

Abstract
Evidence suggests that male rats are protected against the hypotensive and myocardial depressant effects of ethanol compared with females. We investigated whether E2 modifies the myocardial and oxidative effects of ethanol in male rats. Conscious male rats received ethanol (0.5, 1 or 1.5 g/kg i.v.) 30-min after E2 (1 μg/kg i.v.) or its vehicle (saline), and hearts were collected at the conclusion of hemodynamic measurements for ex vivo molecular studies. Ethanol had no effect in vehicle-treated rats, but it caused dose-related reductions in LV developed pressure (LVDP), end-diastolic pressure (LVEDP), rate of rise in LV pressure (dP/dtmax) and systolic (SBP) and diastolic (DBP) blood pressures in E2-pretreated rats. These effects were associated with elevated (i) indices of reactive oxygen species (ROS), (ii) malondialdehyde (MDA) protein adducts, and (iii) phosphorylated death-associated protein kinase-3 (DAPK3), Akt, and extracellular signal-regulated kinases (ERK1/2). Enhanced myocardial antioxidant enzymes (heme oxygenase-1, catalase and aldehyde dehydrogenase 2) activities were also demonstrated. In conclusion, E2 promotes ethanol-evoked myocardial oxidative stress and dysfunction in male rats. The present findings highlight the risk of developing myocardial dysfunction in men who consume alcohol while receiving E2 for specific medical conditions.
Keywords: Ethanol, estrogen, oxidative stress, LV dysfunction, extracellular signal-regulated kinases, male rats
Introduction
Acutely administered ethanol reduces cardiac output and BP in female, but not in male rats (El-Mas and Abdel-Rahman, 1999b, 1999c) or young men (Bae et al., 2012). Sex-specific data analysis reveals that ethanol reduces BP in young, but not in older women (Klatsky, 1990), and causes cutaneous flushing in women receiving hormone replacement therapy (Nitzan and Dan, 2009). The role of sex hormones, particularly E2, is supported by the observations that the ethanol-evoked hypotension virtually disappears in ovariectomized (OVX) rats and is fully restored after chronic (El-Mas and Abdel-Rahman, 2012b) or acute E2 administration (El-Mas and Abdel-Rahman, 2014). Evidence suggests that the male gender is protected against the direct cardiodepressant/hypotensive effect of ethanol by at least two mechanisms. First, acute ethanol exerts a counterbalancing sympathoexcitatory effect in male rats (Li et al., 2005) and men (Child et al., 1979). Second, ethanol enhances the activity of mitochondrial aldehyde dehydrogenase 2 (mitALDH2), which facilitates the detoxification of myocardial cytotoxic aldehydes (Churchill et al., 2009).
Whether E2 compromises or corroborates the male-dependent resilience against the cardiodepressant effect of ethanol is not known. Addressing this question is clinically relevant because men receive E2 under clinical settings such as prostate cancer (Ho et al., 2011) and osteoporosis (Navaneethan et al., 2011) and when higher E2 levels are also seen in men with endocrine disturbances (Roth et al., 2008) or upregulated aromatase expression (Williams, 2012). Notably, in a limited number of studies, acute E2 administration in male rats enhanced baroreflex sensitivity (Saleh and Connell, 1998) and conferred cardioprotection against trauma-evoked myocardial dysfunction via activation of heme oxygenase (HO) and Akt (Hsu et al., 2009). Notably, E2-dependent Akt phosphorylation is implicated in ethanol-evoked myocardial dysfunction in female rats (El-Mas and Abdel-Rahman, 2014). Whether the E2-dependent phosphorylation of Akt offsets or unravels ethanol-evoked myocardial depression in male rats will determine if the role of this kinase is sex- or intervention (trauma or ethanol)-specific. Further, the effect of E2 on myocardial HO and two other antioxidant enzymes, mitALDH2 and catalase, and their potential roles in the myocardial effects of ethanol in male rats are not known. Importantly, while all 3 enzymes confer cardioprotection (Zhang et al., 2003; Ma et al., 2009) catalase, whose activity is enhanced by E2 (El-Mas and Abdel-Rahman, 2014), catalyzes the metabolic oxidation of ethanol into acetaldehyde (Comporti et al., 2010), and the latter contributes to the cardiotoxic effect of ethanol (Zhang et al., 2004). It is also important to note that recent evidence implicates the death associated protein kinase-3 (DAPK3) in MAPK-evoked oxidative stress and inflammatory responses (Usui et al., 2012).
In the current contribution, we tested the hypothesis that E2 exacerbates ethanol evoked myocardial oxidative stress and LV dysfunction in male rats. We conducted in vivo hemodynamic and ex vivo molecular/biochemical studies to investigate the dose-related effects of ethanol (0.5, 1 or 1.5 g/kg; i.v.) on LV function, BP, and mediators of oxidative stress (MDA adducts and p-ERK1/2) and antioxidant enzymes (catalase, mitALDH2 and HO-1) in conscious male rats pretreated with E2 (1 μg/kg; i.v.) or its vehicle. We also investigated, for the first time, if DAPK3 phosphorylation is induced by ethanol in presence of E2. Here, we show that E2 uncovers dose-related myocardial oxidative stress and dysfunction caused by ethanol in male rats via activation of myocardial Akt/DAPK3-dependent mechanisms. The findings also highlight a role for the E2-dependent enhancement of myocardial catalase activity in a counterintuitive metabolism of ethanol into its more cardiotoxic metabolite acetaldehyde.
Materials and methods
Animals
Male Sprague-Dawley rats (250–275 g; Harlan Laboratories, Indianapolis, IN) were used. Upon arrival, rats were housed individually in standard plastic cages and allowed free access to water and Purina chow and were maintained on a 12-12-hr light-dark cycle with light off at 7:00 p.m. Room temperature was maintained at 22±1°C. Surgical procedures and postoperative care were performed in accordance with, and approved by, the Institutional Animal Care and Use Committee and in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health.
Drugs
Ketaject (ketamine), Xyla-ject (xylazine) (Phoenix Pharmaceuticals Inc., St Joseph, MI), buprenorphine (Rickitt & Colman, Richmond, VA), Durapen (Vedco Inc., Overland Park, KS), 17β-estradiol sulfate (Sigma Chemical Co., St. Louis, MO), and ethanol (Midwest Grain Products Co., Weston, MO) were purchased from commercial vendors.
