation conditions were performed in the same medium with a concentration of 0.5% charcoal/dextran treated FBS.
17-β-estradiol, recombinant IGF-1, and MG132 were obtained from Sigma-Aldrich. Cycloheximide, AKT inhibitor VIII, UO126, and p38 MAP Kinase inhibitor were obtained from Calbiochem. ICI 182780 was obtained from Tocris Biosciences. LY294002 was obtained from Cell Signaling Technology.
Immunoblot, Immunoprecipitation, Antibodies, and Densitometry
Whole cell lysates were prepared by washing cell monolayers twice with PBS and then scraping into ice-cold PBS. Pelleted cells were resuspended in modified RIPA buffer (50 mM Tris base pH=7.4, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.5% Na-deoxycholate, 0.1% SDS, 50 mM NaF, 5 mM Na3VO4, plus Roche protease inhibitor tablets). Lysates were incubated for 30 minutes on ice, clarified by centrifugation, and protein concentration determined by Bradford assay (BioRad). Where indicated, lysates harvested without phosphatase inhibitors were treated with λ-phosphatase (New England Biolabs) according to manufacturer's instructions. 100 μg of total protein was loaded per lane for Tris-glycine SDS-PAGE. Samples were transferred to PVDF membrane (Millipore) for 200 volt-hours at 50 V constant at 16°C. Membranes were blocked in 5% dry non-fat milk (Carnation) dissolved in either PBS or TBS (25mM Tris pH 8.0, 135 mM NaCl, 2.5 mM KCl). Primary antibody incubations were performed overnight at 4°C diluted in either 0.5% dry non-fat milk/PBS-Tween-20 0.1% or in 3% BSA (Santa Cruz Biotechnology)/TBS-Tween-20 0.1%. Antibody suppliers are: BRCA1 (Ab-4 and Ab-1, Calbiochem); BARD1 (Bethyl Laboratories); Lamin A/C, phospho-(Ser/Thr) Akt Substrate, phospho-S473 Akt, and Akt (Cell Signaling Technology); Cyclin D1 (Lab Vision Corp.); Tubulin (Chemicon). Secondary antibodies were obtained from Amersham/GE Healthcare. Densitometry was performed on 600 dpi TIFF scans of Western blot films with Quantity One software (BioRad). Density values for BRCA1 and BARD1 in each sample were normalized by the value of the corresponding loading control.
For immunoprecipitation, cells were harvested by a similar protocol as described above although cells were lysed in IP lysis buffer (20 mM Tris pH 8, 120 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 0.25% Na-deoxycholate, 50 mM NaF, 1 mM Na3VO4, plus Roche protease inhibitor cocktail tablets). 1 mg of total protein was diluted in IP buffer (10 mM Tris pH 7.4, 50 mM NaCl, 5 mM EDTA, 0.5% CHAPS, 50 mM NaF, 1 mM Na3VO4) to a final volume of 1 mL, and 1 μg each of Ab-1 and Ab-4 antibodies was added with Exacta-Cruz immunoprecipitation matrix (Santa Cruz Biotechnology) according to manufacturer's protocols. Control immunoprecipitation was performed with 2 μg of anti-tubulin antibody. SDS-PAGE/WB was peformed as described above with the use of Exacta-Cruz secondary antibodies following manufacturer's protocols (Santa Cruz Biotechnology).
Densitometry was performed on grayscale TIFF images acquired at 600 dpi from the original films. Density analysis was performed using Quantity One Software (BioRad) with global background subtraction. Density values for each immunoreactive band were normalized with division by the density value for the corresponding loading control for that experimental group. The density value for the control group within each experiment was set to 1. Relative density values for each sample are expressed as a fraction or multiple of the corresponding control group.
