

Abstract
The composition of the liver changes dramatically with age. Hepatocytes in young individuals are relatively small and uniform in size. In contrast, adult hepatocytes vary considerably in cell and nuclear size, number of nuclei per cell and DNA content per nucleus (1, 2). Many of these striking age-associated morphological changes are attributed to hepatic polyploidy, a numerical change in the entire complement of chromosomes. Polyploid hepatocytes were recognized over a century ago, but it is unclear whether these cells play a specialized role in liver homeostasis, regeneration or disease. Despite our limited understanding of the physiological role of polyploid hepatocytes, nearly a dozen genes have been implicated in the regulation of polyploidy (2). Notably, Chantal Desdouets’ group previously demonstrated a critical role for insulin in the generation of binucleate polyploid hepatocytes in rats (3). Motivated by these early findings, Gentric, Celton-Morizur, Desdouets and colleagues rationalized that liver diseases frequently associated with dysregulated insulin signaling could have an abnormal ploidy spectrum. Indeed, in recent work the authors identified a connection between abnormal hepatic polyploidy and nonalcoholic fatty liver disease (NAFLD)
Mammalian livers are characterized by alterations in chromosome number. Hepatocytes in young livers are almost exclusively diploid, and polyploid hepatocytes develop in a step-wise manner by a process termed physiological polyploidization. First, a subset of diploid hepatocytes undergoes failed cytokinesis, generating tetraploid daughter cells, each containing two diploid nuclei. Secondly, mono- and binucleate hepatocytes continue to undergo DNA replication and mitosis, either with complete cytokinesis (generating pairs of mononucleate diploid or polyploid cells) or with failed cytokinesis (generating single binucleate polyploid cells) (4). In this way, diploid, tetraploid, octaploid and even higher ploidy hepatocytes are generated, with polyploid hepatocytes existing in mononucleate and binucleate forms. In adults, up to 90% of hepatocytes in mice (5) and 50% of hepatocytes in humans (6) are polyploid. Hepatic polyploidy is also reversible. Proliferating polyploid hepatocytes can form multipolar spindles and undergo nuclear segregation errors, generating reduced-ploidy daughters, as well as daughters with chromosome gains and/or losses (i.e., aneuploidy) (5, 6).
Gentric, Celton-Morizur, Desdouets and colleagues first examined hepatic ploidy in models of NAFLD, including ob/ob mice and wild-type mice fed methionine-choline-deficient diet (MCD) or high-fat diet (HFD). Cell cycle analysis demonstrated a change in ploidy with a significant increase in the percentage of polyploid hepatocytes. Microscopic analysis of liver sections showed dramatic enrichment for mononucleate cells with high nuclear content. Most importantly, a similar phenotype was also seen in patients with nonalcoholic steatohepatitis (NASH), a form of NAFLD. Livers from these patients displayed dramatic enrichment for highly polyploid mononucleate hepatocytes. Taken together, altered polyploidy is strongly associated with NAFLD in both animal models and patients.
Next, the investigators examined the mechanism leading to ploidy changes in NAFLD livers. They examined cell cycle kinetics of proliferating hepatocytes in vitro using ob/ob and control mice, as well as wild-type mice treated with MCD, HFD or control diet. Compared to controls, NAFLD hepatocytes progressed through G1 and entered S-phase similarly, but they delayed exit from S-phase and accumulated/arrested in G2. To further explore the genesis of these highly polyploid mononucleate hepatocytes, wild-type mice were fed MCD to induce NAFLD symptoms and pulsed sequentially with thymidine analogues, CldU and IdU, to mark cells in S-phase; here, only cells passing through two rounds of S-phase would incorporate both analogs. Approximately 40% of tetraploid cells and 10% of highly polyploid cells were positive for both CldU and IdU. Although this observation could be explained in different ways, the data are consistent with the idea that G2-arrested hepatocytes skip mitosis and reenter the cell cycle, becoming even more highly polyploid.
Finally, the investigators connected the dots from NAFLD to hepatic cell cycle arrest and extreme polyploidy. Ob/ob hepatocytes displayed high levels of oxidative stress, as measured by elevated levels of superoxide and increased expression of genes involved in the oxidative stress response. In addition to oxidative damage, proliferating ob/ob hepatocytes, specifically during S-phase and G2, displayed robust phosphorylation of ATR (an indicator of DNA damage response) and markers for cell cycle arrest (phosphorylation of p53 and increased p21 expression). These observations strongly support a scenario in which excessive oxidative stress in livers with NAFLD leads to DNA damage, cell cycle arrest and the emergence of highly polyploid mononucleate hepatocytes. Consistent with this model, NAFLD symptoms were ameliorated in ob/ob hepatocytes treated either in vitro or in vivo with the antioxidant N-acetylcysteine (NAC). NAC treatment led to reduced oxidative stress, dephosphorylation of ATR, near-normal cell cycle and decreased numbers of the highly polyploid subset of mononucleate hepatocytes.