Femoral and left ventricular catheterizations
Catheters were inserted into femoral vessels, for BP and i.v. administration, and into the left ventricle, via the right carotid artery, as in our previous studies (El-Mas and Abdel-Rahman, 1997, 2014). Rats were anesthetized with i.p. injection of a mixture of ketamine (90 mg/kg) and xylazine (10 mg/kg). Catheters were tunneled subcutaneously and exteriorized at the back of the neck between the scapulae. Each rat received i.m. injections of the analgesic buprenorphine hydrochloride (Buprenex, 30 μg/kg) and penicillin G benzathine and penicillin G procaine in an aqueous suspension (Durapen, 100,000 U/kg). Two days later, hemodynamic measurements were made in conscious rats. This 2-day time period has been shown adequate for rats to recover from surgery and restore normal activity and hemodynamics (El-Mas and Abdel-Rahman, 1997, 2014). For example, the baseline values SBP, DBP, HR, and dP/dtmax in the current study (Table 1) are similar to those measured in radiotelemetered rats several weeks after surgery (El-Mas and Abdel-Rahman, 2013).
Table 1.
Average baseline levels of systolic blood pressure (SBP), diastolic blood pressure (DBP), heart rate (HR), rate of rise in left ventricular pressure (LV dP/dtmax), LV developed pressure (LVDP), LV end diastolic pressure (LVEDP) in male rats prior to treatment with i.v. saline or E2. Plasma ethanol concentrations measured 30 min after i.v. infusion of ethanol are also shown.
| Parameter | Saline | Ethanol 0.5 g/kg |
Ethanol 1.0 g/kg |
Ethanol 1.5 g/kg |
|---|---|---|---|---|
| SBP (mmHg) | ||||
| Saline | 139±6 | 131±7 | 131±3 | 125±5 |
| E2 | 140±5 | 127±3 | 127±5 | 132±7 |
| DBP (mmHg) | ||||
| Saline | 95±5 | 91±6 | 90±5 | 83±5 |
| E2 | 92±6 | 79±3 | 97±3 | 92±7 |
| HR (beats/min) | ||||
| Saline | 406±21 | 382±30 | 425±26 | 384±26 |
| E2 | 418±15 | 413±22 | 303±28 | 406±19 |
| dP/dtmax (mmHg/sec) | ||||
| Saline | 6638±1176 | 6626±682 | 8148±1140 | 7748±750 |
| E2 | 9688±578 | 9942±498 | 7707±1087 | 9525±885 |
| LVDP (mmHg) | ||||
| Saline | 166±10 | 165±6 | 183±12 | 170±10 |
| E2 | 185±10 | 174±8 | 172±10 | 195±10 |
| LVEDP (mmHg) | ||||
| Saline | 13±9 | 6±2 | 12±2 | 12±3 |
| E2 | 9±2 | 12±2 | 6±1 | 9±3 |
| Blood ethanol (g/dL) | ||||
| Saline | -- | 0.033±0.002 | 0.089±0.002 | 0.110±0.004 |
| E2 | -- | 0.030±0.002 | 0.089±0.002 | 0.114±0.002 |
Values are means±SEM of 7–8 observations.
Quantification of cardiac ROS
The 2′,7′-dichlorofluorescein (DCF) biochemical assay was used (El-Mas and Abdel-Rahman, 2014). The ventricles were homogenized using Radnoti tissue grinders (Radnoti Glass Technology, Monrovia, CA) to increase protein yield, and kinetic readings (250 μg protein) were taken at 5 min intervals for 60 min at 37°C. ROS levels were calculated as relative DCF fluorescence/μg protein.
Western blots
The rat ventricle was homogenized on ice in a homogenization buffer [50 mM Tris (pH 7.5), 0.1 mM EGTA, 0.1 mM EDTA, 2 μM leupeptin, 1 mM phenylmethylsulfonyl fluoride, 0.1% (vol/vol) Nonidet P-40, 0.1% SDS, and 0.1% deoxycholate]. After centrifugation (12,000 g for 10 min), protein in the supernatant was quantified (Bio-Rad protein assay system; Bio-Rad, Hercules, CA). Protein extracts (50 μg per lane) were run on a 4 to 12% SDS-polyacrylamide gel electrophoresis gel (Invitrogen, Carlsbad, CA) and electroblotted to nitrocellulose membranes. Blots were blocked for 2 hr at room temperature in Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE). They were then incubated overnight at 4°C in Odyssey blocking buffer with mouse antibodies to p-Akt, or ERK1/2 (1:200; Cell Signaling Technology, INC. Danvers, MA), rabbit antibodies to Akt or ERK1/2 (1:200; Cell Signaling Technology; INC. Danvers, MA), DAPK (1:200; Sigma Chemical Co., St. Louis, MO), p-DAPK3 (pThr265, 1:200; Sigma Chemical Co., St. Louis, MO), HO-1 (1:500; Abcam Inc., Cambridge, MA) or MDA protein adducts (1:150; academy biomedical Co., Houston, TX). After 3 washes with PBS-T buffer, the blots were incubated for 50 min at room temperature with a goat anti-mouse secondary antibody IRDye® 800CW or a goat anti-rabbit secondary antibody IRDye® 680CW (LI-COR Biosciences, Lincoln, NE). After 3 washes with PBS-T buffer, the blots were detected by Odyssey Infrared Scanning System. Equivalent sample loading was confirmed by stripping membranes with blot restore membrane rejuvenation solution (SignaGen Laboratories, MD) and reprobing with mouse β-actin antibody (1:5000, ABCAM antibodies, Cambridge, MA). Protein bands were quantified by integrated intensities using Odyssey Infrared Imaging Software, which serve as good surrogates for the relative express levels of targeting proteins. Data were normalized to the corresponding total protein in the same sample and results were then expressed as percent of control (El-Mas et al., 2008, 2009b, El-Mas and Abdel-Rahman, 2014).
Measurement of catalase or mitALDH2 activity
Commercially available colorimetric assay kits for the determination of the activity of catalase (Sigma Chemical Co., St. Louis, MO) and mitALDH2 (Biomedical Research Services Center, University of the State of New York at Buffalo, Buffalo, NY). Catalytic enzyme activity was determined in equal amounts of protein extracted from ventricular tissues according to the manufacturer’s protocol and reported studies (El-Mas and Abdel-Rahman, 2012). ALDH2 (mitochondrial ALDH) activity was measured by a modified protocol reported in previous studies (Yoon et al., 2006) where subcellular fractions were prepared from ventricular tissues to isolate the mitochondrial portion.
Plasma ethanol and E2 levels
The enzymatic and radioimmunoassay (Diagnostic Systems Laboratories, Inc., Webster, TX) methods were used for the measurement of plasma ethanol and E2, respectively, as described in our previous studies (El-Mas and Abdel-Rahman, 1999a, 1999b).