Generation of pS694 BRCA1 Rabbit Polyclonal Antibody
Polyclonal antiserum specific for phospho-serine 694 of BRCA1 was generated and purified by Global Peptide (Fort Collins Colorado). The antiserum was generated using the phosphorylated peptide TSKRHD(p)S-DTFPELK to immunize female New Zealand white rabbits. Phosphospecific antiserum was obtained by first depleting the resulting serum with non-phosphorylated TSKRHDSDTFPELK-sepharose (to remove antibodies not specific for the phosphorylated protein). The flow through was then column purified with TSKRHD(p)S-DTFPELK-sepharose and elution of the bound polyclonal antibodies was performed to yield an antiserum specific for the phosphorylated epitope.
qPCR and Cell Cycle Analysis
RNA was isolated from cells using an RNeasy kit (Qiagen) according to manufacturer's protocols. cDNA was synthesized from 2 μg of RNA using the SuperScript III First Strand Synthesis System (Invitrogen) with random hexamers. Real-time quantitative PCR was performed using Sybr green (ABI) according to the manufacturer's protocols, 5 μl of cDNA template, and the following gene specific primers: BRCA1 forward 5′-GGGTAGTTAGCTATTTCTGGGTGAC-3′, reverse 5′-CTTCCATTGACCACATCTCCTCTG-3′; BARD1 forward 5′-AATCATCCTCAGCTA GCCACTGCT-3′; reverse 5′-TTCTGAAGACAGCCCACTGCCTAT-3′; PS2 forward 5′-AATAAGGGCTGCTGTTTCG ACGAC-3′, reverse 5′-TTCTGGAGGGACGTCGATG GTATT-3′; and Actin forward 5′-GC TCCCTTAAAGATATTTGCTCAGGATGGG-3′, reverse 5′-ATATGTGCCGTCCATCCGGTTCTT-3′. Samples were normalized to internal actin control and analyzed using Opticon II software (MJ Research).
For cell cycle analysis, cells were cultured as described, trypsinized, and recovered in culture medium. Pelleted cells were washed twice with ice-cold PBS and resuspended in Krishan's stain (66). Analysis was performed on a Becton Dickinson FC500 with ModFit LT Software (Verity Software House) by the University of Colorado Cancer Center Flow Cytometry Core.
Recombinant Human Adenovirus
Construction of recombinant human adenovirus expressing wild-type BRCA1 (Ad-BRCA1) or 1853stop truncated BRCA1 (Ad-1853) have been previously described (40). Recombinant human adenovirus expressing myr-AKT or kd-AKT have been previously described (67). High titer stocks were generated by infection of HEK-293 packaging cells and CsCl banding, followed by dialysis into viral storage buffer (10 mM Tris pH 7.4, 10 mM histidine, 75 mM NaCl, 1 mM MgCl2, 0.1 mM EDTA, 0.5% EtOH v/v, 50% glycerol v/v). Viral concentrations were determined by spectrophotometer as previously described (68).
GST-BRCA1 Fusion Proteins
Oligonucleotides containing threonine 509 mutated to alanine or glutamic acid were cloned into pBluescript-BRCA1 digested with Bsa1 and EcoN1. Mutations were confirmed by DNA sequencing. PCR products were produced spanning nucleotides 1323-1886 (T509 and A509), 1926-2538 (S694/T696), 3714-4310 (T1246), and 1323-2538 (T/A/E509+S694). These products were cloned into pGEX 4T-1 (GE Healthcare) digested with EcoR1 and Xho1. Production of fusion proteins was previously described (69).
In vitro Kinase Assay and Electrospray Ionization Mass Spectrometry
The AKT kinase assay was previously described (69). Purified full length BRCA1 protein was obtained from ProteinOne.
