

Abstract
Tumor necrosis factor-α (TNF-α) is a pro-inflammatory cytokine produced by monocytes/macrophage that plays a pathological role in rheumatoid arthritis (RA). In this study, we investigate the effect of thymoquinone (TQ), a phytochemical found in Nigella sativa, in regulating TNF-α-induced RA synovial fibroblast (RA-FLS) activation. Treatment with TQ (1–5 μM) had no marked effect on the viability of human RA-FLS. Pre-treatment of TQ inhibited TNF-α-induced interleukin-6 (IL-6) and IL-8 production and ICAM-1, VCAM-1, and cadherin-11 (Cad-11) expression in RA-FLS (p<0.01). Evaluation of the signaling events showed that TQ inhibited TNF-α-induced phospho-p38 and phospho-JNK expression, but had no inhibitory effect on NF-κB pathway, in RA-FLS (p<0.05; n=4). Interestingly, we observed that selective down-regulation of TNF-α-induced phospho-p38 and phospho-JNK activation by TQ is elicited through inhibition of apoptosis-regulated signaling kinase 1 (ASK1). Furthermore, TNF-α selectively induced phosphorylation of ASK1 at Thr845 residue in RA-FLS, which was inhibited by TQ pretreatment in a dose dependent manner (p<0.01). Pre-treatment of RA-FLS with ASK1 inhibitor (TC ASK10), blocked TNF-α induced expression of ICAM-1, VCAM-1, and Cad-11. Our results suggest that TNF-α-induced ASK1-p38/JNK pathway is an important mediator of cytokine synthesis and enhanced expression of adhesion molecule in RA-FLS and TQ, by selectively inhibiting this pathway, may have a potential therapeutic value in regulating tissue destruction observed in RA.
Keywords: Rheumatoid arthritis, synovial fibroblasts, ASK1, Thymoquinone, TNF-α, MAP kinase
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic inflammatory disorder characterized by cellular infiltration and proliferation of synovium, leading to progressive destruction of the joints (Ahmed et al., 2008; Smolen and Aletaha, 2009). Antigen-activated CD4+ T cells stimulate monocytes, macrophages, and synovial fibroblasts (FLS) to produce cytokines such as interleukin-1β (IL-1β), IL-6, and tumor necrosis factor-α (TNF-α). These proinflammatory cytokines are master regulator of chronic inflammation and tissue destruction in RA (Choy and Panayi, 2001; Iwamoto et al., 2008; Jones et al., 2013). In response to these cytokines, FLS produce chemokines, matrix metalloproteinases (MMPs), and adhesion molecules that further promote inflammation, hyperplasia and cartilage destruction (Ahmed et al., 2006; Vaillancourt et al., 2011; Jones et al., 2013).
TNF- α is a potent cytokine that exerts diverse effects by stimulating a variety of cells (Abdel-Aziz et al., 2013). It is mainly produced by monocytes and macrophages, but also by B-cells, T-cells, and FLS (Zhu et al., 2014). TNF-α acts as a potent inducer of inflammatory responses through up-regulation of many genes, including cytokines, chemokines, and adhesion molecules (Liang et al., 2011; Tsou et al., 2012). TNF-α binds to cell surface receptors (TNFR1) to initiate multiple signal transduction pathways, including mitogen-activated protein (MAP) kinases and nuclear factor kappa B (NF-κB) pathways (Sabio and Davis, 2014). MAP kinase pathway includes central three-tiered core signaling proteins comprising of MAP kinase kinase kinase (MAP3K), MAP kinase kinase (MAP2K), and MAP kinase (MAPK) (Kyriakis and Avruch, 2012). C-Jun N-terminal kinase (JNK), p38 MAPK, and extracellular signal-regulated kinase (ERK) are well characterized sub-groups of a large MAP kinase family. MAP3Ks, as the proteins upstream in the signaling cascade, sense the degree of stress-induced cell damage and determine cell fate by regulation of the downstream MAP kinase pathways (Cuevas et al., 2007). Apoptosis signal regulating kinase 1 (ASK1) is an important member of MAP3K family that activates both the JNK and p38 MAPK pathways in response to TNF-α stimulation (Nishitoh et al., 1998). ASK1 is activated by various types of stress, including oxidative stress, endoplasmic reticulum (ER) stress, calcium overload, and inflammatory cytokines such as TNF-α (Mnich et al., 2010; Philippe et al., 2013). However, the role of ASK1 in TNF-α signaling pathway to regulate IL-6 and IL-8 production, or the expression adhesion molecules in RA-FLS is still unknown.
Thymoquinone (TQ) is the major active compound derived from Nigella sativa (Woo et al., 2012). Recent animal studies support the potential of TQ for the treatment of a variety of inflammatory disorders like inflammatory bowel disease (IBD), RA, and osteoarthritis (OA) (Salem, 2005; Badr et al., 2011). We have previously shown that oral administration of TQ (5 mg/kg/day) significantly reduced the serum levels of IL-1β and TNF-α as well as a number of inflammatory mediators involved in RA pathogenesis (Umar et al., 2012). In this study, we evaluated the intracellular signaling mechanism by which TQ inhibits TNF-α-induced IL-6 and IL-8 production, and the expression of ICAM-1, VCAM-1, and cadherin-11 (Cad-11) in human RA-FLS.