Experimental groups and protocols
A total of 8 groups of conscious male rats (n=7–8 each) pre-instrumented, 48 hr earlier, with intravascular and LV ventricular catheters were used to investigate the impact of acute E2 on the hemodynamic effects of ethanol. On the experiment day, the femoral and LV catheters were connected to Gould-Statham pressure transducers (Oxnard, California, USA). The transducers were then attached through MLAC11 Grass adapter cable to a computerized data acquisition system with LabChart-7 pro software (PowerLab, ADInstruments, Inc., Colorado, USA) as in our previous studies (El-Mas and Abdel-Rahman, 2014). SBP and DBP were monitored continuously and HR was extracted from BP recordings using the LabChart-7 blood pressure analysis module. Indices of the LV function (LV dp/dtmax, LV dp/dtmin, LVESP, LVEDP, and LVDP) were also monitored. All cardiovascular variables were recorded by ML870 (PowerLab 8/30) and analyzed using LabChart 7 software (ADInstruments, Colorado, USA).
At least 30 min stabilization period was allowed at the beginning of the experiment. Then, the rats received i.v. bolus dose of E2 (1 μg/kg) or equal volume of saline 30 min before a 30 min i.v. infusion of ethanol (0.5, 1.0, or 1.5 g/kg) or equal volume of saline. The final concentrations of ethanol, in an equal volume of saline (2 ml), were 6.5%, 13%, or 19.5% for the 3 doses, respectively (see timeline in figure 1). Hemodynamic responses were monitored for 90 min post-ethanol or saline. Changes in SBP, DBP, HR, and indices of LV function (LVDP, LVEDP, and dP/dtmax) were computed at 10 min intervals. The data collected by fluid filled LV catheters are comparable to those produced by the Millar catheter (Wang et al., 2004), and the ethanol and E2 dose regimen were based on previous studies including ours (Saleh and Connell, 1998; El-Mas and Abdel-Rahman, 2014).
Figure 1.
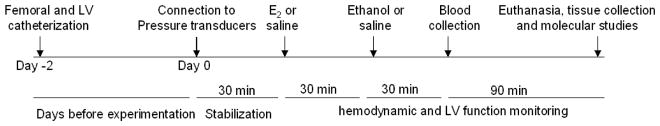
A schematic of surgical, drug administration, and biochemical and molecular schedules employed to investigate the effect of ethanol (0.5, 1, or 1.5 g/kg i.v.) on hemodynamic and left ventricular (LV) function in male Sprague-Dawley rats pretreated, 30 min earlier, with saline or estrogen (E2, 1 μg/kg i.v.).
During hemodynamic measurements, blood (0.5 ml) was drawn, collected into a heparinized tube and centrifuged at 1200 g for 5 min. The plasma was aspirated and stored at −80°C for subsequent measurements of plasma ethanol and E2 levels as described earlier. At the conclusion of the experiment (150 min after E2 or saline treatment, Fig. 1), rats were euthanized with an overdose of ketamine and xylazine mixture. The heart was excised immediately and atria and great vessels were removed. The ventricles were blotted free of blood, and used for biochemical and protein expression studies, which included measurements of myocardial ROS, phosphorylated Akt, ERK1/2 and DAPK3, and the antioxidant enzymes mitALDH, catalase and HO in the treatment and control groups. A schematic of surgical, drug administration, and biochemical and molecular schedules employed in this experiment is depicted in figure 1.
Data analysis and Statistics
Values are presented as means ± S.E.M. The time course effects of ethanol in the absence and presence of E2 on MAP, HR, and LV indices were analyzed by the repeated measures two-way ANOVA followed by a Newman-Keuls post-hoc test. The one-way ANOVA was used for the analysis of multiple comparisons of the Western data. The trend analysis was used to analyze and extract patterns of the dose-response data generated in ethanol (0.5, 1 or 1.5 g/kg) treated rats in the absence or presence of E2. Statistical significance was defined as p<0.05.
Results
Baseline data
The baseline values of SBP, DBP, (HR, LV (LV dp/dtmax, LVDP, and LVEDP in conscious male rats, prior to saline or E2 treatment, were similar (Table 1). Blood ethanol concentrations, measured 30 min after i.v. infusion of ethanol (0.5, 1, or 1.5 g/kg), correlated well with ethanol doses and were similar in rats pretreated with E2 or the vehicle (Table 1). Similarly, plasma E2 levels in male rats, pretreated with E2 (1 μg/kg i.v.), were comparable in the presence or absence of ethanol (71±6 and 65±5 pg/ml). These plasma E2 levels were similar to the levels obtained in OVX rats pretreated with the same dose of E2 in our recent studies (El-Mas and Abdel-Rahman, 2014). No E2 was detected in plasma of saline pretreated male rats.
E2 promotes myocardial depressant and hypotensive effects of ethanol in male rats
In saline-pretreated male rats, i.v. ethanol caused no change in SBP (Fig. 2A) or DBP (Fig. 2B) or LV function (Figs. 3, 4), but caused dose-related tachycardia that reached statistical significance (P<0.05) at the 1.5 g/kg dose (Fig. 2C). By contrast, ethanol infusion (0.5, 1, or 1.5 g/kg) in E2-pretreated male rats caused significant (P<0.05) and dose-related reductions in SBP (Fig. 2D) and DBP (Fig. 2E), and had no effect on HR (Fig. 2F). The hypotensive response started 10 min after ethanol infusion, reached its maximum at 40 min, and remained till the end of the 90-min observation period (Fig. 2D, 2E). The BP lowering effect of ethanol in E2-preated rats was paralleled with time- and dose-related reductions in LV functions including LV dp/dtmax (Fig. 3B), LVDP (Fig. 4C), and LVEDP (Fig. 4D). The trend analysis showed significant dose-response relationships for the ethanol effects only in presence of E2 (see supplemental Fig. 1).
Figure 2.

Changes in systolic (SBP, panels A and D) and diastolic (DBP, panels B and E) blood pressures and heart rate (HR, panels C and F) caused by ethanol (0.5, 1, or 1.5 g/kg i.v.), compared with equal volume of saline, in male Sprague-Dawley rats pretreated 30 min earlier with saline (left panels) or estrogen (E2, 1 μg/kg i.v., right panels). Values are means±S.E.M of 7–8 observations. *P<0.05 versus corresponding saline values.
Figure 3.

Changes in the rate of rise in LV pressure (LV dP/dtmax) caused by ethanol (0.5, 1, or 1.5 g/kg i.v.), compared with equal volume of saline, in male Sprague-Dawley rats pretreated 30 min earlier with saline (panel A) or estrogen (E2, 1 μg/kg i.v.) (panel B). Values are means±S.E.M of 7–8 observations. *P<0.05 versus corresponding saline values.
Figure 4.