pGEX-BRCA1 S694/T696 fusion protein was phosphorylated by AKT1 and prepared for mass spectrometry as previously described (69). Samples were concentrated to dryness and re-suspended in 1% formic acid to a total volume of 15 μL. The sample (2 μL) was injected onto a reverse-phase column using a cooled (8 °C) autosampler (Eksigent) connected to a HPLC system run at 14-18 μL/min before the split and ∼400 nL/min post-split (Eldex). A gradient of 5% to 30% acetonitrile over thirty two minutes was employed for peptide separation. Along with aqueous and organic washes, the total LC run time was sixty minutes. The column effluent was coupled directly via fused silica capillary transfer line to a QSTAR Pulsar™ Q-TOF tandem mass spectrometer (Sciex/Applied Biosystems) with a nanospray ion source. Data acquisition was performed using the instrument supplied Analyst software. The sixty minute LC runs were monitored by sequentially recording the precursor scan (MS, 1s) followed by one collision-induced dissociation (CID) acquisitions (MS/MS, 4s each). Singly charged ions were excluded from CID selection. Normalized collision energies were employed using nitrogen as the collision gas. The mass spectrometer control software Analyst™ was used to create de-isotoped centroided peak lists from raw spectra (.mgf format). These peak lists were searched against databases using an in-house developmental Protein Prospector™, LC Batch-Tag Web™ (Version 4.25.2, UCSF) and an in-house Mascot™ server (Version 2.0, Matrix Science). For searches mass tolerances were +/-100 ppm for MS peaks, and +/- 0.3 Da for MS/MS fragmentions. Trypsin specificity was used allowing for 1 missed cleavage. The modifications of Met oxidation, protein N-terminal acetylation, peptide N-terminal pyro-glutamic acid formation and Ser, Thr, Tyr phosphorylation were allowed for. Samples were searched against all entries in the full NCBInr.
Immunofluorescence Microscopy
Cells were transduced with the indicated adenovirus vectors at the following MOI's: Ad-BRCA1=25; Ad-myr-AKT, Ad-kd-AKT, and Ad-LacZ=100. Cells were plated on cell culture grade glass cover slips (Fisher Scientific) and cultured in CSS medium for 48 hours prior to fixation with 10% neutral buffered formalin. Cells were permeabilized for 5 min. in 0.2% Triton X-100/PBS, washed in PBS, and then blocked in 2% BSA/PBS. Dual BRCA1 staining utilized an N-terminal mouse monoclonal antibody Ab-2 (1:50; Calbiochem) and an exon 11 directed rabbit polyclonal antibody (1:1250; BD Biosciences) diluted in blocking buffer. Anti-mouse 594 and anti-rabbit 488 (Molecular Probes) secondary antibodies were also diluted in blocking buffer. Nuclei were stained with DAPI (Sigma-Aldrich). Microscopic images were captured at 600× using a Nikon Eclipse 80i microscope and deconvolution was performed with Slidebook software (v4.1, Intelligent Imaging Innovations, Inc.).
Colony Formation
Cells were transduced with the indicated adenoviral vectors in normal culture medium at the following MOI's: 10 for Ad-BRCA1 and Ad-1853, 100 for Ad-myr-AKT, Ad-kd-AKT, and Ad-GFP. Mock transduction groups were not exposed to adenovirus but were otherwise handled identically. 5000 cells per transduction were plated in triplicate in six-well plates. Cells were allowed to attach overnight and then each well was washed twice with PBS and incubated in CSS medium for 24 hours. Ionizing radiation treatment groups were exposed to 1, 2, or 4 Gy generated by a RS2000 irradiator (Rad Source Technologies, Inc.). All groups, including non-irradiated controls, were maintained in CSS medium for an additional 48 hours with medium refreshed at 24 hours. All groups were then changed back into normal culture medium and incubated for three weeks to allow colony outgrowth with medium changes every third day.
Cells were fixed in 10% neutral buffered formalin and stained with 0.2% crystal violet. Percentage survival for each group at each dose was determined by dividing the number of surviving colonies by the average number of colonies formed in the non-irradiated control for each transduction group (N=3). Error bars = SEM and a student's t-test was performed to determine significance (p ≤ 0.01).
Sequence Alignment/Analysis
Human, mouse, rat, and chimpanzee sequences were accessed from Swis-Prot (# P38398, P48754, O54952, and Q9GKK8 respectively). Protein sequences were aligned with MegAlign software (v5.05, DNASTAR, Inc.) by the Clustal W method. AKT consensus recognition sequences were identified with Scansite (mit.scansite.edu).