MATERIALS AND METHODS
Antibodies and Reagents
Recombinant human TNF-α, goat polyclonal antibodies against human ICAM-1 and VCAM-1, IL-6 and IL-8 Duoset ELISA kits, and ASK1 inhibitor (TC ASK10) were purchased from R&D Systems (Minneapolis, MN). Rabbit polyclonal antibodies against phosphorylated ERK1/2, JNK/SAPK, and p38, cadherin-11, and anti-rabbit and anti-mouse horseradish peroxide-linked secondary antibodies were purchased from Cell Signaling Technologies (Beverly, MA). Mouse anti p-ASK1 (Thr845) and TRAF-2 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and a rabbit polyclonal anti-ASK1, Mcl-1 from Abcam (Cambridge, MA). Thymoquinone (>98.5% purity) and rabbit anti-β-actin were purchased from Sigma-Aldrich (St. Louis, MO).
Isolation and culture of RA-FLS
FLS were isolated from RA synovium obtained according to the Institutional Review Board (IRB) approved protocol in compliance with the Helsinki Declaration from patients who had undergone total joint replacement surgery or synovectomy and processed as described previously (Ahmed et al., 2008). RA-FLS were grown in RPMI containing 2 mM L-glutamine with 10% FBS, at 37 °C, in a humidified atmosphere with 5% CO2. Cells were used between passages 7–11.
Cell viability assay
To study the effect of TQ on cell viability, RA-FLS (2 × 104/well) were plated in 96-well, flat-bottomed tissue culture plates (Corning, Corning, NY) and cultured in RPMI 1640 plus 10% FBS for 6 hrs. This was then replaced with fresh medium and culture continued for 24 hrs. TQ (1–5 μM) alone or with TNF-α (20 ng/ml) was added to RA-FLS in serum-free medium and the culture was incubated at 37 °C for another 24 hrs. Two hrs prior to termination of each time point, 20 μl of MTT dye (5 mg/ml in sterile phosphate buffered saline) was added to each well and further incubated at 37 °C. At the end of incubation, cells were washed twice with PBS, solubilized in 100 μl of DMSO at 37 °C for 5 min, and read at an optical density of 570 nm.
Treatment of RA-FLS
To evaluate the activation of TNF-α signaling pathway and inhibition by TQ treatment, RA-FLS (2 × 105/well) were plated in 6-well, flat-bottomed tissue culture plates and cultured in RPMI 1640 plus 10% FBS until greater than 80% confluent. RA-FLS were serum-starved for 12 hrs and pretreated with or without TQ (1–5 μM) for 2 hrs, followed by TNF (20 ng/ml) stimulation for 30 min (for signaling studies) or 24 hrs to evaluate the production of IL-6 and IL-8 in the conditioned media or cellular expression of the adhesion molecules ICAM-1, VCAM-1, and Cad-11.
Western immunoblotting
To study the effect of TQ on TNF-α signaling, cells were lysed in lysis buffer (100 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM NaP2O4, 2 mM Na3VO4, 1% Triton X-100, 10% glycerol, 0.1% SDS, 0.5% deoxycholate, 1 mM phenylmethylsulfonyl fluoride), and protease and phosphatase inhibitors (Roche Diagnostics Corporation, IN; 1 tablet/10 ml), and protein was measured using BCA protein assay kits (Pierce Biotechnology Inc., Rockford, IL). Equal amounts of protein (25–30 μg) were separated by SDS/PAGE and transferred onto nitrocellulose membranes (Bio-Rad). Blots were probed with primary antibodies for the specific protein of interest, followed by incubation with the appropriate horseradish peroxidase-conjugated secondary antibody. Immunoreactive protein bands were visualized by enhanced chemiluminescence using Chemidoc (Bio-Rad) and densitometrically analyzed using Image lab 4.2 software (Bio-Rad). Blots were stripped and reprobed for β-actin to ensure equal protein loading. To study the impact of ASK1 in signaling mechanism induced by TNF-α, RA-FLS were incubated with a chemical inhibitor of ASK1 (TC ASK 10; 12.5 and 25 μM), for 2 hrs, followed by stimulation with TNF-α for 24 hrs. After 24 hrs, the expression of ICAM-1, VCAM-1, and Cad-11 were evaluated in the cell lysates.
IL-6 and IL-8 ELISA
Cells were pretreated with TQ (1–5 μM) and stimulated by TNF-α at 20 ng/ml for 24 hrs at 37°C and condition media was collected for IL-6 and IL-8 estimation. IL-6 and IL-8 were measured using ELISA Kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturers’ instructions.