Changes in the LV developed pressure (LVDP, top panels) or LV end diastolic pressure (LVEDP, bottom panels) caused by ethanol (0.5, 1, or 1.5 g/kg i.v.), compared with equal volume of saline, in male Sprague-Dawley rats pretreated 30 min earlier with saline (left panels) or estrogen (E2, 1 μg/kg i.v.) (right panels). Values are means±S.E.M of 7–8 observations. *P<0.05 versus corresponding saline values.
Acute E2 exacerbates LV oxidative stress caused by ethanol in male rats
Compared with saline, E2 had no effect on myocardial ROS level (Figs. 5A, B). Although higher doses of ethanol caused slight elevation in myocardial ROS of saline-pretreated rats (Fig. 5A), the rate of ROS generation was similar in ethanol and saline treated rats (Fig. 5C). In E2-pretreated male rats, ethanol significantly increased myocardial ROS levels in a dose-related manner when expressed as time-course (Fig. 5B) or slope of the ROS generation line (Fig. 5C). In rats pretreated with E2 30 min earlier, ethanol caused dose-related increases in LV expression of myocardial MDA adducts (Fig. 6A), p-Akt (Fig. 7A), p-ERK1/2 (Fig. 7B), and p-DAPK3 (Fig. 7C). By contrast, the abundance of these proteins was not affected by ethanol in saline-pretreated rats (Figs. 6 and 7). LV antioxidant enzymes, HO-1 protein expression (Fig. 6B) and catalase activity (Fig. 7A) and mitALDH2 activity (Fig. 8B), were also dose-dependently increased by ethanol in E2-treated rats. E2 alone enhanced the activity of LV mitALDH2 and catalase and the increase reached statistical significance in case of catalase (Fig. 8). None of the measured biochemical responses was influenced by ethanol in the saline-pretreated male rats (Figs. 6–8).
Figure 5.

The 2′,7′-dichlorofluorescein biochemical assay of the generation of reactive oxygen species (ROS) evoked by ethanol (0.5, 1, or 1.5 g/kg i.v.), compared with equal volume of saline, in male Sprague-Dawley rats pretreated 30 min earlier with saline (panel A) or estrogen (E2, 1 μg/kg i.v.) (panel B). Slopes (regression coefficients) of the regression lines are shown in panel C. Values are means±S.E.M of 7–8 observations. *P<0.05 versus corresponding saline values.
Figure 6.

Protein expressions of MDA adducts and HO-1 in ventricular homogenates obtained from male Sprague-Dawley rats 120 min after ethanol (0.5, 1, or 1.5 g/kg); estrogen (E2, 1 μg/kg, i.v.) or saline was administered 30 min before ethanol. Western blot bands of MDA adducts or HO-1 depicting ventricular protein expression are shown. Values are means±S.E.M of 7–8 observations. *P<0.05 versus corresponding saline values.
Figure 7.

Protein expression of phosphorylated and total ERK1/2 (panel A), Akt (panel B), and DAPK3 (panel C) in ventricular homogenates obtained from male Sprague-Dawley rats 120 min after ethanol (0.5, 1, or 1.5 g/kg); estrogen (E2, 1 μg/kg, i.v.) or saline was administered 30 min before ethanol. Western bands depicting ventricular protein expressions are shown. Values are means±S.E.M of 7–8 observations. *P<0.05 versus corresponding saline values.
Figure 8.
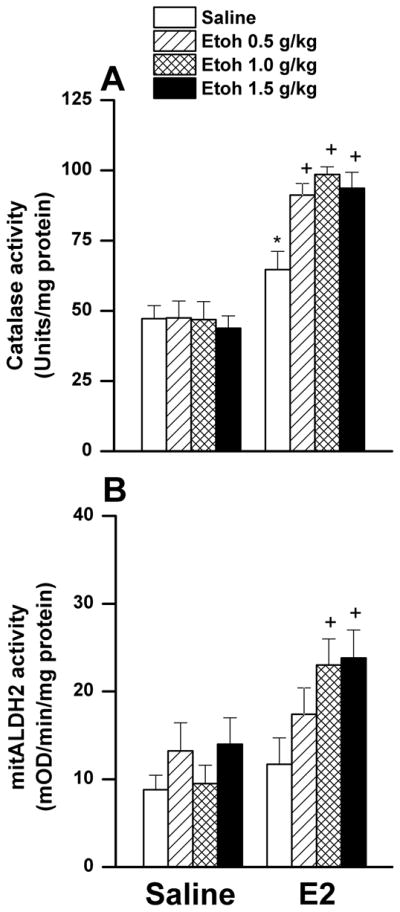
Effect of ethanol (0.5, 1, or 1.5 g/kg i.v.), compared with equal volume of saline, on ventricular catalase (panel A) or mitALDH (panel B) activity in male Sprague-Dawley rats pretreated 30 min earlier with saline or estrogen (E2, 1 μg/kg i.v.). Values are means±S.E.M of 7–8 observations. *P<0.05 versus corresponding values in saline-pretreated rats, +P<0.05 versus corresponding E2 values.
Discussion
Moderate doses of ethanol cause transient (Bae et al., 2012) or no effect on BP in young men (Child et al., 1979) or male (El-Mas and Abdel-Rahman, 1999c) or OVX (El-Mas and Abdel-Rahman, 2014) rats, but cause myocardial dysfunction and hypotension in intact and E2-pretreated OVX rats (El-Mas and Abdel-Rahman, 2014). Here, we show that this sex-dependent protection is compromised in E2-pretreated male rats, which exhibited analogous reductions in BP and LV performance (LV dp/dtmax, LVDP, and LVEDP) to those caused by ethanol in proestrus rats. The detrimental LV effects of ethanol are caused, at least partly, by elevations in oxidative molecules (ROS, and MDA adducts, p-Akt, p-ERK1/2, and p-DAPK3) in the myocardium of E2-pretreated male rats. The ethanol-evoked myocardial dysfunction and oxidative stress could have been more drastic without the concomitant elevations in LV antioxidant defenses mechanisms (HO-1, catalase, and mitALDH2). Nonetheless, the higher catalase activity, caused by E2 alone and its exacerbation by ethanol, might have also contributed to the deleterious effects by catalyzing ethanol metabolism to its cardiotoxic metabolite acetaldehyde. It is concluded that E2 transformation of myocardial cellular environment renders male rats more susceptible to the oxidative stress-dependent myocardial dysfunction and hypotension caused by ethanol. These findings have clinical ramifications in men who consume ethanol along with E2 for the treatment of prostate cancer or osteoporosis or when E2 levels increase due to endocrine disturbances.
