

Abstract
The sexually-transmitted pathogen, Neisseria gonorrhoeae, undergoes natural transformation at high frequency. This property has led to the rapid dissemination of antibiotic resistance markers and to the panmictic structure of the gonococcal population. However, high frequency transformation also makes N. gonorrhoeae one of the easiest bacterial species to manipulate genetically in the laboratory. Techniques have been developed that result in transformation frequencies >50%, allowing the identification of mutants by screening and without selection. Constructs have been created to take advantage of this high frequency transformation, facilitating genetic mutation, complementation, and heterologous gene expression. Techniques are described for genetic manipulation of N. gonorrhoeae, as well as for growth of this fastidious organism.
Keywords: Neisseria gonorrhoeae, natural transformation, electroporation, complementation, genetic transformation, heterologous gene expression
Neisseria gonorrhoeae is a Gram-negative coccus and the causative agent of the sexually transmitted disease, gonorrhea. The disease generally manifests as localized urethritis in men and cervicitis in women. In women, the infection is often asymptomatic initially, but may spread to the uterus and fallopian tubes, causing pelvic inflammatory disease, chronic pelvic pain, and infertility. Untreated infections in men or women may also cause systemic disease (disseminated gonococcal infection), which may be manifested as tenosynovitis, meningitis, or endocarditis. For laboratory workers, it is most important to remember that N. gonorrhoeae causes eye infections. Normally gonococcal eye infections occur in newborns infected during passage through the birth canal of an infected mother; these infections often lead to blindness if left untreated. Laboratory-acquired N. gonorrhoeae eye infections have been reported; therefore, it is important to wear safety glasses when performing procedures that may cause gonococci to be propelled into the air and to seek medical treatment if the eyes or other mucosal surfaces are exposed to N. gonorrhoeae.
Gonococci are naturally transformable and this property has greatly facilitated its study. Gonococci readily take up DNA containing the 10-bp Neisseria DNA uptake sequence (DUS) GCCGTCTGAA (Goodman and Scocca, 1988). The DUS is found frequently in the chromosome of N. gonorrhoeae and other Neisseria species, often in two copies as an inverted repeat. Plasmids used for transformation of N. gonorrhoeae must contain the DUS to be taken up efficiently. An extended 12-bp DUS, atGCCGTCTGAA, was identified by whole genome sequence analysis as the most prevalent form of the DUS in Neisseria genomes(Smith, Gwinn et al. 1999), and this sequence increases gonococcal transformation with plasmids two-fold over the frequency of those carrying the 10-bp DUS (Ambur, Frye et al. 2007). Unlike many transformable bacteria, N. gonorrhoeae does not regulate transformation: gonococci are transformable at all stages of growth (Sparling, 1966). However, only piliated gonococci are naturally transformable, and piliation is a phase-variable phenotype. Therefore, care must be taken to choose piliated colonies for transformation.
This unit describes methods for genetic manipulation of N. gonorrhoeae including various methods of mutagenesis and complementation, as well as methods for growing and purifying DNA from these bacteria.
CAUTION: Neisseria gonorrhoeae is a Biosafety Level 2 (BSL-2) pathogen. Follow all appropriate guidelines and regulations for the use and handling of pathogenic microorganisms. In particular, wear safety glasses as exposure of the eyes to N. gonorrhoeae will cause conjunctivitis (see Critical Parameters and Troubleshooting). See UNIT 1A.1 and other pertinent resources (APPENDIX 1B) for more information.
IMPORTANT NOTE: Unless otherwise indicated, all incubations are to be performed at 37°C, 5% CO2.
STRATEGIC PLANNING
Growth on Agar or in Liquid Medium
Gonococci are fastidious. They have multiple nutritional requirements and are generally grown in rich medium, either gonococcal base medium (GCB), chocolate agar, or a rich chemically-defined medium such as Graver-Wade medium (Wade and Graver 2007). They require CO2 in the atmosphere for growth on agar plates or sodium bicarbonate in liquid medium (Morse and Bartenstein, 1974). Medium must be supplemented with Kellogg’s supplements (Kellogg et al., 1963) or IsoVitalex (Becton Diconson). Recipes for gonococcal base medium and supplements are found in the Reagents and Solution section below. Gonococci should be inoculated from an agar plate into GCBL at a density no less than 107 CFU/ml to reliably obtain a viable liquid culture. The medium must be prewarmed to 37°C. In general, inoculating to a density of 5 × 107 to 1 × 108 (OD540 of 0.2) is the most reliable method. Transfer may be done using a sterile Dacron swab. Do not use cotton, as these swabs may contain fatty acids which are inhibitory to some strains of N. gonorrhoeae. Optimal numbers of live gonococci for inoculation are obtained from agar plates 18- to 20-hr old. The doubling time for laboratory strains in liquid medium is ~60 min.
Antibiotic Concentrations for Selection in N. gonorrhoeae Strains
Some gonococcal strains have much higher levels of antibiotic resistance than others. It is therefore advisable to search the available literature for concentrations of antibiotic used with particular strains. Most strains, however, exhibit either high-level overall resistance levels (e.g., strain MS11) or low-level overall resistance (e.g., strain FA1090); therefore, useful antibiotic concentrations are given for these two commonly used strains (Table 4A.2.1) and may serve as a starting point for the determination of concentrations appropriate for uncharacterized strains.
Table 4A.2.1.
Antibiotic Concentrations Used for Common High- and Low-Level Overall Resistance Strains of N. gonorhoeaea
| ANTIBIOTIC | MS11 (MG/L) | FA1090(MG/L) |
|---|---|---|
| Chloramphenicol | 10 | 2 |
| Erythromycin | 10 | 2 |
| Kanamycin | 80 | 40 |
| Nalidixic Acid | 1.5 | 1.5 |
| Penicillin G | 0.3 | ND |
| Spectinomycin | 75 | 75 |
| Streptomycin | 100 | 100 |
| Tetracycline | 2 | 0.2 |
Abbreviation: ND, not determined.
BASIC PROTOCOL 1 SPOT TRANSFORMATION OF NEISSERIA GONORRHOEAE ON AGAR PLATES
N. gonorrhoeae can be transformed at high efficiency by growing the bacteria on agar plates in the presence of high concentrations of DNA. DNA is soaked into the plate at a few spots and the bacteria to be transformed (recipients) are streaked through the spots. Colonies arising on the spots have a high likelihood of containing transformed gonococci, often as high as 20% to 50% of total colony forming units. This method is often used for transformation with plasmids containing mutation constructs created in E. coli. Gonococcal DNA may be cloned into an E. coli plasmid and then interrupted with the gene for a selectable marker. Gonococcal DNA flanking the marker on both sides facilitates double-crossover homologous recombination and insertion of the marker into the gonococcal chromosome. To prevent the recovery of bacteria that have undergone a single-crossover recombination event and integrated the entire plasmid into the chromosome, the plasmid should be linearized before transformation. Commonly used restriction enzymes and buffers do not inhibit transformation. Therefore, the restriction digest mixture can be used directly without further purification. If the plasmid is 8 kb or larger, it is not necessary to linearize the plasmid, since N. gonorrhoeae processes large DNA fragments prior to DNA uptake during natural transformation (reviewed in Hamilton, 2006).
Alternatively, the ability of N. gonorrhoeae to incorporate plasmids into the chromosome can be used for making mutations (Hamilton et al., 2001). A single fragment of gonococcal DNA can be cloned into a plasmid in E. coli. Transformation with the circular plasmid, followed by selection for the antibiotic resistance marker on the plasmid will result in the isolation of transformants that have integrated the plasmid by single-crossover recombination. Since this method results in duplication of the cloned sequence, the method is often referred to as insertion-duplication mutagenesis.
Due to the high efficiency of transformation observed with this method, it is possible to introduce mutations that do not contain an antibiotic resistance gene or other selectable marker. Plasmids or PCR products containing deletions, insertions, or point mutations can be used for transformation. If plasmids are used, they should be linearized to prevent single-crossover recombination. Potential transformants can be screened by PCR for an increase or decrease in the size of the PCR product amplified from the gene of interest. If small mutations are introduced (e.g., point mutations, epitope tag insertions), it is helpful to also introduce or eliminate a restriction site, allowing potential transformants to be identified by PCR and restriction digest.
Materials
Recipient N. gonorrhoeae strain, frozen
GCB plates (see recipe), room temperature and 37°C
≥10 ng/μl plasmid DNA
Appropriate restriction enzyme and buffer (for double-crossover only)
GCB plate containing appropriate antibiotic (Table 4A.2.1; for fragments containing selectible markers only)
Dacron swabs (Fisher Scientific), sterile
Additional reagents and equipment for PCR (see Support Protocol)
-
1
Streak recipient N. gonorrohoeae strain from frozen stock onto a GCB plate. Incubate overnight (<24 hr).
Prepare DNA
-
2a
For inserting a construct into the chromosome by double-crossover event: Linearize the plasmid unless it is large (>8 kb). Be sure not to cut the transforming DNA away from the gonococcal DNA uptake sequence.
-
2b
For performing insertion-duplication mutagenesis: Do not linearize the plasmid.
-
3
Draw two 1-cm circles on the back of a 37°C GCB agar plate, where, upon streaking, the colony density will be medium and thin, respectively.
Appropriate places would be one-third and two-thirds of the way down the middle of the plate. -
4
Spot ~10 to 20 μl DNA solution (step 2a or 2b) on the agar inside the circles. Maximize the DNA concentration but do not exceed 10 μg. Allow DNA to soak into the plate.