Co-immunoprecipitation assay
To test the effect of TQ treatment on TRAF2-ASK1 association in RA-FLS, >80% confluent cells in 150 mm dishes were starved overnight, pretreated with 5μM TQ for 2 hrs followed by TNF-α stimulation for 30 mins. Cells were washed 2 time with ice cold PBS followed by lysed using RIPA buffer (50mM Tris pH 7.6, 150 mM NaCl, 1 mM EDTA, 1% Triton X100, 0.1% SDS, 0.5% deoxycholate, and 1 tablet each for protease inhibitor and Phos Stop (Roche) per 10 ml) and sonicated using Branson Sonicator 450 for complete cell lysis. After protein estimation, 500 μg protein from each sample was incubated with 2 μg of mouse monoclonal anti-ASK-1 antibody overnight on the rotar in the cold room. Next day antigen antibody complex were captured using Protein G beads (Roche) for addition 3 hrs. Finally, the IP’ed proteins were eluted with 2X SDS sample buffer by boiling for 5 min. The eluted proteins were resolved on a 4–15% SDS-PAGE gel (Bio-Rad) and probed for TRAF-2 and ASK1.
Immunostaining
To determine the effect of TQ on association of TRAF-2 and ASK1 in TNF-α signaling, RA-FLS (2 × 104 cells/well) were plated in 8-well chamber slides (Corning, NY). At 70% confluence, cells were starved overnight and pretreated for 2 hrs with or without TQ 5 μM followed by stimulation with 20 ng/ml TNF-α for 30 min. Cells were washed twice with ice cold PBS and fixed in 3% paraformaldehyde PBS solution for 10 min at room temperature. Cells were washed 3 time with ice cold 1X PBS and stored in serum free RPMI for overnight. Cells were permeabilized by lysis buffer (20 mM PIPES-pH 6.8, 100 mM NaCl, 3 mM MgCl2, 1 mM EDTA, and 0.5% triton X100) for 10 min at room temperature followed by washing with PBS. Coverslips were blocked using 5% protease free BSA for 1 hrs at room temperature. Primary antibodies mouse monoclonal TRAF-2 (1:250) or rabbit polyclonal ASK1 (1: 100) were used at room temperature for 2 hrs, followed by 3 washes using 1% BSA PBS. Donkey anti mouse Alexa Flour 594 or Donkey anti rabbit Alexa Flour 488 (Invitrogen, CA) as 1:400 dilution were used as secondary staining for 60 min at room temperature. After 3 washes in 1% BSA PBS slides were mounted using Prolong gold anti-fade with DAPI (Invitrogen, CA). Images (40X magnification) were captured using Leica microscope.
Statistical analysis
Statistical analysis was performed using Graph pad prism 6.0. Data was analysed by applying the analysis of variance (ANOVA), followed by Dunnett’s t tests were performed to evaluate the statistical significance of group differences. P values less than 0.05 (2-tailed) were considered significant.
RESULTS
Effect of TQ on RA-FLS viability
The results of an MTT-based viability assay showed that TQ (0.1–10 μM) had only modest effect on the cell viability of cultured RA-FLS (Fig. 1A; n=4). We observed that TQ in combination with TNF-α also had slight change in the viability of RA-FLS in vitro (Fig. 1B; n=4). Furthermore, studied the effects of TQ on expression of Mcl-1, as its overexpression in RA-FLS is a major cause of their resistance to TNF-α-induced apoptosis (Ahmed et al., 2009). We found that the upregulating expression of Mcl-1 after stimulation with TNF-α and is inhibited by TQ in a dose dependent, indicate that TQ induced apoptosis and further sensitizes RA-FLS to TNFα-induced apoptosis by blocking Mcl-1 expression (supplementary Fig. 1).
Fig. 1. Effect of Thymoquinone on human RA-FLS viability.
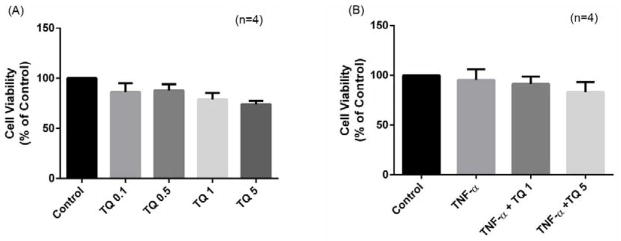
RA-FLS were treated with (A) Thymoquinone (TQ) (0.1–5 μM) for 24 hrs; (B) TQ (1–5 μM) with or without TNF-α (20 ng/ml).