In this study, we opted to limit the doses of ethanol to levels that are compatible with moderate human consumption (Child et al., 1979; Abdel-Rahman et al., 1987). The blood ethanol concentration achieved ranged from 42–138% of the legal blood limit with the middle dose producing slightly higher BAC (0.089 g/dL) than the legal blood limit (Tremblay et al., 2015). Our findings that ethanol had no effect on BP or LV function in vehicle-treated male rats are consistent with reported findings (El-Mas and Abdel-Rahman, 1999a, 1999c). In humans, comparable acute regimens of ethanol caused no changes (Child et al., 1979) or transient decreases in BP (Bae et al., 2012). Notably, a remarkable deterioration of indices of myocardial contractile function (LV dP/dtmax and LVDP) occurs in a mouse model of binge drinking in which male mice received a substantially higher dose of ethanol (3 g/kg) (Ma et al., 2009) or in the contractility of isolated cardiac myocyte from male ALDH2 KO mice (Ma et al., 2010). Therefore, the net effect of ethanol on cardiac function is largely influenced by the ethanol dose and the myocardium redox status.
Although E2 modulation of cardiovascular and autonomic functions in men and male rats has been established (Saleh et al., 2005 ; Choi and McLaughlin, 2007), no information is available on whether E2 modulates the hemodynamic effects of ethanol in males. The present findings support this hypothesis because in E2-pretreated male rats, ethanol caused dose-dependent suppression of LV performance indices (Figs. 3 and 4) and its tachycardic effect was abrogated (Fig. 2F). Because E2 was given acutely, E2 exacerbation of the negative influences of ethanol on LV performance in male rats likely engages rapid nongenomic pathways. This notion is supported by the observation that E2, administered within a similar time frame in OVX rats, lowered BP via a nontranscriptional activation of PI3K/Akt signaling (Wu et al., 2012). Notably, plasma E2 levels seen in male rats after a single E2 dose of 1 μg/kg was similar to those established in proestrus or in E2-treated OVX rats (El-Mas et al., 2009a). The clinical relevance of the current study is likely because comparable plasma E2 levels exist in women (T’Sjoen et al., 2005) as well as in men with endocrine disorders (Cailleux-Bounacer et al., 2009).
It was also important to investigate the role of myocardial redox modulators in the hemodynamic effects of ethanol in male rats for the following reasons. First, despite ethanol-evoked accumulation of cardiotoxic aldehydes, the male rat myocardium is rescued by a counterbalancing protective effect of ethanol via activation of mitochondrial mitALDH2 (Churchill et al., 2009). Second, ethanol suppressed the contractility of myocytes isolated from male mice lacking ALDH2 (Ma et al., 2010) or catalase, another myocardial antioxidant enzyme (Zhang et al., 2003). Third, E2 enhances myocardial ALDH2 and catalase activities (Lagranha et al., 2010; El-Mas and Abdel-Rahman, 2014). Based on these findings, we reasoned that E2 might corroborate the reported ALDH2-dependent cardioprotection in male rats (Churchill et al., 2009). Contrary to our hypothesis, and despite the significant and dose-related increases in LV ALDH2 caused by ethanol in E2-treated male rats (Fig. 8B), we report dose-related myocardial dysfunction that was coupled with increases in ventricular ROS generation and MDA adducts (Figs. 5 and 6) caused by the E2/ethanol regimen. The accumulation of these oxidative products might damage cellular carbohydrates, proteins, lipids, and nucleic acids (Saravanan and Pugalendi, 2006). Additionally, the cellular and molecular basis for the pro-oxidant capacity of ethanol was illustrated by the concomitant elevation in LV p-Akt, and p-ERK1/2 (Fig. 7). The latter contribute to the formation of lipid aldehydes such as MDA (Ceravolo et al., 2013). Notably, these molecular responses resemble those caused by ethanol in proestrus rats (highest endogenous E2 level). Further, pharmacologic inhibition of MAPKs phosphorylation rescued the myocardium from the deleterious effects of ethanol in female rats (El-Mas and Abdel-Rahman, 2014). Collectively, the present findings suggest that oxidative damage due to the facilitation of LV Akt/ERK1/2 signaling and MDA accumulation might constitute important molecular mechanisms for LV dysfunction and hypotension caused by ethanol in E2-pretreated male rats.
DAPK3 is a Ca2+/calmodulin-regulated serine/threonine kinase whose activation in response to oxidative stress enhances cell death via apoptosis (Eisenberg-Lerner and Kimchi, 2012). Evidence suggests that the DAPK phosphorylation at Thr265 is critical for its apoptotic activity (Sato et al., 2006) whereas its phosphorylation at Ser308 causes deactivation (Bialik and Kimchi, 2012). In the present study, we report the novel observation that while neither E2 nor ethanol affected DAPK phosphorylation, their combination (particularly the higher ethanol dose) significantly increased the level of p-DAPK3/Thr265 in the myocardium (Fig. 7C). Because the ROS/DAPK pathway modulates the cardiac contractile function (Dirkx et al., 2012) and bidirectional signaling between DAPK and ERK is critically important for apoptosis promotion (Chen et al., 2005), data of the current study suggest a key role for the ROS/DAPK3/Akt/ERK cascade in the E2-dependent LV dysfunction caused by ethanol in male rats. Notably, this is the first demonstration of the role of this cascade in the E2-dependent myocardial dysfunction caused by ethanol.
The failure of the E2-dependent enhancement of antioxidant defense system in protecting the myocardium against the deleterious effects of ethanol deserves a comment. This is particularly important given the ability of E2 to protect: (i) the heart against trauma-induced myocardial damage in male rats (Hsu et al., 2009), (ii) the developing rat brain against the detrimental effects of ethanol (Ramezani et al., 2011), (iii) against vascular oxidative stress (Ceravolo et al., 2013), and (iv) against retinal oxidative stress in male and female rats (Wang et al., 2014). While our study is the first to show significant enhancement of LV catalase by E2 in male rats, the additional enhancement of catalase and mitALDH activities in E2/ethanol treated rats (Fig. 8) is consistent with reported findings (Zhang et al., 2003; Ma et al., 2009; El-Mas and Abdel-Rahman, 2014). We also extended our previous findings in female rats by demonstrating significant enhancement of LV HO-1 expression by ethanol in presence of E2 (Fig. 6B). Together, these heightened defense mechanisms may protect the heart against further deterioration in redox status and function.