Supplementary Material
Acknowledgments
This work was funded by grants from the Department of Defense Breast Cancer Research Program W81XWH-06-1-0335 to ACN, DAMD17-02-1-0351 to TRL, W81XWH-06-1-0423 to CDY, and from the Public Health Service DK063674 to SMA. The authors wish to thank: Ms. Maria Wong and Ms. Rita Lieberman for technical laboratory assistance, Ms. Lisa Litzenberger for preparation of the figures, Dr. Jerome Schaack for assistance with recombinant adenovirus production, Drs. David Orlicky and James McManaman for assistance with microscopy, Ms. Lauren Kiemele for assistance with mass spectroscopy, and Mr. Michael Rudolph and Dr. Wilbur Franklin for assistance with qPCR. Bortezomib was provided by Dr. Stuart Lind. The Proteomics and Flow Cytometry Core Laboratories at the University of Colorado Cancer Center contributed to this work.
Grant Support: This work was funded by grants from the Department of Defense Breast Cancer Research Program W81XWH-06-1-0335 to ACN, DAMD17-02-1-0351 to TRL, W81XWH-06-1-0423 to CDY, and from the Public Health Service DK063674 to SMA.
Footnotes
Disclosure Statement: TRL, CDY, and KCH have nothing to declare. ACN, SMA, and JTH are listed as inventors on USPTO # 83025-378844.
References
- 1.Hall JM, Lee MK, Newman B, et al. Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990;250:1684–9. doi: 10.1126/science.2270482. [DOI] [PubMed] [Google Scholar]
- 2.Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 3.Thompson ME, Jensen RA, Obermiller PS, Page DL, Holt JT. Decreased expression of BRCA1 accelerates growth and is often present during sporadic breast cancer progression. Nature Genetics. 1995;9:444–450. doi: 10.1038/ng0495-444. [DOI] [PubMed] [Google Scholar]
- 4.Abbott DW, Thompson ME, Robinson-Benion C, Tomlinson G, Jensen RA, Holt JT. BRCA1 Expression Restores Radiation Resistance in BRCA1-defective Cancer Cells Through Enhancement of Transcription-coupled DNA Repair. Journal of Biological Chemistry. 1999;274:18808–18812. doi: 10.1074/jbc.274.26.18808. [DOI] [PubMed] [Google Scholar]
- 5.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–8. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 6.Scully R, Ganesan S, Vlasakova K, Chen J, Socolovsky M, Livingston DM. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol Cell. 1999;4:1093–9. doi: 10.1016/s1097-2765(00)80238-5. [DOI] [PubMed] [Google Scholar]
- 7.Zhong Q, Chen CF, Li S, et al. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science. 1999;285:747–50. doi: 10.1126/science.285.5428.747. [DOI] [PubMed] [Google Scholar]
- 8.Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol. 2004;24:9478–86. doi: 10.1128/MCB.24.21.9478-9486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, Brody LC. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet. 2002;30:285–9. doi: 10.1038/ng837. [DOI] [PubMed] [Google Scholar]
- 10.Anderson SF, Schlegel BP, Nakajima T, Wolpin ES, Parvin JD. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat Genet. 1998;19:254–6. doi: 10.1038/930. [DOI] [PubMed] [Google Scholar]
- 11.Scully R, Anderson SF, Chao DM, et al. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc Natl Acad Sci U S A. 1997;94:5605–10. doi: 10.1073/pnas.94.11.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Welcsh PL, Lee MK, Gonzalez-Hernandez RM, et al. BRCA1 transcriptionally regulates genes involved in breast tumorigenesis. Proc Natl Acad Sci U S A. 2002;99:7560–5. doi: 10.1073/pnas.062181799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu LC, Wang ZW, Tsan JT, et al. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet. 1996;14:430–40. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- 14.Koonin EV, Altschul SF, Bork P. BRCA1 protein products … Functional motifs. Nat Genet. 1996;13:266–8. doi: 10.1038/ng0796-266. [DOI] [PubMed] [Google Scholar]