Effect of TQ on IL-6 and IL-8 production and ICAM-1, VCAM-1, and Cad-11 expression in RA-FLS
RA-FLS stimulation with TNF-α (20 ng/ml) resulted in more than 10- and 44-fold increase in IL-6 and IL-8 production, respectively (Fig. 2A & 2B; p<0.001). However, pretreatment with TQ (1–5 μM) inhibited the production of IL-6 and IL-8 in a dose-dependent manner (Fig. 2A and B). ICAM-1 and VCAM-1 expressed by RA-FLS play an important role in the perpetuation of inflammation by facilitating adhesion of circulating inflammatory cells to the walls of endothelial cells and consequently their extravasation at the site of inflammation or tissue injury (Jones et al., 2013). More importantly, Cad-11 expressed by RA-FLS plays an important role in cell-to-cell communication in RA pathogenesis to propagate inflammation and tissue destruction (Lee et al., 2007; Chang et al., 2011). To study the effect of TQ on TNF-α-induced ICAM-1, VCAM-1 and Cad-11 expression, we pretreated RA-FLS with or without TQ followed by TNF-α stimulation for 24 hrs. Stimulation of RA-FLS with TNF-α (20 ng/ml) resulted in a significant 32-, 10-, and 2-fold increase in ICAM-1, VCAM-1, and Cad-11 expression, respectively, when compared to the expression in untreated samples (Fig. 2C; p<0.01). Interestingly, pretreatment of RA-FLS with TQ (1 and 5 μM) resulted in a significant decrease in TNF-α-induced ICAM-1, VCAM-1, and Cad-11 expression, when compared to the expression level with TNF-α alone treatment (p<0.01).
Fig. 2. Effect of TQ on IL-6 and IL-8 production and ICAM-1/VCAM-1 and Cad-11 expression in RA-FLS.

(A and B) RA-FLS were pre-treated with TQ (1–5 μM) for 2 hrs, followed by TNF-α (20 ng/ml) stimulation for 24 hrs. IL-6 and IL-8 production was determined in the conditioned media using commercially available ELISA kits. (C) Expression of adhesion molecule (ICAM-1, VCAM-1 and Cad-11) were analyzed in cell lysates using Western blotting. Densitometric analysis was performed and values are presented as mean ± SEM of n=4 experiments using different donors. ## p<0.01 for NS vs TNF-α; *p<0.05 **p<0.01 for TNF-α vs TQ.
TQ inhibits TNF-α-induced activation of p38 and JNK, but not ERK, in RA-FLS
To understand the molecular mechanism through which TQ modulates TNF-α response in RA-FLS, we studied the effect of TQ on TNF-α-induced MAPK pathways, which are integral signaling mediators of TNF-α-induced downstream inflammatory proteins in RA pathogenesis (Ahmed et al., 2006; Ahmed et al., 2008; Ahmed et al., 2013). RA-FLS were pretreated with TQ stimulated with TNF-α for 30 min. Western blot analysis of the phosphorylated MAPKs showed that TQ selectively inhibits TNF-α-induced phosphorylation of p38 and JNK in a dose-dependent manner (1–5 μM) (Fig. 3A and B; p<0.05 and p<0.01, respectively), with no significant change in TNF-α-induced ERK expression (Fig. 3B). Interestingly, TQ pretreatment had no effect on TNF-α-induced I-κBα degradation suggesting that TQ selectively inhibits p38 and JNK, without interfering NF-κB pathway (Fig. 3A and B). We showed in our previous study that p38 and JNK played an important role in regulating TNF-α-induced ICAM-1 and VCAM-1 expression, whereas there is no evident change with ERK inhibition in RA-FLS (Ahmed et al., 2013).
Fig. 3. TQ inhibits TNF-α-induced activation of p38 and JNK, but not ERK or IκBα, in RAFLS.
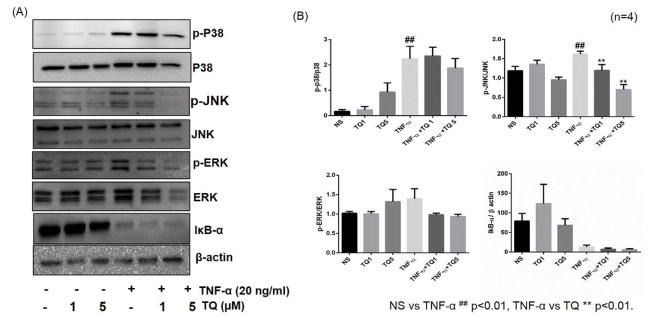
RA-FLS were pre-treated with TQ (1–5 μM) for 2 hrs, followed by TNF-α (20 ng/ml) stimulation for 30 min. Cell lysates were evaluated for the total and phosphorylated form of p38, JNK, ERK, IκB-α and β-actin by Western blotting (A). Densitometric analysis were done by Image lab 5.1 and values are represented as mean ±SEM of n=4 experiments using different donors (B). ## p<0.01 for NS vs TNF-α; **p<0.01 for TNF-α vs TQ.