It is also imperative to consider the risk associated with additive enhancement of myocardial catalase caused by combining E2 and ethanol (Fig. 8B). Catalase catalyzes the metabolic oxidation of ethanol into acetaldehyde, which contributes to the cardiotoxic effect of ethanol (Zhang et al., 2004; Comporti et al., 2010). Similarly, the enhanced generation of acetaldehyde might be re-enforced by the E2-evoked upregulation of alcohol dehydrogenase, another enzyme that breaks down ethanol into acetaldehyde (Dembić Dembić et al., 1996). Therefore, it is likely that E2 enhancement of catalase and alcohol dehydrogenase activities would increase acetaldehyde accumulation and tip the balance towards oxidative stress and myocardial dysfunction in our model system. This view gains credence from the study by Eriksson et al. (1996), which established positive correlation between blood E2 and acetaldehyde concentration after alcohol drinking. More importantly, we have recently shown that microinjection of ethanol into the rostral ventrolateral medulla of spontaneously hypertensive rats, which exhibits higher catalase activity than its control counterpart, results in a greater pressor response, and the latter was attenuated by prior inhibition of local catalase activity (El-Mas and Abdel-Rahman, 2012a). In addition to its role in ethanol cardiovascular toxicity, acetaldehyde has also been implicated in behavioral, reinforcing, and neurotoxic effects of ethanol (Lee et al., 2005; Pastor et al., 2008; Karahanian et al., 2011). It should be remembered, however, that the expected rise in acetaldehyde formation due to heightened catalase and alcohol dehydrogenase activities might be counterbalanced by the concomitant increases in mitALDH2 activity (Fig. 8B), which detoxifies acetaldehyde into acetate. Overall, more studies are needed to discern the precise roles of the oxidative enzymes and products of ethanol, particularly acetaldehyde, to the adverse E2-dependent cardiac dysfunction elicited by acute, and probably chronic, ethanol administration.
The issue whether the E2-dependent oxidative and LV effects of ethanol and underlying molecular mechanisms can be replicated upon long-term ethanol/E2 exposures cannot be ascertained from the current study. Typically, chronic regimens might permit the development of some adaptive changes that would alter the pattern or the magnitude of the cardiovascular consequences of the interacting molecules. It should be remembered, however, that the 90-min observation period was employed here because the same time course was adopted in our previous studies that evaluated the BP and LV effects of ethanol in female rats (El-Mas and Abdel-Rahman, 1999b, 1999c, 2014). Moreover, similar to the acute setting, our previous reports showed that 3-month ethanol treatment caused hypotension, myocardial depression, and autonomic dysfunction in female rats, and these effects that were drastically reduced in ovariectomized rats and restored after simultaneous administration of E2 (El-Mas and Abdel-Rahman, 2001, 2012b).
In conclusion, the present findings reveal important E2-dependent cellular events that might render the myocardial cellular environment more conducive to the ethanol-evoked oxidative damage and subsequent LV dysfunction and hypotension in male rats. These events include the increase in ROS generation and lipid peroxidation and phosphorylation (activation) of pro-oxidant and apoptotic proteins, such as DAPK3, Akt, and ERK1/2, in the myocardium. These data illustrate that men undergoing E2 therapy for medical conditions could be more susceptible to the cardiotoxic effects of alcohol.
Supplementary Material
Highlights.
Ethanol lowers blood pressure and causes LV dysfunction in E2-treated rats
E2/ethanol aggravates cardiac oxidative state via of DAPK3/Akt/ERK activation
E2/ethanol causes a feedback increase in cardiac HO-1, catalase and ALDH2
Alcohol might increase risk of myocardial dysfunction in men treated with E2
Acknowledgments
This work was supported by the National Institute on Alcohol Abuse and Alcoholism [2R01 AA014441-08].
The authors thank Ms. Kui Sun for her technical assistance.
Footnotes
Conflict of Interest Statement
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Dembić Z, Sabolić I. Alcohol dehydrogenase activity in rat kidney cortex stimulated by oestradiol. Biochim Biophys Acta. 1982;714:331–336. doi: 10.1016/0304-4165(82)90341-5. [DOI] [PubMed] [Google Scholar]
- Eriksson CJ, Fukunaga T, Sarkola T, Lindholm H, Ahola L. Estrogen-related acetaldehyde elevation in women during alcohol intoxication. Alcohol Clin Exp Res. 1996;20:1192–1195. doi: 10.1111/j.1530-0277.1996.tb01110.x. [DOI] [PubMed] [Google Scholar]
- Abdel-Rahman ARA, Merrill RH, Wooles WR. Effect of acute ethanol administration on the baroreceptor reflex control of heart rate in normotensive human volunteers. Clin Sci. 1987;72:113–122. doi: 10.1042/cs0720113. [DOI] [PubMed] [Google Scholar]
- Bae KY, Kim SW, Shin HY, Kim JM, Shin IS, Kim SJ, Kim JK, Yoon JS. The acute effects of ethanol and acetaldehyde on physiological responses after ethanol ingestion in young healthy men with different ALDH2 genotypes. Clin Toxicol (Phila) 2012;50:242–249. doi: 10.3109/15563650.2012.672743. [DOI] [PubMed] [Google Scholar]
- Bialik S, Kimchi A. Biochemical and functional characterization of the ROC domain of DAPK establishes a new paradigm of GTP regulation in ROCO proteins. Biochem Soc Trans. 2012;40:1052–1057. doi: 10.1042/BST20120155. [DOI] [PubMed] [Google Scholar]
- Cailleux-Bounacer A, Rohmer V, Lahlou N, Lefebvre H, Roger M, Kuhn JM. Impact level of dihydrotestosterone on the hypothalamic-pituitary-leydig cell axis in men. Int J Androl. 2009;32:57–65. doi: 10.1111/j.1365-2605.2007.00818.x. [DOI] [PubMed] [Google Scholar]
- Ceravolo GS, Filgueira FP, Costa TJ, Lobato NS, Chignalia AZ, Araujo PX, Tostes RC, Dantas AP, Fortes ZB, Carvalho MH. Conjugated equine estrogen treatment corrected the exacerbated aorta oxidative stress in ovariectomized spontaneously hypertensive rats. Steroids. 2013;78:341–346. doi: 10.1016/j.steroids.2012.11.018. [DOI] [PubMed] [Google Scholar]