Amounts of DNA in the hundreds of nanograms are appropriate.If the spots are not dry within 10 min, it may be necessary to dry the plate in a biosafety hood with the lid of the plate removed, 20 min or less. -
5
Pick a piliated colony of the recipient from the plate prepared in step 1. Streak through the DNA spots and incubate overnight.
See Background Information for a discussion of appraising the piliation state of N. gonorrhoeae. -
6a
For transformations using fragments containing selectable markers: Using a sterile Dacron swab, transfer and uniformly spread the colonies that grew on the spots onto a GCB plate containing the appropriate antibiotic.
-
6b
For transformations using fragments without selectable markers: Streak individual colonies to be screened onto GCB plates using an inoculating loop.
CAUTION: See Critical Parameters and Troubleshooting for important safety considerations.
-
7
Incubate transformants overnight.
If using no selection, colonies will be ready for screening in 24 hr or less. Antibiotic selection will significantly slow the appearance of transformants. Transformants selected with chloramphenicol or erythromycin may not be apparent until 36 to 48 hr. -
8
Streak individual transformants onto GCB plates and screen for nonselectable mutations by PCR (see Support Protocol).
Due to transformation occurring within only some members of a colony, nonselected mutants identified by this method should be regarded as likely mixed populations and should be restreaked; the presence of the mutation in individual colonies should be verified.
BASIC PROTOCOL 2 TRANSFORMATION OF NEISSERIA GONORRHOEAE IN LIQUID CULTURE
Transformation of N. gonorrhoeae has traditionally been done in liquid culture. This method is not as efficient as transformation on agar plates as described in Basic Protocol 1; however, it can be quicker, particularly if frozen cultures of piliated bacteria are prepared ahead of time. Also, this method is less likely to give colonies representing mixed populations.
Materials
Recipient N. gonorrhoeae strain, frozen
GCB plate (see recipe)
≥ 10 ng/μl plasmid DNA
GCBL medium (see recipe) containing 5 mM MgSO4, room temperature and 37°C
GCBL medium containing Kellogg’s supplements (see recipe)
60% (v/v) glycerol
Dacron swabs, sterile
T-25 tissue culture flask, 37°C
-
1
Streak recipient N. gonorrhoeae strain from frozen stock onto a GCB plate. Incubate overnight.
-
2
Pick a piliated colony. Streak onto a GCB plate and incubate overnight (18 to 20 hr).
See Background Information for a discussion of appraising the piliation state of N. gonorrhoeae. -
3
Add 20 μl plasmid DNA to 200 μl GCBL medium containing 5 mM MgSO4 in a 1.5-ml microcentrifuge tube. Warm mixture to 37°C in a water bath. Maximize DNA amount (not to exceed 10 μg) and minimize DNA volume (not to exceed 20 μl).
Typical DNA amounts are in the hundreds of nanograms (~200 ng to 1 μg). Plasmids smaller than 8 kb should be linearized by restriction enzyme digestion prior to transformation unless insertion of the entire plasmid into the chromosome is desired. -
4
Using a Dacron swab, prepare a recipient cell suspension by transfering 1/4 to 1/2 of all of the colonies resulting from step 2 into a 1.5-ml microcentrifuge tube containing
1 ml GCBL medium/5 mM MgSO4, 37°C. Swirl the swab to suspend bacteria.
-
5
Prepare transformation mix by adding 20 μl recipient cells to the DNA solution (step 3). Incubate 5 to 15 min at 37°C.
-
6
If it is possible this strain will again be transformed, add 60% glycerol to a final concentration of 15% (v/v) to the remaining recipient cell suspension (without DNA). Flash freeze using liquid nitrogen or a dry ice/ethanol bath.
-
7
Transfer entire transformation mix to 2 ml GCBL containing Kellogg’s supplements in a T-25 tissue culture flask (already at 37°C). Incubate flask with the lid loosened 2 to 6 hr.
Plate transformants
-
8a
If selecting for antibiotic resistance: Prepare 10-fold serial dilutions in GCBL over a range of 100 to 10−4. Plate 100 μl of each dilution onto an individual GCB plate with the appropriate antibiotic. Incubate until colonies are observed (24 to 48 hr).
-
8b
If screening without selection: Prepare 10-fold serial dilutions in GCBL over a range of 10−3 to 10−6. Plate 100 μl of each dilution onto individual GCB plates without antibiotics. Incubate until colonies are observed (18 to 24 hr).
-
9
Screen for desired mutation by PCR (Support Protocol).
BASIC PROTOCOL 3 ELECTROPORATION OF NEISSERIA GONORRHOEAE
Since the efficiency of electroporation is low and that of natural transformation is high, gonococci are almost never transformed by electroporation. However, there are cases where electroporation is the only reasonable option. For example, transformation of a recA mutant with a replicating plasmid, transformation of a nonpiliated, nonreverting strain, or transformation with DNA lacking a gonococcal DNA uptake sequence (DUS) may only be possible by electroporation. Gonococci will begin to undergo death and lysis under the washing conditions required to remove salts prior to electroporation. Different methods have been described for this purpose (Genco et al., 1991). However, it is most important to perform the washing steps and electroporation quickly in order to get the gonococci back into growth medium before too many of the recipients have died.
Materials
Recipient N. gonorrhoeae strain, frozen
GCB plates (see recipe) with and without antibiotic (Table 4A.2.1)
0.3 M sucrose: pass through a 0.22-μm filter to sterilize; store up to 1 year at room temperature
10 ng/μl DNA solution
GCBL medium with Kellogg’s supplements (see recipe)
GCBL medium (see recipe)
Dacron swabs (Fisher Scientific), sterile
Electroporation cuvette, 2-mm gap length
Electroporatator
-
1
Streak recipient N. gonorrhoeae strain from frozen stock onto a GCB plate. Incubate overnight.
-
2
Using a sterile Dacron swab, transfer 50% to 100% of the colonies on the plate into 1 ml of 0.3 M sucrose at room temperature.
Sucrose provides osmotic stabilization. Suspension of N. gonorrhoeae in low osmotic strength buffer results in autolysis and cell death. -
3
Microcentrifuge 30 sec at 12,000 rpm, room temperature. Resuspend cells in 1 ml of 0.3 M sucrose and microcentrifuge again. Repeat suspension and microcentrifugation a third time. Suspend cells in 100 μl of 0.3 M sucrose.
-
4
Add 1 μl DNA solution.
Plasmid DNA prepared by standard miniprep method and suspended in TE or water works well for electroporation. If arcing occurs during electroporation or if the addition of >3 μl of DNA is desired, the DNA should be concentrated by ethanol precipitation and washed twice with 70% ethanol to remove salts. -
5
Transfer cell/DNA mixture into an electroporation cuvette. Electroporate with the pulse controller set at 2.5 kV, 200 Ω, and 25 μF.
-
6
Resuspend cells in 1 ml GCBL medium with Kellogg’s supplements. Transfer culture to a GCB plate without antibiotics.
-
7
Incubate the plate right-side up 4 to 6 hr.
-
8
Wash the bacteria off the plate with 1 ml GCBL medium. Plate 50 μl onto a GCB plate containing appropriate antibiotic. Repeat for a second plate if desired. Incubate until colonies are observed (~ 24 to 48 hr).
BASIC PROTOCOL 4 CREATION OF UNMARKED MUTATIONS USING A POSITIVE AND NEGATIVE SELECTION CASSETTE
It is often desirable to create unmarked mutations in the chromosome, but it is not always easy to introduce unmarked mutations by simple transformation and screening, i.e., it may be highly labor intensive to find the desired mutant by screening. However, it is possible to use a cassette carrying a selectable marker and a counterselectable marker to select for introduction of an unmarked mutation in a two-step process. An ermC/rpsL cassette has been successfully used in N. gonorrhoeae, as in other organisms, for this purpose (Johnston and Cannon 1999; Cloud and Dillard 2002). Erythromycin can be used to select for transformants carrying the insertion. In order to take advantage of the couter-selectable marker, a streptomycin resistant N. gonorrhoeae strain should be used. Transformants that incorporate the insertion containing rpsL will become streptomycin sensitive, since when both rpsL alleles are expressed, streptomycin sensitivity is dominant over resistance (Lederberg 1951). In a subsequent transformation an unmarked mutation construct can be used to replace the ermC/rpsL cassette, and transformants that have incorporated this mutation can be selected using streptomycin.
Although many E. coli plasmids would work for the initial cloning of the gene of interest, the use of a plasmid carrying a DUS ensures that the construct carrying the unmarked mutation will be able to transform N. gonorrhoeae at high frequency. Plasmid pUP1 and its deriviatives, including pKH6, carry one copy of the 12-bp extended DUS and one copy of the 10-bp DUS and are thus efficiently taken up into N. gonorrhoeae (Figure 1). For the streptomycin resistant N. gonorrhoeae strain, many gonococcal strains would serve, as a significant number of strains naturally carry the rpsL allele conferring resistance. Commonly-used strains MS11 and FA1090 are streptomycin resistant. Furthermore, since a single point mutation confers resistance, spontaneously arising streptomycin resistant isolates may be easily selected for any strain.
Figure 4A.2.1.
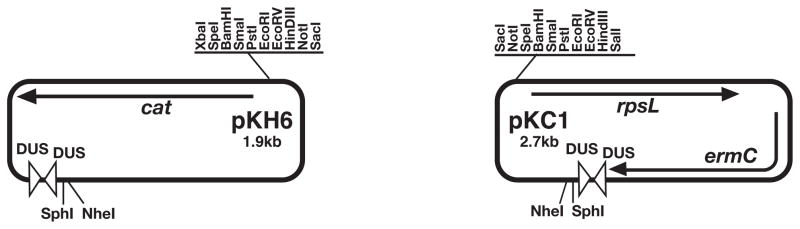
Plasmids for use in creation of unmarked mutations in Neisseria gonorrhoeae.