Involvement of ASK1 in TNF-α stimulated RA fibroblast and its abrogation by TQ
ASK1 belongs to the MAP3K family and activates the p38 and JNK pathways via MKK3/6 and MKK4/7, respectively. Previous studies have shown that JNK pathway is tightly regulated by ASK1 via association and further activation by TRAF-2 (Nishitoh et al., 1998). Western blotting analysis of ASK1 and its phosphorylation forms showed an increase expression of phosphorylation of ASK1 Thr845 (~3 fold) upon TNF-α stimulation, which was markedly inhibited by TQ dose dependently (Fig. 4, p<0.01). However, we observed no significant changes in the total ASK1 expression levels. Since both p38 and JNK MAPKs are involved in phosphorylating c-Jun, we determined the expression levels of phospho-c-Jun to confirm the impact of ASK1 inhibition by TQ. As evident from the Western blotting analysis, TNF-α stimulation resulted in a significant induction of phospho-c-Jun, which was completely inhibited by TQ pretreatment in a dose-dependent fashion (Fig. 4; p<0.01). These results validate that TNF-α signaling in RA-FLS is mediated through activation of ASK1 to induce cytokines and adhesion molecules expression and that TQ inhibits TNF-α-induced ASK1 activation thereby suppressing p38/JNK-mediated phospho-c-Jun expression.
Fig. 4. Involvement of ASK1 in TNF-α stimulated RA fibroblast and their abrogation by TQ.

RA-FLS were treated with TQ (1–5 μM) for 2 hrs, followed by stimulation with TNF-α (20 ng/ml) for 30 min were lysed and probed for the expression of phospho-ASK1 (Thr845), ASK1, and phospho-c-jun (A). Densitometric analysis normalized with β-actin was done by Image Lab 5.1 and values are the mean ± SEM of experiments performed in four different RA-FLS donors (B). # p<0.05, ## p<0.01 for NS vs TNF-α; *p<0.05, **p<0.01 for TNF-α vs TQ.
TQ treatment has no effect on TRAF-2 and ASK1 association in RA-FLS
Studies suggest that the association of TRAF-2 with ASK1 is an important step in TNF-α signaling pathway to activate ASK1-p38/JNK pathway (Nishitoh et al., 1998). To evaluate the effect of TQ on the TRAF-2/ASK1 association step in TNF-α signaling, cell lysates prepared from RA-FLS treated with TQ (5 μM) and/or with TNF-α (20 ng/ml) stimulation were used to IP ASK1 followed by Western blotting for the detection of TRAF-2 (Fig. 5A). The results of the IP assay showed that TQ had no effect on the association of TRAF-2/ASK1 in RA-FLS treated with TNF-α, implicating that TQ inhibits ASK1 phosphorylation and subsequent activation to decelerate TNF-α signaling in RA-FLS. To further confirm this phenomenon, we employed immunofluorescence as a secondary method to determine this interaction and the impact of TQ. As observed in Fig. 5B, there is no significant change in the localisation patterns of TRAF-2 and ASK1 with TQ and/or TNF-α treatment in RA-FLS. Overall, these results suggest that TRAF-2 being an ubiquitin E3 ligase may not be a valid therapeutic target in TNF-α signaling and highlight the role of ASK1 in intimately mediating this process.
Fig. 5. Effect of TQ on TRAF-2 and ASK1 association in RA-FLS.

RA-FLS were treated with TQ (1–5 μM) for 2 hrs, followed by stimulation with TNF-α (20 ng/ml) for 30 min were lysed and IP ASK1 followed by Western blotting for the detection of TRAF-2 (A). RA-FLS were grown to >80% confluence in 8 chamber slides for pre-treatment with TQ (5 μM) for 2 hrs, followed by TNF-α (20 ng/ml) stimulation for 30 min. Cells were fixed and stained with mouse monoclonal TRAF-2 or rabbit polyclonal ASK1 antibody (B), followed by Alexa staining. Images were acquired using 40X magnification.
ASK1 effectively inhibits TNF-α-induced inflammatory mediators in RA-FLS
To verify whether ASK1 indeed plays a role in regulating these inflammatory mediators in TNF-α-induced RA-FLS, cells were pretreated with ASK1 inhibitor (TC-ASK10; 12.5 and 25 μM) for 2 hrs followed by TNF-α stimulation for 24 hours. Western blot analysis on the cell lysates showed a marked inhibitory effect of TC-ASK10 on TNF-α-induced expression of ICAM-1, VCAM-1, and Cad-11 in RA-FLS (Fig. 6; p<0.05–0.01). However, upon determination of IL-6 and IL-8 production in the condition media from these treatment, we observed only modest inhibitory effect of TC-ASK10, suggesting that ASK1 may partly be involved in TNF-α-induced IL-6 and IL-8 production in RA-FLS (data not shown).
Fig. 6. Inhibition of TNF-α-induced expression of adhesion molecules (ICAM-1, VCAM-1, and Cad-11) by ASK1 inhibitor in RA-FLS.

RA-FLS were pre-treated with TC-ASK 10 (ASK1 inhibitor; 12.5–25 μM) for 2 hrs, followed by stimulation with TNF-α (20 ng/ml) for 24 hrs. Cells were lysed and probed for the expression of ICAM-1, VCAM-1 and Cad-11 (A). Densitometric analysis normalized with β-actin was done by Image Lab 5.1 and values are represented as mean ±SEM of 4 independent experiments using different donors (B). # p<0.05, ## p<0.01 for NS vs TNF-α; *p<0.05, **p<0.01 for TNF-α vs ASK inhibitor.