- Chen CH, Wang WJ, Kuo JC, Tsai HC, Lin JR, Chang ZF, Chen RH. Bidirectional signals transduced by DAPK-ERK interaction promote the apoptotic effect of DAPK. EMBO J. 2005;24:294–304. doi: 10.1038/sj.emboj.7600510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Child JS, Kovick RB, Levisman JA, Pearce ML. Cardiac effects of acute ethanol ingestion unmasked by autonomic blockade. Circulation. 1979;59:120–125. doi: 10.1161/01.cir.59.1.120. [DOI] [PubMed] [Google Scholar]
- Choi BG, McLaughlin MA. Why men’s hearts break: cardiovascular effects of sex steroids. Endocrinol Metab Clin North Am. 2007;36:365–377. doi: 10.1016/j.ecl.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Churchill EN, Disatnik MH, Mochly-Rosen D. Time-dependent and ethanol-induced cardiac protection from ischemia mediated by mitochondrial translocation of εPKC and activation of aldehyde dehydrogenase 2. J Mol Cell Cardiol. 2009;46:278–284. doi: 10.1016/j.yjmcc.2008.09.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comporti M, Signorini C, Leoncini S, Gardi C, Ciccoli L, Giardini A, Vecchio D, Arezzini B. Ethanol-induced oxidative stress: basic knowledge. Genes Nutr. 2010;5:101–109. doi: 10.1007/s12263-009-0159-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirkx E, Schwenk RW, Coumans WA, Hoebers N, Angin Y, Viollet B, Bonen A, van Eys GJ, Glatz JF, Luiken JJ. Protein kinase D1 is essential for contraction-induced glucose uptake but is not involved in fatty acid uptake into cardiomyocytes. J Biol Chem. 2012;287:5871–5881. doi: 10.1074/jbc.M111.281881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg-Lerner A, Kimchi A. PKD is a kinase of Vps34 that mediates ROS-induced autophagy downstream of DAPk. Cell Death Differ. 2012;19:788–797. doi: 10.1038/cdd.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mas MM, Abdel-Rahman AA. An association between the estrogen-dependent hypotensive effect of ethanol and an elevated brainstem c-jun mRNA in female rats. Brain Res. 2001;912:79–88. doi: 10.1016/s0006-8993(01)02727-5. [DOI] [PubMed] [Google Scholar]
- El-Mas MM, Abdel-Rahman AA. Cardiovascular autonomic modulation by nitric oxide synthases accounts for the augmented enalapril-evoked hypotension in ethanol-fed female rats. Alcohol. 2013;47:339–346. doi: 10.1016/j.alcohol.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mas MM, Abdel-Rahman AA. Aortic barodenervation upregulates α2-adrenoceptors in the nucleus tractus solitarius and rostral ventrolateral medulla: an autoradiographic study. Neuroscience. 1997;79:581–590. doi: 10.1016/s0306-4522(96)00648-3. [DOI] [PubMed] [Google Scholar]
- El-Mas MM, Abdel-Rahman AA. Acute hemodynamic effects of ethanol in conscious spontaneously hypertensive and normotensive rats. Alcohol Clin Exp Res. 1999a;23:285–292. [PubMed] [Google Scholar]
- El-Mas MM, Abdel-Rahman AA. Estrogen-dependent hypotensive effects of ethanol in conscious female rats. Alcohol Clin Exp Res. 1999b;23:624–632. [PubMed] [Google Scholar]
- El-Mas MM, Abdel-Rahman AA. Sexually dimorphic hemodynamic effects of intragastric ethanol in conscious rats. Clin Exp Hypertens. 1999c;21:1429–1445. doi: 10.3109/10641969909070858. [DOI] [PubMed] [Google Scholar]
- El-Mas MM, Abdel-Rahman AA. Enhanced catabolism to acetaldehyde in rostral ventrolateral medullary neurons accounts for the pressor effect of ethanol in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2012a;302:H837–844. doi: 10.1152/ajpheart.00958.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mas MM, Abdel-Rahman AA. Exacerbation of myocardial dysfunction and autonomic imbalance contribute to the estrogen-dependent chronic hypotensive effect of ethanol in female rats. Eur J Pharmacol. 2012b;679:95–100. doi: 10.1016/j.ejphar.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mas MM, Abdel-Rahman AA. Nongenomic effects of estrogen mediate the dose-related myocardial oxidative stress and dysfunction caused by acute ethanol in female rats. Am J Physiol Endocrinol Metab. 2014;306:E740–747. doi: 10.1152/ajpendo.00465.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mas MM, El-gowilly SM, Gohar EY, Ghazal AM. Sex and hormonal influences on the nicotine-induced attenuation of isoprenaline vasodilations in the rat perfused kidney. Can J Physiol Pharmacol. 2009a;87:539–548. doi: 10.1139/y09-038. [DOI] [PubMed] [Google Scholar]
- El-Mas MM, Fan M, Abdel-Rahman AA. Endotoxemia-mediated induction of cardiac inducible nitric-oxide synthase expression accounts for the hypotensive effect of ethanol in female rats. J Pharmacol Exp Ther. 2008;324:368–375. doi: 10.1124/jpet.107.127498. [DOI] [PubMed] [Google Scholar]
- El-Mas MM, Fan M, Abdel-Rahman AA. Facilitation of myocardial PI3K/Akt/nNOS signaling contributes to ethanol-evoked hypotension in female rats. Alcohol Clin Exp Res. 2009b;33:1158–1168. doi: 10.1111/j.1530-0277.2009.00939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SM, Lee MT, Lam HM, Leung YK. Estrogens and prostate cancer: etiology, mediators, prevention, and management. Endocrinol Metab Clin North Am. 2011;40:591–614. doi: 10.1016/j.ecl.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JT, Kan WH, Hsieh CH, Choudhry MA, Bland KI, Chaudry IH. Mechanism of salutary effects of estrogen on cardiac function following trauma-hemorrhage: Akt-dependent HO-1 up-regulation. Crit Care Med. 2009;37:2338–2344. doi: 10.1097/CCM.0b013e3181a030ce. [DOI] [PubMed] [Google Scholar]
- Karahanian E, Quintanilla ME, Tampier L, Rivera-Meza M, Bustamante D, Gonzalez-Lira V, Morales P, Herrera-Marschitz M, Israel Y. Ethanol as a prodrug: brain metabolism of ethanol mediates its reinforcing effects. Alcohol Clin Exp Res. 2011;35:606–612. doi: 10.1111/j.1530-0277.2011.01439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klatsky AL. Blood pressure and alcohol intake. In: Laragh JH, Brenner BM, editors. Hypertension: Pathophysiology, Diagnosis, and Management. Raven Press Ltd; 1990. pp. 277–294. [Google Scholar]