Careful attention should be paid to the screening steps in this method. Be certain that the gonococcal transformant with the cassette insertion is streptomycin sensitive. Streptomyin resistant mutants will arise in the population carrying the two rpsL alleles. They tend to arise at a frequency higher than the spontaneous mutation rate, suggesting that the presence of the two rpsL alleles is detrimental to the bacteria. Results have shown that gonococci will both acquire spontaneous mutations in the introduced rpsL allele and undergo a gene conversion event that incorporates the resident rpsL mutation into the introduced allele (Kohler, Cloud et al. 2005). Thus, many streptomycin resistant isolates will be obtained in the transformation with the unmarked mutation construct, and it will be necessary to screen these for erythromycin sensitivity. Nevertheless, this selection and screening procedure can be easier and more successful for the introduction of some mutations into gonococci than screening alone.
Materials
Gene of interest
pKH6, pUP1 (Elkins, Thomas et al. 1991), or similar plasmid carrying a DUS and a marker conferring resistance other than erythromycin
pKC1 (Cloud and Dillard 2002) or similar plasmid carrying an ermC/rpsL cassette
PCR primers for introducing an unmarked mutation into gene of interest
E. coli
N. gonorrhoeae, streptomycin resistant strain
GCB plate (see recipe) containing erythromycin (Table 4A.2.1)
GCB plate (see recipe) containing streptomycin (Table 4A.2.1)
Dacron swab, sterile (Fisher Scientific)
Additional reagents and equipment for preparing a miniprep (Engebrecht et al., 1991), spot transformation (Basic Protocol 1), and PCR (Support Protocol)
-
1
Clone gene of interest with ~1kb of flanking DNA into pKH6 or similar plasmid and transform into E. coli.
-
2
Create an unmarked mutation such as a deletion or point mutation in the gene of interest using inverse PCR or the quick-change method (Stratagene) with mutagenic PCR primers, and transform into E. coli.
-
3
Excise the ermC/rpsL cassette from pKC1 and clone it into the site of the gene of interest.
Note that it is often useful to introduce the ermC/rpsL cassette at the site of the unmarked deletion or point mutation, although other sites very close to the mutation would also work. -
4
Purify both plasmids using standard miniprep procedure (Engebrecht et al., 1991).
-
5
Use 200 to 500 ng of the plasmid carrying the ermC/rpsL cassette and the streptomycin resistant N. gonorrhoeae strain to perform spot tranformation (Basic Protocol 1) on a GCB plate.
-
6
Use a sterile Dacron swab to transfer all of the resulting colonies from a DNA spot onto a GCB plate containing erythromycin. Incubate 24 to 48 hr.
-
7
Pick individual colonies and streak them simultaneously on a GCB plate containing erythromycin and a GCB plate containing streptomycin. Choose a transformant that is resistant to erythromycin and sensitive to streptomycin. This transformant should have an insertion mutation in the gene of interest in the chromosome.
-
8
Use 200 to 500 ng of the plasmid carrying the unmarked mutation in the gene of interest and the erythromycin-resistant, streptomycin-sensitive N. gonorrhoeae transformant to perform spot tranformation (Basic Protocol 1) on a GCB plate.
-
9
Use a sterile Dacron swab to transfer all of the resulting colonies from a DNA spot onto a GCB plate containing streptomycin. Incubate 24 to 48 hr.
-
10
Pick individual colonies and streak them simultaneously on a GCB plate containing erythromycin and a GCB plate containing streptomycin. Choose a transformant that is resistant to streptomycin and sensitive to erythromycin. This transformant should have replaced the insertion mutation in the gene of interest with the unmarked mutation.
BASIC PROTOCOL 5 SHUTTLE MUTAGENESIS OF NEISSERIA GONORRHOEAE DNA CLONED INTO PHSS6 PLASMIDS USING MTNCMNS
One of the most common methods of mutagenesis in N. gonorrhoeae is insertional mutagenesis using an antibiotic resistance marker to interrupt a gene or replace part or all of its coding sequence. The number of selectable markers for N. gonorrhoeae is limited. Researchers have reported using cat (chloramphenicol resistance), ermC (erythromycin resistance), tetM (tetracycline resistance), aph3 (kanamycin resistance), bla TEM-type (penicillin resistance), and the omega cassette (spectinomycin and streptomycin resistance). These markers are often introduced directly into the gene of interest cloned on a plasmid in E. coli and introduced into the gonococcal chromosome by natural transformation and allelic exchange. However, mutations may also be produced using plasmid or transposon insertions in the chromosome as described below.
Transposons generally do not transpose in N. gonorrhoeae. However, researchers wishing to make transposon mutants of gonococci have circumvented this problem in two ways. In the first method, gonococcal DNA is cloned into plasmids and mutagenized with transposons in E. coli, then introduced back into gonococci by natural transformation, a technique known as shuttle mutagenesis. Derivatives of Tn3 and Tn1721 have been adapted for use in gonococci, including derivatives containing reporters lacZ, or phoA (Seifert et al., 1990; Boyle-Vavra and Seifert, 1993, 1994; Haas et al., 1993). A second method is to purify gonococcal DNA and mutagenize it with transposons in vitro. Both Tn5 and Mariner derivatives have been used for this purpose in N. gonorrhoeae and N. meningitidis (Pelicic et al., 2000; Sechman et al., 2005).
Materials
E. coli strains RDP146(pTCA), RDP146(pOX38::mTnCmNS), and NS2114Sm (Boyle-Vavra and Seifert, 1993)
LB medium (APPENDIX 4A)
1 mM HEPES, pH 7 (optional): sterilize by autoclaving; store up to 1 year at room temperature
10 ng/μl target plasmid
LB plates (APPENDIX 4A) containing 40 μg/ml kanamycin
LB plates containing 40 μg/ml kanamycin and 12 μg/ml tetracycline
LB plates containing 40 μg/ml kanamycin and 25 μg/ml chloramphenicol
LB plates containing 100 μg/ml streptomycin, 40 μg/ml kanamycin, and 25 μg/ml chloramphenicol
GCB plates (see recipe) containing chloramphenicol (Table 4A.2.1)
Electoporation cuvette, 2-mm gap length
Electroporator
Additional reagents and solutions for PCR (Support Protocol; optional) and spot transformation with N. gonorrohoeae (Basic Protocol 1)
-
1
Incubate E. coli strain RDP146(pTCA) in LB medium several hours to overnight at 37°C.
pTCA encodes the transposase for Tn3. -
2
Microcentrifuge desired volume of cells (generally ~1.5 ml) for 30 sec at 12,000 rpm. Resuspend cells in 1 ml low ionic strength buffer, such as sterile 1 mM HEPES, pH 7. Repeat wash two more times.
-
3
Resuspend cells in 40 μl 1mM HEPES per planned electroporation.
-
4
Add target plasmid to cell suspension.
-
5
Transfer cell/DNA mixture into an electroporation cuvette. Electroporate with the pulse controller set at 2.5 kV, 200 Ω, and 25 μF.
-
6
Plate 10 and 100 μl electrotransformation mix onto LB plates containing 40 μg/ml kanamycin. Incubate overnight at 37°C.
-
7
Use sterile toothpicks to transfer several potential transformants onto LB plates
-
containing 40 μg/ml kanamycin and 12 μg/ml tetracycline.
Transformants should be resistant to both antibiotics and show two plasmids when examined by whole-cell lysis (cracking) gel (Kado and Liu, 1981; Dillard and Yother, 1994). Such transformants will serve as the primary recipient. -
8
Grow E. coli strain RDP146(pOX38::mTnCmNS) and primary recipient (step 7) to
mid-log phase (OD600 of 0.5) without antibiotics. Mate at a ratio of 1:1 for 1 hr at
-
37°C without shaking.
This step brings the transposon into the strain carrying the transposase and the target plasmid. -
9
Plate 100, 10, and 1 μl onto individual LB plates containing 40 μg/ml kanamycin and 25 μ/ml chloramphenicol to select for KnRCmR transformants. Incubate ~16 hr at 37°C.
Any RDP146 cells not carrying all three plasmids will be killed in this step. -
10
Grow NS2114Sm to mid-log phase (OD600 of 0.5) in LB medium without antibiotics
-
(~4 hr).
This step can be started towards the end of the incubation described in step 9. -
11
Wash or scrape transconjugates from plates into LB medium and mix 1:1 with mid-log phase NS2114Sm.
-
12
Mate 15 min in a 37°C water bath. Incubate 30 min in rotator at 37°C.
-
13
Plate 100, 10, and 1 μl onto individual LB plates containing 100 μg/ml streptomycin, 40 μg/ml kanamycin, and 25 μg/ml chloramphenicol. Incubate ~16 hr at 37°C.
-
14
If desired, screen for transposon location and orientation by PCR (see Support Protocol).
-
15
Transform plasmid DNA into gonococci by spot transformation (Basic Protocol 1).
Select for transformants on GCB plates containing 25μg/ml chloramphenicol.
BASIC PROTOCOL 6 CHEMICAL MUTAGENESIS
Chemical mutagenesis has been used in N. gonorrhoeae. Its use is infrequent since directed mutagenesis is highly efficient. Chemical mutagens that directly act on the DNA are effective (Campbell and Yasbin, 1984). However, chemical mutagenesis methods that rely on error-prone repair are not, since N. gonorrhoeae has no SOS-type system (Black et al., 1998). The procedure below describes use of ethylmethane sulphonate (EMS). N-Methyl-N′-nitro-N-nitrosoguanidine (MMNG) could be used instead, at 5 μg per 108 cfu.