- 15.Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol. 2001;8:833–7. doi: 10.1038/nsb1001-833. [DOI] [PubMed] [Google Scholar]
- 16.Baer R, Ludwig T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr Opin Genet Dev. 2002;12:86–91. doi: 10.1016/s0959-437x(01)00269-6. [DOI] [PubMed] [Google Scholar]
- 17.Henderson BR. Regulation of BRCA1, BRCA2 and BARD1 intracellular trafficking. Bioessays. 2005;27:884–93. doi: 10.1002/bies.20277. [DOI] [PubMed] [Google Scholar]
- 18.Choudhury AD, Xu H, Baer R. Ubiquitination and proteasomal degradation of the BRCA1 tumor suppressor is regulated during cell cycle progression. J Biol Chem. 2004;279:33909–18. doi: 10.1074/jbc.M403646200. [DOI] [PubMed] [Google Scholar]
- 19.Ruffner H, Joazeiro CA, Hemmati D, Hunter T, Verma IM. Cancer-predisposing mutations within the RING domain of BRCA1: loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc Natl Acad Sci U S A. 2001;98:5134–9. doi: 10.1073/pnas.081068398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narod SA. Hormonal prevention of hereditary breast cancer. Ann N Y Acad Sci. 2001;952:36–43. doi: 10.1111/j.1749-6632.2001.tb02726.x. [DOI] [PubMed] [Google Scholar]
- 21.Bachelier R, Xu X, Li C, et al. Effect of bilateral oophorectomy on mammary tumor formation in BRCA1 mutant mice. Oncol Rep. 2005;14:1117–20. [PubMed] [Google Scholar]
- 22.Poole AJ, Li Y, Kim Y, Lin SC, Lee WH, Lee EY. Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science. 2006;314:1467–70. doi: 10.1126/science.1130471. [DOI] [PubMed] [Google Scholar]
- 23.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spillman MA, Bowcock AM. BRCA1 and BRCA2 mRNA levels are coordinately elevated in human breast cancer cells in response to estrogen. Oncogene. 1996;13:1639–45. [PubMed] [Google Scholar]
- 25.Marks JR, Huper G, Vaughn JP, et al. BRCA1 expression is not directly responsive to estrogen. Oncogene. 1997;14:115–21. doi: 10.1038/sj.onc.1200808. [DOI] [PubMed] [Google Scholar]
- 26.Edwards DP. Regulation of signal transduction pathways by estrogen and progesterone. Annu Rev Physiol. 2005;67:335–76. doi: 10.1146/annurev.physiol.67.040403.120151. [DOI] [PubMed] [Google Scholar]
- 27.Castoria G, Migliaccio A, Bilancio A, et al. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. Embo J. 2001;20:6050–9. doi: 10.1093/emboj/20.21.6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–41. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katzenellenbogen BS. Estrogen receptors: bioactivities and interactions with cell signaling pathways. Biol Reprod. 1996;54:287–93. doi: 10.1095/biolreprod54.2.287. [DOI] [PubMed] [Google Scholar]
- 30.Kahlert S, Nuedling S, van Eickels M, Vetter H, Meyer R, Grohe C. Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway. J Biol Chem. 2000;275:18447–53. doi: 10.1074/jbc.M910345199. [DOI] [PubMed] [Google Scholar]
- 31.Lee AV, Weng CN, Jackson JG, Yee D. Activation of estrogen receptor-mediated gene transcription by IGF-I in human breast cancer cells. J Endocrinol. 1997;152:39–47. doi: 10.1677/joe.0.1520039. [DOI] [PubMed] [Google Scholar]
- 32.Altiok S, Batt D, Altiok N, et al. Heregulin induces phosphorylation of BRCA1 through phosphatidylinositol 3-Kinase/AKT in breast cancer cells. J Biol Chem. 1999;274:32274–8. doi: 10.1074/jbc.274.45.32274. [DOI] [PubMed] [Google Scholar]