DISCUSSION
Rheumatoid arthritis (RA) is an autoimmune disease that leads to inflammation and destruction of synovial joints (Jones et al., 2013). Curing RA is still out of our reach, despite the broad spectrum of anti-rheumatic drugs (Koenders and van den Berg, 2015). The inflammatory process is mediated through a complex cytokine network which is not yet completely understood. Current treatment strategies for RA include nonsteroidal anti-inflammatory drugs (NSAIDs), corticosteroids, disease-modifying anti-rheumatic drugs (DMARDS) and biologic response modifiers (Ahmed et al., 2005). Despite the availability of biological therapies to treat RA, rigours ongoing research for orally active small-molecule drugs are highly desirable to fulfil unmet medical need. Mitogen-activated protein kinases (MAPKs) p38 in particular, have attracted considerable attention as potential therapeutic targets due to their ability to suppress the production of key inflammatory mediators (Thalhamer et al., 2008). The success of first p38 inhibitors to advance to phase IB clinical trials was hampered by hepatotoxicity and preclinical safety studies (Hammaker and Firestein, 2010).
Apoptosis signal regulating kinase 1 (ASK1) is a member of MAP3K family that activates both the JNK and p38 MAPK pathways through a variety of mechanism, including oxidative stress, ER stress, calcium overload, and inflammatory cytokines such as tumor necrosis factor α (TNF-α) (Matsuzawa and Ichijo, 2008; Choi et al., 2011). Our finding suggest that ASK1 may serve as a promising therapeutic target in the regulation of pro-inflammatory cytokines release in autoimmune disorders especially in RA. TNF-α is a major pathologic mediator and a target of anti TNF-α therapies for RA (Choy and Panayi, 2001). In this study, we further identified TQ as a therapeutic molecule that regulates TNF-α signaling pathways to suppress the inflammatory cellular response. ASK1 activity is controlled by phosphorylation and interactions with other proteins (Yu et al., 2009). A previous study showed that ROS induces dephosphorylation of ASK1 at Ser967 as well as phosphorylation of Thr845 in the ASK1 activation loop, both of which are correlated with ASK1 activity (Valko et al., 2006). Here, we found that TNF-α enhanced phosphorylation of ASK1 at Thr845 and TQ was effective in inhibiting that phosphorylation, thereby decelerating downstream signalling pathways. TNF-α signaling also relies on TRAF-2 association with ASK1 in order to phosphorylate it in a TNF-α-dependent manner (Nitshitoh et al., 1998). Our immunoprecipitation and immunofluorescence results also confirmed that TQ does not alter TRAF-2 and ASK1 interaction, but effectively inhibits ASK1 activation in RA-FLS. Using ASK1 inhibitor (TC-ASK10), we also validate a novel regulatory role of ASK1 in TNF-α-induced expression of adhesion molecules (ICAM-1/VCAM-1 and Cad-11). Our present study may have two-pronged relevance in the field of RA pathogenesis. First, we show that the therapeutic regulation of ASK1 is important in regulating adhesion molecules that are relevant in disease pathogenesis (Lee et al.,2007; Chang et al., 2001). Second, we also provide an evidence that TQ, a natural phytochemical, inhibits ASK1 activation may have potential for its testing in pre-clinical models of RA and further development in this regard.
Activated MAP kinases transform the stimulus into the pathophysiological responses by phosphorylating of downstream substrates, including transcription factors, cytoskeletal proteins involved in mRNA translation (Li et al., 2015). Among the numerous effectors that intervene TNF-α action is activator protein (AP-1), which comprises of transcription factors belonging to the jun and fos families (Adiseshaiah et al., 2006). In response to stress and cytokine, JNK1/2, and p38 kinases binds to transcription factors that regulate c-Jun (Tsou et al., 2012). We showed that there is an increase in phosphorylation of c-Jun expression in TNF-α stimulated RA-FLS and the pretreatment of TQ is able to reduced TNF-α induced expression of p-c-Jun.
In conclusion, we validated the signaling pathway involved in TNF-α-induced proinflammatory cytokines (IL-6 and IL-8) and adhesion molecules (ICAM-1, VCAM-1, and Cad-11) expression in human RA-FLS. We found that TNF-α activated ASK1 plays an important role in the enhanced production of proinflammatory mediators in RA and TQ may be a potential small-molecule inhibitor to modulate TNF-α induced signaling in RA-FLS to minimize inflammation and tissue destruction in RA.