Materials
N. gonorrhoeae strain, frozen
GCB plates (see recipe)
GCBL medium with Kellogg’s supplements and sodium bicarbonate (see recipe), 37°C
Ethyl methanesulfonate (EMS)
GCBL medium
GCBL medium containing 15% glycerol
GCB plates containing nalidixic acid (Table 4A.2.1)
Dacron Swab
-
1
Streak N. gonorrhoeae strain from frozen stock onto a GCB plate. Incubate overnight.
-
2
Using a sterile Dacron swab, transfer cells from 1/4 to 1/2 of the plate into 3 ml GCBL medium with Kellogg’s supplements and sodium bicarbonate, 37°C, to an
OD540 of 0.2. Retain a 0.5-ml aliquot.
-
3
Add EMS to a final concentration of 1% (v/v), e.g., 20 μl to a 2-ml culture.
-
4
Incubate with aeration and remove 0.5-ml aliquots at 10, 20, 30, and 40 min. For each time point, immediately microcentrifuge aliquot 30 sec at 12,000 rpm and remove supernatant. Resuspend cells with 0.5 ml GCBL.
Do not forget to include the untreated culture (0-min time point) taken in step 2. -
5
Prepare serial dilutions over a range of 100 to 10−6. To determine killing frequency, plate 100 μl of each dilution onto an individual GCB plate for quantification of colony forming units. Plate 100 μl of each dilution onto an individual GCB plate containing nalidixic acid to determine mutation frequency.
Point mutations in the gyrB gene result in nalidixic acid resistance (NalR). Thus, measuring the frequency of mutations conferring nalidixic acid resistance gives a measure of the effectiveness of the chemical mutagenesis procedure. -
6
Microcentrifuge the remainder of each undiluted aliquot 30 sec at 12,000 rpm. Resuspend cells in GCBL containing 15% glycerol and freeze at −70°C.
-
7
After overnight growth, identify time-points exhibiting 99% to 99.9% killing and those exhibiting the highest frequency of NalR colonies.
Typically 30- and 40-min time points are optimal. -
8
Streak bacteria onto GCB plates from the frozen, mutagenized stock from step 6 that gave the highest mutation frequency or 99% to 99.9% killing frequency, then screen for phenotype.
BASIC PROTOCOL 7 COMPLEMENTATION ON THE NEISSERIA GONORRHOEAE CHROMOSOME
Complementation in N. gonorrhoeae is usually done by insertion of the gene of interest onto the gonococcal chromosome. Because a majority of DNA that enters the cell during natural transformation is single-stranded, it is not restricted, and is therefore much more efficiently incorporated than replicating plasmids. Chromosomal sites that have been used for complementation include within or adjacent to the iga gene encoding IgA1 protease, and an intergenic region between lctP and aspC (Mehr et al., 2000). IgA protease is not essential for growth of N. gonorrhoeae; therefore, it can be interrupted without affecting in vitro phenotypes (Koomey et al., 1982). However, intergenic insertions are preferred since they are not known to affect any phenotypes. In theory, most any location that does not interrupt a gene or operon will work for this process. However, significant work has gone into making the lctP-aspC constructs useful for gonococcal complementation. A series of plasmids was created containing more than 3 kb of the lctP-aspC region, interrupted by a polylinker, an antibiotic resistance marker, and in some cases, a regulatable promoter. Transformation of gonococci followed by selection for the antibiotic resistance marker leads to selection of bacteria that have incorporated the construct by double cross-over recombination. Genes cloned into the construct are thus introduced into the chromosome with the antibiotic resistance marker between lctP and aspC. Plasmid pGCC6 contains two copies of the lac promoter-operator, Lac repressor gene lacIQ, allowing for inducible expression of the gene of interest, and cat, conferring chloramphenicol resistance (Mehr et al., 2000). Plasmid pKH35 is a smaller pGCC6 derivative with a more extensive polylinker (Hamilton et al., 2005). Plasmid pGCC4 is similar to pGCC6 but carries an ermC marker instead of cat, and pGCC2 carries only the ermC marker (Stohl et al., 2003). New vectors for making insertions between iga and trpB were recently created, pMR32 and pMR33. These plasmids carry either the regulatable lac promoter-operator like the pGCC plasmids or carry the consititutive opaB promoter (Ramsey and Dillard 2010). Plasmids for N. gonorrhoeae complementation are shown in Figure 2.
Figure 4A.2.2.

Plasmids for expression of genes from the Neisseria gonorrhoeae chromosome.
Materials
Gene of interest
pKH35 (Hamilton et al., 2005) or similar plasmid
E. coli
N. gonorrhoeae mutant of interest
GCB plate (see recipe) containing chloramphenicol (Table 4A.2.1)
1 M IPTG solution
Dacron swab, sterile (Fisher Scientific)
Additional reagents and equipment for preparing a miniprep (Engebrecht et al., 1991), spot transformation (Basic Protocol 1), and PCR (Support Protocol)
-
1
Clone gene of interest into pKH35 or similar plasmid and transform into E. coli.
-
2
Purify plasmid using standard miniprep procedure (Engebrecht et al., 1991).
-
3
Use 200 to 500 ng plasmid and the N. gonorrhoeae mutant of interest to perform spot tranformation (Basic Protocol 1) on a GCB plate.
-
4
Use a sterile Dacron swab to transfer all of the resulting colonies from a DNA spot onto a GCB plate containing chloramphenicol. Incubate 24 to 48 hr.
-
5
Pick individual colonies and screen by PCR for presence of gene of interest adjacent to lctP (see Support Protocol).
-
6
Induce expression of the gene of interest by adding of 1 M IPTG to a final concentration of 1 mM either in liquid culture or on solid medium.
ALTERNATE PROTOCOL 1 COMPLEMENTATION WITH REPLICATING PLASMIDS
Complementation with replicating plasmids is rarely performed with N. gonorrhoeae. During transformation, plasmid DNA is cut by a nuclease prior to entry into the cell. Unless the incoming DNA is homologous to the gonococcal chromosome or a resident plasmid, and the cut occurs in the homologous segment, the incoming plasmid cannot reform a circle. A circle can be reformed if the cell brings in more than one copy of the plasmid. Thus, transformation with replicating plasmids follows “two-hit” kinetics. Additionally, the introduced plasmid is subject to restriction, further reducing the transformation frequency. The introduced plasmid is subject to restriction, further reducing the transformation frequency. Use of replicating plasmids would be necessary for complementation of certain recombination- or transformation-deficient mutants that would be unable to take up or incorporate DNA by natural transformation. The complementing plasmid could be introduced by electroporation. Also, higher copy numbers for some replicating plasmids would facilitate increased expression of introduced genes. Plasmids have been developed for complementation in gonococci and commonly rely on the replication origin of the gonococcal β-lactamase plasmids or the cryptic plasmid. Two such plasmids are pLES2 (Stein et al., 1983) and pFP10 (Pagotto et al., 2000). A shuttle plasmid was also developed from an N. flavescens cryptic plasmid (O’Dwyer, Langford et al. 2005). In addition, broad-host range plasmids of the IncP and IncQ groups will replicate in gonococci and can be used for complementation. A complementation method based on the conjugative plasmid ptetM25.2 from N. gonorrhoeae has been described (Kupsch et al., 1996).
BASIC PROTOCOL 8 PREPARATION OF CHROMOSOMAL DNA FROM NEISSERIA GONORRHOEAE GROWN ON SOLID MEDIUM
Purified chromosomal DNA is required for Southern analysis of mutants and complemented strains. For PCR amplification of gonococcal genes for cloning, use of purified chromosomal DNA as template gives cleaner results with fewer false-priming products. Gonococci are less likely to autolyse when grown on agar plates as compared to growth in liquid medium. Therefore, preparation from gonococci grown on agar plates is the most reliable method for chromosomal DNA purification.
Materials
N. gonorrhoeae, overnight culture
GCB plates (see recipe)
TES buffer (see recipe)
10 mg/ml RNase A
10% SDS
Tris-buffered phenol (APPENDIX 2A)
Chloroform
3 M sodium acetate
95% and 70% ethanol
Dacron swab, sterile
13-ml snap-cap polypropylene tubes
1-ml pipette tips with wide bore
-
1
Using an inoculating loop, streak three to five colonies of N. gonorrhoeae onto each of eight to ten GCB plates. Incubate overnight.
-
2
Using one sterile Dacron swab per plate, tranfer and suspend cells in 5 ml TES buffer in a 13-ml snap-cap polypropylene tube.
-
3
Add 1 μl of 10 mg/ml RNase A (10 mg/ml) and 50 μl of 10% SDS. Incubate 10 min at 37°C to digest RNA.
Lysis should occur instantly. -
4
Extract with an equal volume of tris-buffered phenol. Mix by gentle inversion twenty times. Centrifuge 10 min at 6000 × g (7000 rpm), 4°C.
-
5
Transfer aqueous (top) phase to a new tube using a micropipette equipped with a wide-bore 1-ml pipette tip and repeat extraction.
-
6
Extract once with chloroform. Invert and centrifuge as in step 4.
-
7
Transfer aqueous (top) phase to a new tube. Add 1/10 vol 3 M sodium acetate and 2 vol of 95% ethanol. Mix and incubate 30 min at −20°C.
-
8
Centrifuge 10 min at 12,000 × g, 4°C.