The implications of this new work are numerous. On a cell biology level, there are two separate but distinct types of polyploidy in the liver. As we have long appreciated, physiological polyploidization is a form of liver maturation occurring in postnatal rodents and adolescent humans that is associated with increased numbers of polyploid hepatocytes. Proliferating diploid hepatocytes undergo cytokinesis failure to generate binucleate tetraploids. These binucleate tetraploids continue dividing, producing mononucleate tetraploids that can progress through additional rounds of mitosis with cytokinesis failure to yield binucleate octaploids and so on. New findings from the investigators identified an alternate mechanism regulating ploidy that is associated with liver injury and disease, pathological polyploidization (Fig. 1). Liver damage in adults is frequently associated with compensatory hepatic proliferation, and, in contrast to young livers dividing hepatocytes most often complete cytokinesis; thus, populations of regenerating hepatocytes become increasingly mononucleate (7). In cases of injury caused by NAFLD, livers are damaged by reactive oxygen species and proliferating mononucleate hepatocytes go through S-phase and arrest in G2. Intriguingly, a subset of these cells may even skip mitosis altogether, a process known as endoreplication (8), re-enter the cell cycle, proceed through S-phase and either arrest in G2 or continue cycling. Overall, pathological polyploidization leads to changes in the composition of the liver and is characterized by enrichment for highly polyploid mononucleate hepatocytes.
Fig. 1.
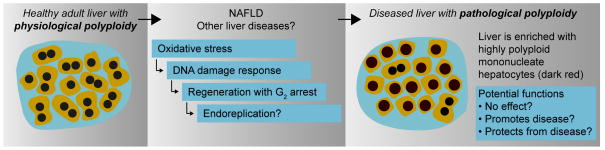
Model for pathological polyploidization in the liver. Question marks indicate areas for future investigation.
The relationship between pathological polyploidy and liver injury is unclear. The investigators demonstrated that an outcome of NAFLD is emergence of highly polyploid mononucleate hepatocytes. But what are these cells doing? One possibility is that liver damage generates pathologically polyploid hepatocytes that contribute minimally, if at all, to disease. A more interesting possibility is that these highly polyploid mononucleate hepatocytes somehow maintain or promote disease progression. Does cell cycle arrest of hepatocytes in G2 contribute to the poor regenerative capacity of cirrhotic livers? Alternatively, in contrast to contributing to disease, pathological polyploidization could serve as a natural defense mechanism, slowing pathogenesis and protecting the liver. The presence of multiple genomes could “buffer” polyploid hepatocytes from genotoxic damage, increase functional capacity or even promote the formation of injury-resistant aneuploid daughters (1).
It is intriguing to consider whether pathological polyploidization is common to liver diseases besides NAFLD. Pathological polyploidization could be part of a conserved response to oxidative stress, involving DNA damage response, G2 arrest and possibly endoreplication. In support of this idea, it is well documented that liver injury resulting from partial hepatectomy, hepatitis C infection, iron overload and copper overload leads to increased numbers of highly polyploid hepatocytes (1, 2). A feature shared by each of these forms of injury is elevated oxidative stress in the liver. Hence, it is tempting to hypothesize that oxidative stress, in general, functions as a primary driver of pathological polyploidization.
In summary, the discovery of pathological polyploidization proves that hepatic polyploidy is even more complicated than originally believed. Currently, there are more questions than answers about these fascinating highly polyploid mononucleate hepatocytes. Future work will unravel which diseases are associated with pathological polyploidization, how these cells behave in damaged livers and whether they can influence disease progression.
Acknowledgments
Support
This work was supported to grants to AWD by the NIH (R01 DK103645) and the Commonwealth of Pennsylvania.
Footnotes
Potential conflict of interest
Nothing to report.
References
- 1.Duncan AW. Aneuploidy, polyploidy and ploidy reversal in the liver. Semin Cell Dev Biol. 2013;24:347–356. doi: 10.1016/j.semcdb.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Pandit SK, Westendorp B, de Bruin A. Physiological significance of polyploidization in mammalian cells. Trends Cell Biol. 2013;23:556–566. doi: 10.1016/j.tcb.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Celton-Morizur S, Merlen G, Couton D, Margall-Ducos G, Desdouets C. The insulin/Akt pathway controls a specific cell division program that leads to generation of binucleated tetraploid liver cells in rodents. J Clin Invest. 2009;119:1880–1887. doi: 10.1172/JCI38677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Margall-Ducos G, Celton-Morizur S, Couton D, Bregerie O, Desdouets C. Liver tetraploidization is controlled by a new process of incomplete cytokinesis. J Cell Sci. 2007;120:3633–3639. doi: 10.1242/jcs.016907. [DOI] [PubMed] [Google Scholar]
- 5.Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, Olson SB, Finegold MJ, et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature. 2010;467:707–710. doi: 10.1038/nature09414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duncan AW, Hanlon Newell AE, Smith L, Wilson EM, Olson SB, Thayer MJ, Strom SC, et al. Frequent aneuploidy among normal human hepatocytes. Gastroenterology. 2012;142:25–28. doi: 10.1053/j.gastro.2011.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyaoka Y, Ebato K, Kato H, Arakawa S, Shimizu S, Miyajima A. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr Biol. 2012;22:1166–1175. doi: 10.1016/j.cub.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 8.Fox DT, Duronio RJ. Endoreplication and polyploidy: insights into development and disease. Development. 2013;140:3–12. doi: 10.1242/dev.080531. [DOI] [PMC free article] [PubMed] [Google Scholar]