Supplementary Material
Acknowledgments
This study was supported by the NIH grant AR063104 (S.A.), the Arthritis Foundation Innovative Research Grant (S.A.), the start-up funds from Washington State University (S.A.), and the ASPET summer undergraduate research fellowship (SURF) award (O.H.). Authors also thank the National Disease Research Interchange (NDRI), Philadelphia and Cooperative Human Tissue Network (CHTN) for providing the synovial tissue for research.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- Abdel-Aziz H, Wadie W, Abdallah DM, Lentzen G, Khayyal MT. Novel effects of ectoine, a bacteria-derived natural tetrahydropyrimidine, in experimental colitis. Phytomedicine. 2013;20:585–591. doi: 10.1016/j.phymed.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Adiseshaiah P, Kalvakolanu DV, Reddy SP. A JNK-independent signaling pathway regulates TNF alpha-stimulated, c-Jun-driven FRA-1 protooncogene transcription in pulmonary epithelial cells. J Immunol. 2006;177:7193–7202. doi: 10.4049/jimmunol.177.10.7193. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Anuntiyo J, Malemud CJ, Haqqi TM. Biological basis for the use of botanicals in osteoarthritis and rheumatoid arthritis: a review. Evid Based Complement Alternat Med. 2005;2:301–308. doi: 10.1093/ecam/neh117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S, Marotte H, Kwan K, Ruth JH, Campbell PL, Rabquer BJ, Pakozdi A, Koch AE. Epigallocatechin-3-gallate inhibits IL-6 synthesis and suppresses transsignaling by enhancing soluble gp130 production. Proc Natl Acad Sci U S A. 2008;105:14692–14697. doi: 10.1073/pnas.0802675105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S, Silverman MD, Marotte H, Kwan K, Matuszczak N, Koch AE. Down-regulation of myeloid cell leukemia 1 by epigallocatechin-3-gallate sensitizes rheumatoid arthritis synovial fibroblasts to tumor necrosis factor alpha-induced apoptosis. Arthritis Rheum. 2009;60:1282–1293. doi: 10.1002/art.24488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed S, Pakozdi A, Koch AE. Regulation of interleukin-1beta-induced chemokine production and matrix metalloproteinase 2 activation by epigallocatechin-3-gallate in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2006;54:2393–2401. doi: 10.1002/art.22023. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Riegsecker S, Beamer M, Rahman A, Bellini JV, Bhansali P, Tillekeratne LM. Largazole, a class I histone deacetylase inhibitor, enhances TNF-alpha-induced ICAM-1 and VCAM-1 expression in rheumatoid arthritis synovial fibroblasts. Toxicol Appl Pharmacol. 2013;270:87–96. doi: 10.1016/j.taap.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badr G, Alwasel S, Ebaid H, Mohany M, Alhazza I. Perinatal supplementation with thymoquinone improves diabetic complications and T cell immune responses in rat offspring. Cell Immunol. 2011;267:133–140. doi: 10.1016/j.cellimm.2011.01.002. [DOI] [PubMed] [Google Scholar]
- Chang SK, Noss EH, Chen M, Gu Z, Townsend K, Grenha R, Leon L, Lee SY, Lee DM, Brenner MB. Cadherin-11 regulates fibroblast inflammation. Proc Natl Acad Sci USA. 2011;108:8402–8407. doi: 10.1073/pnas.1019437108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi TG, Lee J, Ha J, Kim SS. Apoptosis signal-regulating kinase 1 is an intracellular inducer of p38 MAPK-mediated myogenic signalling in cardiac myoblasts. Biochim Biophys Acta. 2011;1813:1412–1421. doi: 10.1016/j.bbamcr.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344:907–916. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]
- Cuevas BD, Abell AN, Johnson GL. Role of mitogen-activated protein kinase kinase kinases in signal integration. Oncogene. 2007;26:3159–3171. doi: 10.1038/sj.onc.1210409. [DOI] [PubMed] [Google Scholar]
- Hammaker D, Firestein GS. “Go upstream, young man”: lessons learned from the p38 saga. Ann Rheum Dis. 2010;69(Suppl 1):i77–82. doi: 10.1136/ard.2009.119479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto T, Okamoto H, Toyama Y, Momohara S. Molecular aspects of rheumatoid arthritis: chemokines in the joints of patients. FEBS J. 2008;275:4448–4455. doi: 10.1111/j.1742-4658.2008.06580.x. [DOI] [PubMed] [Google Scholar]
- Jones BA, Riegsecker S, Rahman A, Beamer M, Aboualaiwi W, Khuder SA, Ahmed S. Role of ADAM-17, p38 MAPK, cathepsins, and the proteasome pathway in the synthesis and shedding of fractalkine/CX(3) CL1 in rheumatoid arthritis. Arthritis Rheum. 2013;65:2814–2825. doi: 10.1002/art.38095. [DOI] [PubMed] [Google Scholar]