-
9
Wash pellet twice with 0.5 ml 70% ethanol.
-
10
Invert tube and allow pellet to dry until white or clear.
-
11
Suspend DNA in ~200 to 500 μl water.
ALTERNATE PROTOCOL 2 PREPARATION OF CHROMOSOMAL DNA FROM NEISSERIA GONORRHOEAE GROWN IN BROTH
Growth of N. gonorrhoeae in liquid culture makes the preparation steps for DNA purification easier than those for preparing DNA from bacteria grown on plates. The bacteria can be harvested by centrifugation (instead of swabbing) and the recovery of bacteria is more efficient. The drawback to this method is that the cultures must be monitored closely to ensure that the bacteria are still in the growth phase and have not begun to autolyse before chromosome purification begins. Autolysed gonococcal cultures still exhibit significant optical density (OD540 of ~1.2), but purification of DNA from these cultures gives low yields and mostly degraded DNA. However, if growth is monitored to ensure growing cells are used, preparation of DNA from 200 ml of late-log phase gonococci (in this protocol) provides significantly more DNA than the preparation from ten agar plates. This method does not have an RNase step. Rather, the RNA functions as carrier nucleic acid, allowing the DNA and large RNA to be easily recovered with a glass hook.
Materials
N. gonorrhoeae, frozen stock
GCB plate (see recipe)
GCBL medium with Kellog’s supplements and sodium bicarbonate (see recipe), 37°C
TE buffer (APPENDIX 2A)
10% SDS
5 M potassium acetate
95% and 70% ethanol
Dacron swab
13-ml polypropylene tube
Glass rod
65°C water bath
-
1
Streak N. gonorrhoeae from frozen stock onto a GCB plate and incubate overnight.
-
2
Using a sterile Dacron swab, transfer 1/4 to 1/2 of colonies on the plate to 3 ml GCBL medium, 37°C, with Kellogg’s supplements and sodium bicarbonate.
-
3
Incubate until culture reaches late log phase (OD540 of 1.0 – 1.4; ~4 to 6 hr). Transfer
to 200 ml GCBL, 37°C, with supplements and sodium bicarbonate.
-
4
Incubate until culture reaches late log phase (OD540 of 1.4; <20 hr).
-
5
Centrifuge culture 10 min at 3,000 × g, 4°C. Resuspend in 2.5 ml TE buffer on ice.
-
6
Transfer to a 13-ml polypropylene tube. Add 10% SDS to 1% (~300 μl of 10%) and lyse the cells 15 min at 65°C.
-
7
Add 1/5 vol of 5 M potassium acetate (~700 μl) and incubate an additional 15 min in a 65°C water bath. Transfer to ice and incubate 60 min.
-
8
Remove cell debris by centrifuging 15 min at 12,000 × g, 4°C. Add supernatant to 2 vol of 95% ethanol. Hook out the DNA with a glass rod.
Addition of the cleared lysate to ethanol will cause the DNA and RNA to come out of solution and appear as long white strands or gobs floating in the liquid. A glass hook can easily be fashioned from a Pasteur pipette using a Bunsen burner. The hook should be sealed and contain at least a slight bend. If the DNA cannot be removed with a hook it can still be recovered by centrifugation. -
9
Wash DNA once with 0.5 ml 70% ethanol, allow it to dry, and resuspend in 200 to 500 microliters TE buffer.
SUPPORT PROTOCOL PREPARING PCR TEMPLATES FROM NEISSERIA GONORRHOEAE COLONIES
It is often necessary to screen gonococcal transformants for the presence of the introduced mutation or construct. This process can often be done by PCR or PCR followed by restriction digest. One gonococcal colony contains sufficient DNA for this process, although it will work better with three to five colonies from the isolate. The resulting PCR product can then be examined for its size, size following digestion, or simply its presence using agarose gel electrophoresis.
Materials
N. gonorrhoeae growing on solid medium
Colony lysis solution (see recipe)
Appropriate restriction enzyme(s) and buffer(s) (optional)
Thermocycler and appropriate PCR tubes
Additional reagents and equipement for PCR (Kramer and Coen, 2001)
-
1
Scrape an amount of N. gonorrhoeae equal to one to five colonies and transfer to 50 to 100 μl colony lysis in a PCR tube.
-
2
Lyse cells 15 min at 94°C in a thermocycler followed by incubation for 5 min at 25°C.
-
3
Use 2.5 μl lysate as template to a 25-μl PCR reaction (Kramer and Coen, 2001).
-
4
Use 5 μl PCR product in 30 μl restriction digest, if identification of a restriction site is necessary.
See Bloch (1995) for more information about restriction mapping.
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see APPENDIX 2A; for suppliers, see SUPPLIERS APPENDIX.
Colony lysis solution
40 μl 500 mM EDTA
200 μl Tris·Cl, pH 8.5 (APPENDIX 2A)
100 μl Triton X-100
9.66 ml H2O
Store up to 1 year at room temperature
Scale as needed.
GC medium base
42% (w/w) Proteose peptone #3 (15.10 g)
3% (w/w) cornstarch (1.01 g)
11% (w/w) K2HPO4 (4.03 g)
3% (w/w) KH2PO4 (1.01 g)
14% (w/w) NaCl (5.03 g)
28% (w/w) agar (10.07 g)
Weight amounts given in parentheses indicate appropriate mix for preparing GCB plates (see below).
GCB plates
36.25 g GC Medium Base (Difco; also see recipe)
1.25 g agar
1 liter H2O
Autoclave with stir bar
Cool to 50° to 60°C
Add 10 ml filter-sterilized Kellogg’s supplements I (see recipe)
Add 1 ml filter-sterilized Kellogg’s supplement II (see recipe)
Pour into 100-mm plates using 20 ml per plate
Store plates sealed in bags up to 6 months at 4°C
GCBL medium
Dissolve the following in 1 liter H2O:
15 g proteose peptone #3
4 g K2HPO4
1 g KH2PO4
1 g NaCl
Adjust pH to 7.2 with HCl if necessary
Autoclave and store indefinitely at room temperature
Add Kellogg’s Supplements I & II to 1× each from individual or a combined stock (see recipe)
-
Add sodium bicarbonate to 0.042% (final) from a 100× stock (0.42 g in 10 ml H2O; filter sterilize)
Unless the protocol step specifcally indicates that Kellogg’s supplements or sodium bicarbonate are to be included in the medium, these reagents should be omitted.It is only necessary to add Kellogg’s supplements when the bacteria are to be grown in the medium. It is not necessary to add Kellogg’s supplements when the bacteria are diluted or transformed in GCBL, i.e., steps when the bacteria are only in the medium for a short period of time. Sodium bicarbonate is added only when the bacteria are to be grown in liquid culture with aeration (in sealed tubes or flasks) and not when they are grown in static culture in a CO2 incubator. Unless the protocol step specifically indicates that Kellogg’s supplements or sodium bicarbonate are to be included in the medium, these reagents can be omitted.
Kellogg’s supplements
Supplements I and II may be mixed (10 ml supplement I + 1 ml supplement II) and frozen in 11-ml aliquots. One such aliquot is sufficient to supplement 1 liter of GCB or GCBL medium (see recipe). Filter sterilize before use.
Supplement I, 100×
40 g glucose
1 g glutamine
2 mg cocarboxylase (thiamine pyrophosphate)
Adjust volume to 100 ml with H2O
Sterilize by passing through a 0.2-μm filter
Store indefinitely at −20°C
Supplement II, 1000×
50 mg Fe(NO3)3·9H2O
Adjust volume to 100 ml with H2O
Sterilize by passing through a 0.2-μm filter
-
Store indefinitely at −20°C
An alternative to preparing supplements is to purchase them lyophilized. Supplement Iso Vitalex (Becton Dickinson) can be used in place of Supplements I & II.Do not store Kellogg’s supplements at 4°C or room temperature.
Graver Wade (GW) medium
Prepare 1L of M199 cell culture medium (commercially available with Earle’s salts, but without L-glutamine, phenol red, or sodium bicarbonate) in distilled, deionized H2O. A further 500ml H2O is added plus (grams):
| glucose | 10.0 |
| ammonium bicarbonate | 2.0 |
| sodium acetate | 10 |
| glutamine | 0.75 |
| spermidine& | 0.2 |
| arginine | 0.1 |
| hypoxanthine | 0.05* |
| uracil | 0.05* |
| oxaloacetate | 0.05 |
| thiamine HCl | 0.05 |
| NAD | 0.01 |
| ornithine | 0.01 |
| sodium lactate(60% syrup) | 2.5ml |
Adjust pH to 6.8 with 1M NaOH and sterilize through 0.22μ filter.
Dissolve in 5ml 1M NaOH.
Spermidine is not necessary for many N. gonorrhoeae strains.
This medium can be stored at 4 C for up to three weeks.
TES
250 μl 1 M Tris·Cl, pH 8 (APPENDIX 2A)
200 μl 0.5 M EDTA
50 μl 5 M NaCl
4.5 ml H2O
Store up to 1 year at room temperature
COMMENTARY Background Information
Natural transformation of Neisseria gonorrhoeae was first reported by Sparling (1966). Over the years it has been found that the high degree of gonococcal transformation has led to quick spread of antibiotic resistance markers throughout the gonococcal population and has created a problem for the treatment of gonococcal infections. Transformation also leads to variation in genes coding for gonococcal antigens, allowing the bacteria to avoid the host immune response (Cooke et al., 1998), although significant variation of some antigen-encoding genes occurs by intracellular recombination (Stern et al., 1986; Seifert, 1996). As is the case for many naturally transformable bacteria, the process of transformation involves the components of a type IV pilus. Multiple mutations affecting piliation or pilus function in N. gonorrhoeae have been shown to block natural transformation (Chen and Dubnau 2004) (Hamilton and Dillard 2006). The gene for PilE, the pilus subunit, undergoes high frequency recombination. This recombination results in altered pilin protein sequence allowing the bacteria to escape the host immune response. However, some of the recombination events eliminate pilin expression or result in pilin proteins that cannot be assembled into pili. The resulting nonpiliated bacteria are not transformable. Therefore, care should be taken to choose colonies of piliated N. gonorrhoeae for transformation procedures. See Figure 3.