- 33.Ruffner H, Verma IM. BRCA1 is a cell cycle-regulated nuclear phosphoprotein. Proc Natl Acad Sci U S A. 1997;94:7138–43. doi: 10.1073/pnas.94.14.7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruffner H, Jiang W, Craig AG, Hunter T, Verma IM. BRCA1 is phosphorylated at serine 1497 in vivo at a cyclin-dependent kinase 2 phosphorylation site. Mol Cell Biol. 1999;19:4843–54. doi: 10.1128/mcb.19.7.4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orban TI, Olah E. Emerging roles of BRCA1 alternative splicing. Mol Pathol. 2003;56:191–7. doi: 10.1136/mp.56.4.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.ElShamy WM, Livingston DM. Identification of BRCA1-IRIS, a BRCA1 locus product. Nat Cell Biol. 2004;6:954–67. doi: 10.1038/ncb1171. [DOI] [PubMed] [Google Scholar]
- 37.Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 38.Kawakami Y, Nishimoto H, Kitaura J, et al. Protein kinase C betaII regulates Akt phosphorylation on Ser-473 in a cell type- and stimulus-specific fashion. J Biol Chem. 2004;279:47720–5. doi: 10.1074/jbc.M408797200. [DOI] [PubMed] [Google Scholar]
- 39.Zhang F, Cheng J, Hackett NR, et al. Adenovirus E4 gene promotes selective endothelial cell survival and angiogenesis via activation of the vascular endothelial-cadherin/Akt signaling pathway. J Biol Chem. 2004;279:11760–6. doi: 10.1074/jbc.M312221200. [DOI] [PubMed] [Google Scholar]
- 40.Campbell M, Qu S, Wells S, Sugandha H, Jensen RA. An adenoviral vector containing an arg-gly-asp (RGD) motif in the fiber knob enhances protein product levels from transgenes refractory to expression. Cancer Gene Therapy. 2003;10:559–570. doi: 10.1038/sj.cgt.7700599. [DOI] [PubMed] [Google Scholar]
- 41.Romagnolo D, Annab LA, Thompson TE, et al. Estrogen upregulation of BRCA1 expression with no effect on localization. Mol Carcinog. 1998;22:102–9. doi: 10.1002/(sici)1098-2744(199806)22:2<102::aid-mc5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 42.Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19:833–42. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 43.Scully R, Chen J, Ochs R, et al. Dynamic Changes of BRCA1 Subnuclear Location and Phosphorylation State Are Inititated by DNA Damage. Cell. 1997;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- 44.Jin Y, Xu XL, Yang MC, et al. Cell cycle-dependent colocalization of BARD1 and BRCA1 proteins in discrete nuclear domains. Proc Natl Acad Sci U S A. 1997;94:12075–80. doi: 10.1073/pnas.94.22.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hinton CV, Fitzgerald LD, Thompson ME. Phosphatidylinositol 3-kinase/Akt signaling enhances nuclear localization and transcriptional activity of BRCA1. Exp Cell Res. 2007;313:1735–44. doi: 10.1016/j.yexcr.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scully R, Ganesan S, Brown M, et al. Location of BRCA1 in human breast and ovarian cancer cells. Science. 1996;272:123–6. doi: 10.1126/science.272.5258.123. [DOI] [PubMed] [Google Scholar]
- 47.Hakem R, de la Pompa JL, Sirard C, et al. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell. 1996;85:1009–23. doi: 10.1016/s0092-8674(00)81302-1. [DOI] [PubMed] [Google Scholar]
- 48.Narod SA, Offit K. Prevention and management of hereditary breast cancer. J Clin Oncol. 2005;23:1656–63. doi: 10.1200/JCO.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 49.Jones LP, Tilli MT, Assefnia S, et al. Activation of estrogen signaling pathways collaborates with loss of Brca1 to promote development of ERalpha-negative and ERalpha-positive mammary preneoplasia and cancer. Oncogene. 2007 doi: 10.1038/sj.onc.1210674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gudas JM, Nguyen H, Li T, Cowan KH. Hormone-dependent regulation of BRCA1 in human breast cancer cells. Cancer Res. 1995;55:4561–5. [PubMed] [Google Scholar]
- 51.Fan S, Wang JA, Yuan R, et al. BRCA1 Inhibition of Estrogen Receptor Signaling in Transfected Cells. Science. 1999;284:1354–1356. doi: 10.1126/science.284.5418.1354. [DOI] [PubMed] [Google Scholar]