- Lagranha CJ, Deschamps A, Aponte A, Steenbergen C, Murphy E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ Res. 2010;106:1681–1691. doi: 10.1161/CIRCRESAHA.109.213645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RD, An SM, Kim SS, Rhee GS, Kwack SJ, Seok JH, Chae SY, Park CH, Choi YW, Kim HS, Cho HY, Lee BM, Park KL. Neurotoxic effects of alcohol and acetaldehyde during embryonic development. J Toxicol Environ Health A. 2005;68:2147–2162. doi: 10.1080/15287390500177255. [DOI] [PubMed] [Google Scholar]
- Li G, Wang X, Abdel-Rahman AA. Brainstem norepinephrine neurons mediate ethanol-evoked pressor response but not baroreflex dysfunction. Alcohol Clin Exp Res. 2005;29:639–647. doi: 10.1097/01.alc.0000160083.72579.ec. [DOI] [PubMed] [Google Scholar]
- Ma H, Li J, Gao F, Ren J. Aldehyde dehydrogenase 2 ameliorates acute cardiac toxicity of ethanol: role of protein phosphatase and forkhead transcription factor. J Am Coll Cardiol. 2009;54:2187–2196. doi: 10.1016/j.jacc.2009.04.100. [DOI] [PubMed] [Google Scholar]
- Ma H, Yu L, Byra EA, Hu N, Kitagawa K, Nakayama KI, Kawamoto T, Ren J. Aldehyde dehydrogenase 2 knockout accentuates ethanol-induced cardiac depression: role of protein phosphatases. J Mol Cell Cardiol. 2010;49:322–329. doi: 10.1016/j.yjmcc.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navaneethan U, Shen L, Venkatesh PG, Hammel J, Patel V, Remzi FH, Kiran RP. Influence of ileal pouch anal anastomosis on bone loss in ulcerative colitis patients. J Crohns Colitis. 2011;5:415–422. doi: 10.1016/j.crohns.2011.04.008. [DOI] [PubMed] [Google Scholar]
- Nitzan Z, Dan M. Disulfiram-like effect of cyproterone acetate. Eur J Obstet Gynecol Reprod Biol. 2009;146:236. doi: 10.1016/j.ejogrb.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Pastor R, Aragon CM. Ethanol injected into the hypothalamic arcuate nucleus induces behavioral stimulation in rats: an effect prevented by catalase inhibition and naltrexone. Behav Pharmacol. 2008;19:698–705. doi: 10.1097/FBP.0b013e328315ecd7. [DOI] [PubMed] [Google Scholar]
- Ramezani A, Goudarzi I, Lashkarbolouki T, Ghorbanian MT, Salmani ME, Abrari K. Neuroprotective effects of the 17β-estradiol against ethanol-induced neurotoxicity and oxidative stress in the developing male rat cerebellum: biochemical, histological and behavioral changes. Pharmacol Biochem Behav. 2011;100:144–151. doi: 10.1016/j.pbb.2011.07.010. [DOI] [PubMed] [Google Scholar]
- Roth MY, Amory JK, Page ST. Treatment of male infertility secondary to morbid obesity. Nat Clin Pract Endocrinol Metab. 2008;4:415–419. doi: 10.1038/ncpendmet0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh TM, Connell BJ. Role of 17β-estradiol in the modulation of baroreflex sensitivity in male rats. Am J Physiol. 1998;275 :R770–R778. doi: 10.1152/ajpregu.1998.275.3.R770. Regulatory Integrative Comp. Physiol. 44. [DOI] [PubMed] [Google Scholar]
- Saleh TM, Connell BJ, Cribb AE. Sympathoexcitatory effects of estrogen in the insular cortex are mediated by GABA. Brain Res. 2005;1037:114–122. doi: 10.1016/j.brainres.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Saravanan R, Pugalendi V. Impact of ursolic acid on chronic ethanol-induced oxidative stress in the rat heart. Pharmacol Rep. 2006;58:41–47. [PubMed] [Google Scholar]
- Sato N, Kamada N, Muromoto R, Kawai T, Sugiyama K, Watanabe T, Imoto S, Sekine Y, Ohbayashi N, Ishida M, Akira S, Matsuda T. Phosphorylation of threonine-265 in Zipper-interacting protein kinase plays an important role in its activity and is induced by IL-6 family cytokines. Immunol Lett. 2006;103:127–134. doi: 10.1016/j.imlet.2005.10.015. [DOI] [PubMed] [Google Scholar]
- Tremblay M, Gallant F, Lavallière M, Chiasson M, Silvey D, Behm D, Albert WJ, Johnson MJ. Driving performance on the descending limb of blood alcohol concentration (BAC) in undergraduate students: a pilot study. PLoS One. 2015;10:e0118348. doi: 10.1371/journal.pone.0118348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- T’Sjoen GG, Beguin Y, Feyen E, Rubens R, Kaufman JM, Gooren L. Influence of exogenous oestrogen or (anti-) androgen administration on soluble transferring receptor in human plasma. J Endocrinol. 2005;186:61–67. doi: 10.1677/joe.1.06112. [DOI] [PubMed] [Google Scholar]
- Usui T, Okada M, Hara Y, Yamawaki H. Death-associated protein kinase 3 mediates vascular inflammation and development of hypertension in spontaneously hypertensive rats. Hypertension. 2012;60:1031–1039. doi: 10.1161/HYPERTENSIONAHA.112.200337. [DOI] [PubMed] [Google Scholar]
- Wang Q, Brunner HR, Burnier M. Determination of cardiac contractility in awake unsedated mice with a fluid-filled catheter. Am J Physiol Heart Circ Physiol. 2004;286:H806–H814. doi: 10.1152/ajpheart.00291.2003. [DOI] [PubMed] [Google Scholar]
- Wang S, Wang B, Feng Y, Mo M, Du F, Li H, Yu X. 17β-Estradiol Ameliorates Light-Induced Retinal Damage in Sprague-Dawley Rats by Reducing Oxidative Stress. J Mol Neurosci. 2014 doi: 10.1007/s12031-014-0384-6. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Williams G. Aromatase up-regulation, insulin and raised intracellular oestrogens in men, induce adiposity, metabolic syndrome and prostate disease, via aberrant ER-α and GPER signalling. Mol Cell Endocrinol. 2012;351:269–278. doi: 10.1016/j.mce.2011.12.017. [DOI] [PubMed] [Google Scholar]
- Wu KL, Chen CH, Shih CD. Nontranscriptional activation of PI3K/Akt signaling mediates hypotensive effect following activation of estrogen receptor β in the rostral ventrolateral medulla of rats. J Biomed Sci. 2012;19:76. doi: 10.1186/1423-0127-19-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M, Madden MC, Barton HA. Developmental expression of aldehyde dehydrogenase in rat: a comparison of liver and lung development. Toxicol Sci. 2006;89:386–398. doi: 10.1093/toxsci/kfj045. [DOI] [PubMed] [Google Scholar]
- Zhang X, Klein AL, Alberle NS, Norby FL, Ren BH, Duan J, Ren J. Cardiac-specific overexpression of catalase rescues ventricular myocytes from ethanol-induced cardiac contractile defect. J Mol Cell Cardiol. 2003;35:645–652. doi: 10.1016/s0022-2828(03)00080-4. [DOI] [PubMed] [Google Scholar]
- Zhang X, Li SY, Brown RA, Ren J. Ethanol and acetaldehyde in alcoholic cardiomyopathy: from bad to ugly en route to oxidative stress. Alcohol. 2004;32:175–186. doi: 10.1016/j.alcohol.2004.01.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.