Figure 4A.2.3. Gonococcal colony morphologies.

Gonococci expressing pili (P+) form smaller colonies that appear to have a dark ring around the edge. This effect is due to the cells aggregating to form a mound. Nonpiliated cells form a pancake-shaped colony that has a greater diameter and a fuzzier edge. In addition, when the colonies are viewed with oblique substage lighting, a range of colony opacities will be detected. Colony opacity is due to presence of outer membrane opacity (Opa) proteins. Translucent (Opa−) colonies appear clear or somewhat blue, whereas opaque colonies may appear yellow and refract the light to give an almost crystalline or granular appearance.
The gonococcal genome sequence was completed for strain FA1090 and is accessible in GenBank (accession number AE004969). A useful annotation is available at the STD genomes website of Northwestern Universtiy (http://stdgen.northwestern.edu). Although there are some significant differences in the genomes of N. gonorrhoeae strains (Gibbs and Meyer, 1996; Hamilton et al., 2005), the sequence information obtained for strain FA1090 is generally useful for searching for genes of interest and for designing primers for cloning gonococcal genes. A second completed genome sequence is available for strain NCCP11945 (GenBank accession number CP001050), and 14 draft genome sequences are available on the Broad institute website (http://www.broadinstitute.org/annotation/genome/neisseria_gonorrhoeae/MultiHome.html).
Critical Parameters and Troubleshooting
Safety considerations
N. gonorrhoeae is a human pathogen. It should be used under biosafety level 2 (BSL-2) conditions (UNIT 1A1). Most importantly, wear safety glasses. This is vital when working with cultures that might splatter or other conditions that might propel gonococci into the eyes of the researcher, such as sonication or streaking colonies with an inoculating loop. Exposure of the eyes will cause conjunctivitis. Exposure of any mucosal surface may result in infection. Gonococcal infections should be considered serious as they can result in septicemia and death. Treatment by a physician should be sought following any mucosal exposure to N. gonorrhoeae. Antibiotics are effective in clearing the infection, but laboratory workers with gonococcal eye infections should request systemic (oral or injection) antibiotic treatment since antibiotic eye drops are not always effective.
Troubleshooting growth, death, and autolysis
One of the most difficult aspects of working with N. gonorrhoeae is its tendency to die in culture or to not grow when inoculated into a culture. To avoid these problems, it is best to keep gonococci under growth conditions at all times or to shorten the length of time they are in nonoptimal conditions (such as centrifugations or washes) as much as possible. It is important to prewarm media to 37°C. Streak bacteria from frozen stock onto prewarmed agar plates that contain no antibiotic. Although the bacteria may exhibit a significant level of resistance to an antibiotic while growing, they appear incapable of surviving some selections when restarting from frozen stocks. Finally it is advisable to inoculate at a density of ~107 to 108 CFU/ml when growing the bacteria in liquid culture. Although the bacteria may grow in liquid culture at lower densities, they often exhibit a growth lag lasting for a considerable period, and the lag is often followed by autolysis.
Gonococci are fastidious, requiring rich medium for growth. Complex media work best, but defined media have also been devised that will support the growth of gonococci (LaScolea and Young, 1974; Morse and Bartenstein, 1980). Furthermore, N. gonorrhoeae will grow in some media designed for tissue culture, particularly if supplemented with a few additional nutrients (Hamilton et al., 2001). The growth rate in the defined media is significantly reduced compared to that in rich media, and doubling time is generally ~120 min. A recently developed defined medium (Graver-Wade medium) combines these approaches, using M199 tissue culture medium as the base and adding 13 additional components. This defined medium is thus simpler to make than other defined media for gonococci. Also, N. gonorrhoeae grows in Graver-Wade medium at a rate similar to that observed for growth in complex media such as GCBL (Wade and Graver 2007).
Gonococci are prone to die under any condition not favorable for growth. Furthermore they appear to have no true stationary phase, and are likely to be actively dying if they are not actively growing (Morse and Bartenstein, 1974; Hebeler and Young, 1975). They cannot be maintained alive on ice. Autolysis generally accompanies death, but there are conditions that can be used prevent autolysis (although the bacteria will still die). Incubation in low pH buffer (usually pH 5.5, 50 mM sodium phosphate or Tris·Cl) and/or on ice will limit autolysis. EDTA and other chelators of divalent cations greatly stimulate autolysis and should be avoided if lysis is not desired (e.g., cell fractionation procedures; Wegener et al., 1977).
Those first starting to work with N. gonorrhoeae often experience contamination of their liquid cultures. With a doubling time of 60 min, gonococci grow significantly slower than bacteria that commonly contaminate laboratory cultures. A useful method for overcoming this difficulty is to use streptomycin (100 μg/ml) in the medium. Many gonococcal strains, including the common laboratory strains FA1090 and MS11, carry a mutation in the rpsL gene conferring streptomycin resistance. The mutation is not universal; some common strains such as F62 do not carry the rpsL mutation.
Mapping insertions or deletions by Southern blotting
Transformants carrying mutations are generally first screened by PCR, however it is often desirable to confirm the mutation by Southern blotting. For this purpose it should be noted that gonococci have extensively methylated DNA, and many restriction enzymes do not cut gonococcal DNA efficiently. Restriction enzymes ApaI, BamHI, BclI, NotI, and XbaI have been problematic for N. gonorrhoeae strain MS11.
Colony variants
Gonococci undergo spontaneous high-frequency variation of surface molecules by well described mechanisms (Seifert, 1996; Stern et al., 1986). This variation is seen in colony variants that appear on agar plates. For the purposes of growing gonococci, it should be noted that expression of pili and/or opacity proteins results in clumping during growth in liquid culture. The clumps are difficult to disperse and make it difficult to follow culture growth using OD measurements or plating. Therefore it is advisable to use nonpiliated (P ), transparent (Opa−) variants unless these molecules are required for other purposes (e.g., transformation or adhesion and invasion assays). Piliated variants form colonies that are small and convex and thus appear to have a dark ring around them (Kellogg et al., 1963). Nonpiliated variants form larger, flatter colonies. Opacity protein expression is judged using a stereo dissecting microscope and oblique light. Opa− variants may appear transparent or slightly blue. Opa+ variants are opaque, somewhat yellow, and refractory (Swanson et al., 1971). Nonpiliated variants will come to predominate in liquid culture due to a slightly faster growth rate (Salgado-Pabón, Du et al. 2010). Nearly all colonies will appear opaque on an old (>24-hr) plate due to variation occurring in a proportion of the cells in a colony at an appreciable rate.
Constructs for controlling pilin antigenic variation
Gonococci are notoriously variable, spontaneously altering pilin sequence as well as expression of genes for opacity proteins, lipooligosaccharide types, and many others. Much of the variation occurs by slipped-strand mispairing and cannot be controlled (Belland, 1991). However, pilin variation can be controlled, thereby preventing variation that might affect adherence and transformation. Two types of genetic alterations have been described that prevent pilin variation. The first, and most widely used constructs, are mutations in recA. Since pilin variation requires homologous recombination, mutations in recA or eliminating recA transcription eliminates pilin variation (Koomey et al. 1987). Both recA knockouts and recA-inducible constructs have been created. The recA6 allele carries a lac promoter-operator upstream of recA, allowing recombination to occur in the presence of IPTG with virtually no recombination (and pilin variation) in the absence of induction (Seifert, 1997). It was recently shown that a 16-bp region upstream of pilE (the gene for the pilin subunit) is necessary for pilin variation (Cahoon and Seifert 2009). The sequence forms a G-quartet structure, and mutations that block the formation of this structure block pilE recombination. An insertion mutation in this region (pilE::Tn5#1) was described for strain FA1090 that prevents pilE variation but does not affect production of PilE protein (Sechman, Rohrer et al. 2005; Sechman, Kline et al. 2006). This strain carries a kanamycin resistance marker in the transposon and thus the insertion might easily be moved to other gonococcal strains by transformation with selection. Multiple point mutations were also identified that prevent pilE variation, and these are also in the 16-bp region. Such mutants would be useful in situations where unmarked strains are desired. Both types of mutations allow pilin expression but prevent variation (Sechman et al., 2005)(Cahoon and Seifert 2009). Such strains are more useful than the recA strains since they allow normal homologous recombination required for natural transformation and genetic manipulation.
Troubleshooting transformation
If no transformants are obtained, the problem is often with the donor DNA. Impurities introduced in the preparation of plasmids from E. coli, such as salt or ethanol, will kill the potential N. gonorrhoeae recipients. In the spot transformation procedure, the bacteria will either not grow on the spot where the DNA was added to the plate or will only form very tiny colonies. Reprecipitation of the DNA followed by two washes with 70% ethanol is usually sufficient to remove the impurities. Adding more DNA to the transformation can also be helpful. Increasing the usual 100- to 200-ng amount of DNA to 1 μg may significantly increase the transformation frequency without damaging viability of the gonococcal recipients. Since DNA must contain the gonococcal DNA uptake sequence to act efficiently in transformation, it is important to make sure the DNA uptake sequence is present in the plasmid and was not cut away from the DNA of interest in preparation for transformation. Plasmids used for transformation should be linearized prior to transformation if they are smaller than ~8 kb (unless plasmid insertion in the chromosome is desired). Care should be taken to ensure that the enzyme chosen does not separate the uptake sequence from the DNA of interest. As a control for transformation a plasmid carrying a point mutation conferring antibiotic resistance is frequently used. The plasmid pSY6 conferring nalidixic acid resistance is useful for this purpose (Stein, 1991).