- Koenders MI, van den Berg WB. Novel therapeutic targets in rheumatoid arthritis. Trends Pharmacol Sci. 2015 doi: 10.1016/j.tips.2015.02.001. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GF, Chisaka O, Takeichi M, Brenner MB. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315:1006–1010. doi: 10.1126/science.1137306. [DOI] [PubMed] [Google Scholar]
- Li D, Chen D, Zhang X, Wang H, Song Z, Xu W, He Y, Yin Y, Cao J. c-Jun N-terminal kinase and Akt signalling pathways regulating tumour necrosis factor-alpha-induced interleukin-32 expression in human lung fibroblasts: implications in airway inflammation. Immunology. 2015;144:282–290. doi: 10.1111/imm.12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CJ, Wang SH, Chen YH, Chang SS, Hwang TL, Leu YL, Tseng YC, Li CY, Chen YL. Viscolin reduces VCAM-1 expression in TNF-alpha-treated endothelial cells via the JNK/NF-kappaB and ROS pathway. Free Radic Biol Med. 2011;51:1337–1346. doi: 10.1016/j.freeradbiomed.2011.06.023. [DOI] [PubMed] [Google Scholar]
- Matsuzawa A, Ichijo H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim Biophys Acta. 2008;1780:1325–1336. doi: 10.1016/j.bbagen.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Mnich SJ, Blanner PM, Hu LG, Shaffer AF, Happa FA, O’Neil S, Ukairo O, Weiss D, Welsh E, Storer C, Mbalaviele G, Ichijo H, Monahan JB, Hardy MM, Eda H. Critical role for apoptosis signal-regulating kinase 1 in the development of inflammatory K/BxN serum-induced arthritis. Int Immunopharmacol. 2010;10:1170–1176. doi: 10.1016/j.intimp.2010.06.023. [DOI] [PubMed] [Google Scholar]
- Nagai H, Noguchi T, Homma K, Katagiri K, Takeda K, Matsuzawa A, Ichijo H. Ubiquitin-like sequence in ASK1 plays critical roles in the recognition and stabilization by USP9X and oxidative stress-induced cell death. Mol Cell. 2009;36:805–818. doi: 10.1016/j.molcel.2009.10.016. [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H. ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell. 1998;2:389–395. doi: 10.1016/s1097-2765(00)80283-x. [DOI] [PubMed] [Google Scholar]
- Philippe L, Alsaleh G, Pichot A, Ostermann E, Zuber G, Frisch B, Sibilia J, Pfeffer S, Bahram S, Wachsmann D, Georgel P. MiR-20a regulates ASK1 expression and TLR4-dependent cytokine release in rheumatoid fibroblast-like synoviocytes. Ann Rheum Dis. 2013;72:1071–1079. doi: 10.1136/annrheumdis-2012-201654. [DOI] [PubMed] [Google Scholar]
- Sabio G, Davis RJ. TNF and MAP kinase signalling pathways. Semin Immunol. 2014;26:237–245. doi: 10.1016/j.smim.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem ML. Immunomodulatory and therapeutic properties of the Nigella sativa L. seed. Int Immunopharmacol. 2005;5:1749–1770. doi: 10.1016/j.intimp.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Smolen JS, Aletaha D. Developments in the clinical understanding of rheumatoid arthritis. Arthritis Res Ther. 2009;11:204. doi: 10.1186/ar2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalhamer T, McGrath MA, Harnett MM. MAPKs and their relevance to arthritis and inflammation. Rheumatology. 2008;47:409–414. doi: 10.1093/rheumatology/kem297. [DOI] [PubMed] [Google Scholar]
- Tsou HK, Chen HT, Chang CH, Yang WY, Tang CH. Apoptosis signal-regulating kinase 1 is mediated in TNF-alpha-induced CCL2 expression in human synovial fibroblasts. J Cell Biochem. 2012;113:3509–3519. doi: 10.1002/jcb.24227. [DOI] [PubMed] [Google Scholar]
- Umar S, Zargan J, Umar K, Ahmad S, Katiyar CK, Khan HA. Modulation of the oxidative stress and inflammatory cytokine response by thymoquinone in the collagen induced arthritis in Wistar rats. Chem Biol Interact. 2012;197:40–46. doi: 10.1016/j.cbi.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Vaillancourt F, Silva P, Shi Q, Fahmi H, Fernandes JC, Benderdour M. Elucidation of molecular mechanisms underlying the protective effects of thymoquinone against rheumatoid arthritis. J Cell Biochem. 2011;112:107–117. doi: 10.1002/jcb.22884. [DOI] [PubMed] [Google Scholar]
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Woo CC, Kumar AP, Sethi G, Tan KH. Thymoquinone: potential cure for inflammatory disorders and cancer. Biochem Pharmacol. 2012;83:443–451. doi: 10.1016/j.bcp.2011.09.029. [DOI] [PubMed] [Google Scholar]
- Yu CC, Hsu MJ, Kuo ML, Chen RF, Chen MC, Bai KJ, Yu MC, Chen BC, Lin CH. Thrombin-induced connective tissue growth factor expression in human lung fibroblasts requires the ASK1/JNK/AP-1 pathway. J Immunol. 2009;182:7916–7927. doi: 10.4049/jimmunol.0801582. [DOI] [PubMed] [Google Scholar]
- Zhu W, Jiang C, Xu J, Geng M, Wu X, Sun J, Ma J, Holmdahl R, Meng L, Lu S. Pristane primed rat T cells enhance TLR3 expression of fibroblast-like synoviocytes via a TNF-alpha initiated p38 MAPK and NF-kappaB pathways. Clin Immunol. 2014 doi: 10.1016/j.clim.2014.11.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.