- 52.Katiyar P, Ma Y, Fan S, Pestell RG, Furth PA, Rosen EM. Regulation of progesterone receptor signaling by BRCA1 in mammary cancer. Nucl Recept Signal. 2006;4:e006. doi: 10.1621/nrs.04006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rowzee AM, Lazzarino DA, Rota L, Sun Z, Wood TL. IGF ligand and receptor regulation of mammary development. J Mammary Gland Biol Neoplasia. 2008;13:361–70. doi: 10.1007/s10911-008-9102-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kleinberg DL, Ruan W. IGF-I, GH, and sex steroid effects in normal mammary gland development. J Mammary Gland Biol Neoplasia. 2008;13:353–60. doi: 10.1007/s10911-008-9103-7. [DOI] [PubMed] [Google Scholar]
- 55.Belfiore A, Frasca F. IGF and insulin receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia. 2008;13:381–406. doi: 10.1007/s10911-008-9099-z. [DOI] [PubMed] [Google Scholar]
- 56.Fagan DH, Yee D. Crosstalk between IGF1R and estrogen receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia. 2008;13:423–9. doi: 10.1007/s10911-008-9098-0. [DOI] [PubMed] [Google Scholar]
- 57.Eakin CM, Maccoss MJ, Finney GL, Klevit RE. Estrogen receptor alpha is a putative substrate for the BRCA1 ubiquitin ligase. Proc Natl Acad Sci U S A. 2007;104:5794–9. doi: 10.1073/pnas.0610887104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Razandi M, Pedram A, Rosen EM, Levin ER. BRCA1 Inhibits Membrane Estrogen and Growth Factor Receptor Signaling to Cell Proliferation in Breast Cancer. Molecular and Cellular Biology. 2004;24:5900–5913. doi: 10.1128/MCB.24.13.5900-5913.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma Y, Katiyar P, Jones LP, et al. The breast cancer susceptibility gene BRCA1 regulates progesterone receptor signaling in mammary epithelial cells. Mol Endocrinol. 2006;20:14–34. doi: 10.1210/me.2004-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma Y, Hu C, Riegel AT, Fan S, Rosen EM. Growth factor signaling pathways modulate BRCA1 repression of estrogen receptor-alpha activity. Mol Endocrinol. 2007;21:1905–23. doi: 10.1210/me.2006-0397. [DOI] [PubMed] [Google Scholar]
- 61.Xiang T, Ohashi A, Huang Y, et al. Negative Regulation of AKT Activation by BRCA1. Cancer Res. 2008;68:10040–4. doi: 10.1158/0008-5472.CAN-08-3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hedenfalk I, Duggan D, Chen Y, et al. Gene-expression profiles in hereditary breast cancer. N Engl J Med. 2001;344:539–48. doi: 10.1056/NEJM200102223440801. [DOI] [PubMed] [Google Scholar]
- 63.Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100:8418–23. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4:814–9. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 65.Zheng W, Luo F, Lu JJ, et al. Reduction of BRCA1 expression in sporadic ovarian cancer. Gynecol Oncol. 2000;76:294–300. doi: 10.1006/gyno.1999.5664. [DOI] [PubMed] [Google Scholar]
- 66.Krishan A. Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J Cell Biol. 1975;66:188–93. doi: 10.1083/jcb.66.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Limesand KH, Barzen KA, Quissell DO, Anderson SM. Synergistic suppression of apoptosis in salivary acinar cells by IGF1 and EGF. Cell Death Differ. 2003;10:345–55. doi: 10.1038/sj.cdd.4401153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Orlicky DJ, Schaack J. Adenovirus transduction of 3T3-L1 cells. J Lipid Res. 2001;42:460–6. [PubMed] [Google Scholar]
- 69.Lyons TR, Thorburn J, Ryan PW, Thorburn A, Anderson SM, Kassenbrock CK. Regulation of the Pro-apoptotic scaffolding protein POSH by Akt. J Biol Chem. 2007;282:21987–97. doi: 10.1074/jbc.M704321200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.