Anticipated Results
Transformation of N. gonorrhoeae is highly efficient. If using the spot transformation method to introduce mutations into the gonococcal chromosome (Basic Protocol 1), frequencies of 10% to 50% transformant/total CFUs or greater may be expected (Gunn and Stein, 1996). Screening of ten potential transformants is generally sufficient to identify the mutant of interest, even without selection. Using liquid transformation (Basic Protocol 2), frequencies ~1% may be expected. In general, the frequency will depend on the length of the homologous region targeting the mutation and the length of any heterologous sequence being inserted. Lengths of homologous DNA of ~500 to 1 kb on each side of the mutation or insertion to be introduced are sufficient to give efficient transformation. Long lengths of heterologous sequence to be inserted will greatly reduce the transformation frequency, although insertions >6 kb have been obtained (Boyle-Vavra and Seifert, 1993). The insertion of plasmids into the chromosome (Basic Protocol 1) also depends on the length of the homologous sequence with homologous regions of 300 to 500 bp resulting in transformation frequencies in the 10−7 to 10−6 range (Hamilton et al., 2001).
The frequency of transformation by electroporation (Basic Protocol 3) is very low (~10−9) and will only produce a few transformants per electroporation. Shuttle mutagenesis (Basic Protocol 4) works well and has been used to produce multiple mutations in single genes or random mutations in the gonococcal chromosome (Haas et al., 1993; Boyle-Vavra and Seifert, 1993, 1994; Mehr and Seifert, 1997). The constructs for complementation on the chromosome by double-crossover recombination (Basic Protocol 6) are effective for complementation; however, as these contain significant stretches of heterologous DNA, the transformation frequencies are significantly lower than those of simple insertion mutations. The lac promoter used in some of the constructs gives relatively high levels of transcription of introduced genes, approaching those of strong gonococcal promoters (Seifert, 1997)(Salgado-Pabón, Du et al. 2010).
Time Considerations
Although the transformation step can take as little as 5 min, the entire procedure from streaking the bacteria to freezing down the transformed strain may take five days. Most of the time is spent waiting for the bacteria to grow. In particular, selection with chloramphenicol or erythromycin—two of the most useful antibiotics for gonococcal manipulation significantly slows growth of the bacteria such that transformants will take one and a half to two days to form a colony.
Table 4A.2.2 provides time requirements for many of the protocols presented in this unit.
Table 4A.2.2.
Time Considerations for Select Protocols
| PROTOCOL | DESCRIPTION | TIME REQUIRED |
|---|---|---|
| Basic Protocol 1 or 2 | Gonococcal transformation | 4–5 days |
| Basic Protocol 3 | Electroporation | 4 days |
| Basic Protocol 4 | Gonococcal mutagenesis using selection and counter selection | 16–20 days |
| Basic Protocol 5 | Shuttle mutagenesis | 12–14 days |
| Basic Protocol 6 | Chemical mutagenesis | 2 days |
| Basic Protocol 7 or Alternate Protocol 2 | Complementation | 4–5 days |
| Basic Protocol 8 | Chromosome preparation | 2 days |
Acknowledgments
This work was supported by National Institutes of Health grant R01 AI047958.
Literature Cited
- Ambur OH, Frye SA, Tonjum T. New functional identity for the DNA uptake sequence in transformation and its presence in transcriptional terminators. J Bacteriol. 2007;189(5):2077–85. doi: 10.1128/JB.01408-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belland RJ. H-DNA formation by the coding repeat elements of neisserial opa genes. Mol Microbiol. 1991;5:2351–2360. doi: 10.1111/j.1365-2958.1991.tb02081.x. [DOI] [PubMed] [Google Scholar]
- Black CG, Fyfe JA, Davies JK. Absence of an SOS-like repair system in Neisseria gonorrhoeae. Gene. 1998;208:61–66. doi: 10.1016/s0378-1119(97)00653-7. [DOI] [PubMed] [Google Scholar]
- Bloch KD. Mapping by Multiple Endonuclease Digestions. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. John Wiley & Sons; Hoboken, N.J: 1995. pp. 3.2.1–3.2.5. [DOI] [PubMed] [Google Scholar]
- Boyle-Vavra S, Seifert HS. Shuttle mutagenesis: Two mini-transposons for gene mapping and for lacZ transcriptional fusions in Neisseria gonorrhoeae. Gene. 1993;129:51–57. doi: 10.1016/0378-1119(93)90695-y. [DOI] [PubMed] [Google Scholar]
- Boyle-Vavra S, Seifert HS. Shuttle mutagenesis: A minitransposon for producing PhoA fusions in Neisseria gonorrhoeae. Gene. 1994;155:101–106. doi: 10.1016/0378-1119(94)00890-5. [DOI] [PubMed] [Google Scholar]
- Cahoon LA, Seifert HS. An alternative DNA structure is necessary for pilin antigenic variation in Neisseria gonorrhoeae. Science. 2009;325(5941):764–7. doi: 10.1126/science.1175653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell LA, Yasbin RE. Mutagenesis of Neisseria gonorrhoeae: Absence of error prone repair. J Bacteriol. 1984;160:288–293. doi: 10.1128/jb.160.1.288-293.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I, Dubnau D. DNA uptake during bacterial transformation. Nat Rev Microbiol. 2004;2:241–249. doi: 10.1038/nrmicro844. [DOI] [PubMed] [Google Scholar]
- Cloud KA, Dillard JP. A lytic transglycosylase of Neisseria gonorrhoeae is involved in peptidoglycan-derived cytotoxin production. Infect Immun. 2002;70(6):2752–2757. doi: 10.1128/IAI.70.6.2752-2757.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke SJ, Jolley K, Ison CA, Young H, Heckels JE. Naturally occuring isolates of Neisseria gonorrhoeae, which display anomalous serovar properties, express PIA/PIB hybrid porins, deletions in PIB or novel PIA molecules. FEMS Microbiol Lett. 1998;162:75–82. doi: 10.1111/j.1574-6968.1998.tb12981.x. [DOI] [PubMed] [Google Scholar]
- Dillard JP, Yother J. Genetic and molecular characterization of capsular polysaccharide biosynthesis in Streptococcus pneumoniae type 3. Mol Microbiol. 1994;12:959–972. doi: 10.1111/j.1365-2958.1994.tb01084.x. [DOI] [PubMed] [Google Scholar]
- Elkins C, Thomas CE, Seifert HS, Sparling PF. Species-specific uptake of DNA by gonococci is mediated by a 10-base-pair sequence. J Bacteriol. 1991;173(12):3911–3. doi: 10.1128/jb.173.12.3911-3913.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engebrecht J, Brent R, Kaderbhai MA. Minipreps of Plasmid DNA. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. John Wiley & Sons; Hoboken, N.J: 1991. pp. 1.6.1–1.6.10. [DOI] [PubMed] [Google Scholar]
- Genco CA, Chen CY, Arko R, Kapczynski D, Morse SA. Isolation and characterization of a mutant of Neisseria gonorrhoeae that is defective in the uptake of iron from transferrin and haemoglobin and is avirulent in mouse subcutaneous chambers. J Gen Microbiol. 1991;137:1313–1321. doi: 10.1099/00221287-137-6-1313. [DOI] [PubMed] [Google Scholar]
- Gibbs CP, Meyer TF. Genome plasticity in Neisseria gonorrhoeae. FEMS Microbiol Lett. 1996;145:173–179. doi: 10.1111/j.1574-6968.1996.tb08574.x. [DOI] [PubMed] [Google Scholar]
- Goodman SD, Scocca JJ. Identification and arrangement of the DNA sequence recognized in specific transformation of Neisseria gonorrhoeae. Proc Natl Acad Sci USA. 1988;85:6982–6986. doi: 10.1073/pnas.85.18.6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn JS, Stein DC. Use of a nonselective transformation technique to construct a multiply restriction/modification-deficient mutant of Neisseria gonorrhoeae. Mol Gen Genet. 1996;251:509–517. doi: 10.1007/BF02173639. See above. This article describes the spot transformation method that has greatly increased the frequency of obtaining nonselected mutations and also reviews the data on the many restriction systems of N. gonorrhoeae. [DOI] [PubMed] [Google Scholar]
- Haas R, Kahrs A, Facius D, Allmeier H, Schmitt R, Meyer TF. TnMax- a versatile mini-transposon for the analysis of cloned genes and shuttle mutagenesis. Gene. 1993;130:23–31. doi: 10.1016/0378-1119(93)90342-z. [DOI] [PubMed] [Google Scholar]
- Hamilton HL, Dillard JP. Natural transformation of Neisseria gonorrhoeae: From DNA donation to homologous recombination. Mol Microbiol. 2006;59:376–385. doi: 10.1111/j.1365-2958.2005.04964.x. [DOI] [PubMed] [Google Scholar]
- Hamilton HL, Schwartz KJ, Dillard JP. Insertion-duplication mutagenesis of Neisseria: Use in characterization of DNA transfer genes in the gonococcal genetic island. J Bacteriol. 2001;183:4718–4726. doi: 10.1128/JB.183.16.4718-4726.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton HL, Dominguez NM, Schwartz KJ, Hackett KT, Dillard JP. Neisseria gonorrhoeae secretes chromosomal DNA via a novel type IV secretion system. Mol Microbiol. 2005;55:1704–1721. doi: 10.1111/j.1365-2958.2005.04521.x. [DOI] [PubMed] [Google Scholar]
- Hebeler BH, Young FE. Autolysis of Neisseria gonorrhoeae. J Bacteriol. 1975;122:385–392. doi: 10.1128/jb.122.2.385-392.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston DM, Cannon JG. Construction of mutant strains of Neisseria gonorrhoeae lacking new antibiotic markers using a two gene cassette with positive and negative selection. Gene. 1999;236:179–184. doi: 10.1016/s0378-1119(99)00238-3. [DOI] [PubMed] [Google Scholar]
- Kado CI, Liu S. Rapid procedure for detection and isolation of large and small plasmids. J Bacteriol. 1981;145:1365–1373. doi: 10.1128/jb.145.3.1365-1373.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg DS, Jr, Peacock WL, Jr, Deacon WE, Brown L, Pirkle CL. Neisseria gonorrhoeaeI Virulence genetically linked to clonal variation. J Bacteriol. 1963;85:1274–1279. doi: 10.1128/jb.85.6.1274-1279.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler PL, Cloud KA, Hackett KT, Beck ET, Dillard JP. Characterization of the role of LtgB, a putative lytic transglycosylase in Neisseria gonorrhoeae. Microbiology. 2005;151:3081–3088. doi: 10.1099/mic.0.28125-0. [DOI] [PubMed] [Google Scholar]
- Koomey JM, Gill RE, Falkow S. Genetic and biochemical analysis of gonococcal IgA1 protease: Cloning in Escherichia coli and construction of mutants of gonococci that fail to produce the activity. Proc Natl Acad Sci USA. 1982;79:7881–7885. doi: 10.1073/pnas.79.24.7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koomey M, Gotschlich EC, Robbins K, Bergstrom S, Swanson J. Effects of recA mutations on pilus antigenic variation and phase transitions in Neisseria gonorrhoeae. Genetics. 1987;117:391–398. doi: 10.1093/genetics/117.3.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MF, Coen DM. Enzymatic Amplification of DNA by PCR: Standard Procedures and Optimization. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. John Wiley & Sons; Hoboken, N.J: 2001. pp. 15.1.1–15.1.14. [DOI] [PubMed] [Google Scholar]
- Kupsch EM, Aubel D, Gibbs CP, Kahrs AF, Rudel T, Meyer TF. Construction of Hermes shuttle vectors: A versatile system useful for genetic complementation of transformable and nontransformable Neisseria mutants. Mol Gen Genet. 1996;250:558–569. doi: 10.1007/BF02174444. [DOI] [PubMed] [Google Scholar]
- LaScolea LJ, Jr, Young FE. Development of a defined minimal medium for the growth of Neisseria gonorrhoeae. Appl Microbiol. 1974;28:70–76. doi: 10.1128/am.28.1.70-76.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederberg J. Streptomycin resistance; a genetically recessive mutation. J Bacteriol. 1951;61(5):549–50. doi: 10.1128/jb.61.5.549-550.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehr IJ, Seifert HS. Random shuttle mutagenesis: Gonococcal mutants deficient in pilin antigenic variation. Mol Microbiol. 1997;23:1121–1131. doi: 10.1046/j.1365-2958.1997.2971660.x. [DOI] [PubMed] [Google Scholar]
- Mehr IJ, Long CD, Serkin CD, Seifert HS. A homologue of the recombination-dependent growth gene, rdgC, is involved in gonococcal pilin antigenic variation. Genetics. 2000;154:523–532. doi: 10.1093/genetics/154.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse SA, Bartenstein L. Factors affecting autolysis of Neisseria gonorrhoeae. Proc Soc Exp Biol Med. 1974;145:1418–1421. doi: 10.3181/00379727-145-38025. [DOI] [PubMed] [Google Scholar]
- Morse SA, Bartenstein L. Purine metabolism in Neisseria gonorrhoeae: The requirement for hypoxanthine. Can J Microbiol. 1980;26:13–20. doi: 10.1139/m80-003. [DOI] [PubMed] [Google Scholar]
- O’Dwyer CA, Langford PR, Kroll JS. A novel neisserial shuttle plasmid: a useful new tool for meningococcal research. FEMS Microbiol Lett. 2005;251(1):143–7. doi: 10.1016/j.femsle.2005.07.036. [DOI] [PubMed] [Google Scholar]
- Pagotto FJ, Salimnia H, Totten PA, Dillon JR. Stable shuttle vectors for Neisseria gonorrhoeae, Haemophilus spp and other bacteria based on a single origin of replication. Gene. 2000;244:13–19. doi: 10.1016/s0378-1119(99)00557-0. [DOI] [PubMed] [Google Scholar]
- Pelicic V, Morelle S, Lampe D, Nassif X. Mutagenesis of Neisseria meningitidis by in vitro transposition of Himar1 mariner. J Bacteriol. 2000;182:5391–5398. doi: 10.1128/jb.182.19.5391-5398.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey ME, Dillard JP. Characterization of putative surface-exposed proteins of the type IV secretion system in Neisseria gonorrhoeae. International Pathogenic Neisseria Conference; Banff, Canada. 2010. p. P155. [Google Scholar]
- Salgado-Pabón W, Du Y, Hackett KT, Lyons KM, Grove-Arvidson C, Dillard JP. Increased expression of the type IV secretion system in piliated Neisseria gonorrhoeae variants. J Bacteriol. 2010;192:1912–1920. doi: 10.1128/JB.01357-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechman EV, Rohrer MS, Seifert HS. A genetic screen identifies genes and sites involved in pilin antigenic variation in Neisseria gonorrhoeae. Mol Microbiol. 2005;57:468–483. doi: 10.1111/j.1365-2958.2005.04657.x. [DOI] [PubMed] [Google Scholar]
- Sechman EV, Kline KA, Seifert HS. Loss of both Holliday junction processing pathways is synthetically lethal in the presence of gonococcal pilin antigenic variation. Mol Microbiol. 2006;61(1):185–93. doi: 10.1111/j.1365-2958.2006.05213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert HS. Questions about gonococcal pilus phase- and antigenic variation. Mol Microbiol. 1996;21:433–440. doi: 10.1111/j.1365-2958.1996.tb02552.x. [DOI] [PubMed] [Google Scholar]
- Seifert HS. Insertionally inactivated and inducible recA alleles for use in Neisseria. Gene. 1997;188:215–220. doi: 10.1016/s0378-1119(96)00810-4. [DOI] [PubMed] [Google Scholar]
- Seifert HS, Ajioka RS, Paruchuri D, Heffron F, So M. Shuttle mutagenesis of Neisseria gonorrhoeae: Pilin null mutations lower DNA transformation competence. J Bacteriol. 1990;172:40–46. doi: 10.1128/jb.172.1.40-46.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HO, Gwinn ML, Salzberg SL. DNA uptake signal sequences in naturally transformable bacteria. Res Microbiol. 1999;150(9–10):603–16. doi: 10.1016/s0923-2508(99)00130-8. [DOI] [PubMed] [Google Scholar]
- Sparling PF. Genetic transformation of Neisseria gonorrhoeae to streptomycin resistance. J Bacteriol. 1966;92:1364–1371. doi: 10.1128/jb.92.5.1364-1371.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein DC. Transformation of Neisseria gonorrhoeae: Physical requirements of the transforming DNA. Can J Microbiol. 1991;37:345–349. doi: 10.1139/m91-056. [DOI] [PubMed] [Google Scholar]
- Stein DC, Silver LE, Clark VL, Young FE. Construction and characterization of a new shuttle vector, pLES2, capable of functioning in Escherichia coli and Neisseria gonorrhoeae. Gene. 1983;25:241–247. doi: 10.1016/0378-1119(83)90228-7. [DOI] [PubMed] [Google Scholar]
- Stern A, Brown M, Nickel P, Meyer TF. Opacity genes in Neisseria gonorrhoeae: Control of phase and antigenic variation. Cell. 1986;47:61–71. doi: 10.1016/0092-8674(86)90366-1. [DOI] [PubMed] [Google Scholar]
- Stohl EA, Brockman JP, Burke KL, Morimatsu K, Kawalczykowski SC, Seifert HS. Eschericihia coli RecX inhibits RecA recombinase and coprotease activities in vitro and in vivo. J Biol Chem. 2003;278:2278–2285. doi: 10.1074/jbc.M210496200. [DOI] [PubMed] [Google Scholar]
- Swanson J, Kraus SJ, Gotschlich EC. Studies on gonococcus infection I. Pili and zones of adhesion: Their relation to gonococccal growth patterns. J Exp Med. 1971;134:886–906. doi: 10.1084/jem.134.4.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade JJ, Graver MA. A fully defined, clear and protein-free liquid medium permitting dense growth of Neisseria gonorrhoeae from very low inocula. FEMS Microbiol Lett. 2007;273(1):35–7. doi: 10.1111/j.1574-6968.2007.00776.x. [DOI] [PubMed] [Google Scholar]
- Wegener WS, Hebeler BH, Morse SA. Cell envelope of Neisseria gonorrhoeae: Relationship between autolysis in buffer and the hydrolysis of peptidoglycan. Infect Immun. 1977;18:210–219. doi: 10.1128/iai.18.1.210-219.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]