

Abstract
Beyond the well-defined role of the Eph receptor tyrosine kinases in developmental processes, cell motility, cell trafficking/adhesion and cancer, nothing is known about their involvement in liver pathologies. During blood-stage rodent malaria infection we have found that EphB2 transcripts and proteins were upregulated in the liver, a result likely driven by elevated surface expression on immune cells including macrophages. This was significant for malaria pathogenesis because EphB2−/− mice were protected from malaria-induced liver fibrosis despite having a similar liver parasite burden compared with littermate control mice. This protection was correlated with a defect in the inflammatory potential of hepatocytes from EphB2−/− mice resulting in a reduction in adhesion molecules, chemokines/chemokines receptors RNA levels and infiltration of leukocytes including macrophages/Kupffer cells which mediate liver fibrosis during rodent malaria infections. These observations are recapitulated in the well-established carbon tetrachloride (CCL4) model of liver fibrosis in which EphB2−/− CCL4-treated mice showed a significant reduction of liver fibrosis compared to CCL4-treated littermate mice. Depletion of macrophages by clodronate-liposome abrogates liver EphB2 mRNA and proteins up-regulation and fibrosis in malaria-infected mice. Conclusion: During rodent malaria, EphB2 expression promotes malaria-associated liver fibrosis. To our knowledge, our data is the first to reveal the implication of the EphB family of receptor tyrosine kinases in liver fibrosis or in the pathogenesis of malaria infection.
Keywords: Plasmodium, C57BL/6, inflammation, Kupffer cells, NFκB, hepatic stellate cells, macrophages
Liver fibrosis occurs after acute or chronic liver injury and is characterized by an accumulation of extracellular matrix substances, in particular collagen. This liver phenotypic transformation can result from various conditions including infection with pathogens such as malaria (1). Most of the pathology of malaria infection results from parasite multiplication and egress from infected red blood cells (iRBCs) and the sequestration of iRBCs in various organs of the body, including the liver. An aspect of malaria pathogenesis that is being extensively studied is cerebral malaria (CM) a diffuse encephalopathy involving breakdown of the blood brain barrier (BBB) and herniation that is thought to arise, in part, from sequestration of iRBCs on the brain endothelium (2). Acute liver damage from iRBCs sequestered on the liver endothelium is now emerging as another player that may impact the pathogenesis of CM (3). Hepatic injury and fibrosis are seen in mouse models of erythrocytic malaria infection (4) indicating that mouse models can be used to probe possible mechanisms of liver fibrosis in malaria infection.
A crucial step in the initiation of liver fibrosis is a strong inflammatory response. Liver resident macrophages (Kupffer cells) become activated and release transforming growth factor β1 (TGFβ1) as well as pro-inflammatory mediators that activate quiescent hepatic stellate cells (HSCs) into extracellular matrix (ExCM)-producing, α-smooth muscle actin (α-SMA)-positive myofibroblasts (5, 6). Several studies have shown that recruitment of macrophages in the inflamed liver and hepatic activation of IKK/NFκB signalling (a master regulator of inflammatory gene expression) promotes hepatic fibrosis (6–9). Inflammatory macrophages containing-hemozoin are observed in liver biopsies from malaria-infected individuals and are thought to be involved in the induction of liver fibrosis (10, 11). Contrary to the pathogenesis of experimental CM (ECM) in mice where IFN-γ, CD8+ T cells(12) and perforin (13) are required for breakdown of the BBB, examination of livers specimen from C57BL/6 mice infected with the lethal rodent malaria parasite Plasmodium berghei ANKA (PbA) suggests that liver injury and fibrosis occurs independently of CD8+T cells and correlates with hepatic parasite burden (4). Hemolysis and heme-mediated activation of NFκB in the liver have also been shown to play a role in facilitating accumulation of leukocytes including neutrophils and macrophages in rodent malaria (14).
The Eph (Erythropoietin producing hepatocellular) receptors and their corresponding ephrin ligands (Eph receptor interacting) are cell surface molecules that were first isolated in hepatocellular cell line (15). Originally identified as neuronal path-finding molecules, Eph receptors/ephrin system constitute the largest subfamily of receptor tyrosine kinases (RTKs) and ligation of Eph receptors with ephrin ligands initiates a bidirectional signalling cascade affecting diverse biological processes including actin cytoskeleton remodelling, cell-cell adhesion, migration, proliferation and maintenance of intercellular junctions (16). There is a growing body of work suggesting a role for Eph receptors in inflammation, the migration of immune cells, and in the biology of T cell responses (17–19). Some members of the EphB receptors, including EphB2, are expressed on macrophages and have been shown to mediate adhesion to ephrin B2- expressing endothelial cells (20).
Although migration of immune cells to organs where iRBCs are sequestered is intimately linked with the pathogenesis of malaria infection, nothing is known about the role of the EphB/EphrinB molecules in the pathogenesis of malaria infection. Here, using two models of rodent malaria infection, we show that EphB receptors are up-regulated in the liver of C57BL/6 mice infected with PbA, a commonly used model of ECM (21) and P. chabaudi chabaudi AS (PccAS), a non-lethal infection in C57BL/6 mice. We further show that EphB2 exacerbates liver fibrosis in mouse blood-stage malaria infection as well as in the CCL4 model of hepatic fibrosis because it is a molecule that promotes an optimal pro-inflammatory microenvironment required for fibrogenesis. Finally, depletion of liver macrophages abolished up-regulation of EphB2 expression and malaria-associated hepatic fibrosis.
Materials and methods
Rodent malaria infection
Female C57BL/6 wild type (WT) mice aged 6–12 weeks were bred in-house or purchased from The Jackson laboratory (Bar Harbor, ME, USA). EphB2−/− mice on a C57BL/6 background were re-derived and bred in-house under a heterozygous breeding system. Mice were given water and food (LabDiet, MO, USA: chow 5001) ad libitum and housed under standard conditions. Infections were initiated intraperitoneally with 1×106 PbA iRBCs (clone15cy1) or 1×105 PccAS iRBCs obtained from donor mice. Parasitemia was monitored by counting 300–500 RBC of Giemsastained blood smears and in tissues by quantitative PCR. All experiments were approved and carried out according to protocols approved by the Institutional Animal Care University Committee at Emory University.
CCL4 model of hepatic fibrosis
Mice aged 6–12 weeks were injected intraperitoneally with 2μl/g of CCL4 (Sigma) (adjusted at 10% concentration in olive oil) or olive oil three times per week for 4 weeks. Mice were sacrificed by CO2 inhalation 72 hours after the last dose of CCL4 and the livers removed and processed for further analysis.
Assessment of liver injury
Mice were sacrificed by CO2 inhalation and blood samples collected into 20μl of heparin and centrifuged for collection of plasma within 1 hour of blood collection and frozen at −80°C until analysis. Plasma samples from naive and infected mice were processed in a single batch for determination of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels using a DC Element chemistry analyser (HESKA).
Depletion of Kupffer cells and neutrophils/monocytes
Kupffer cells were depleted by intravenous injection of 200μl of clodronate-loaded liposomes or empty liposome-PBS in control mice on day −1 of malaria infection (ClodronateLiposomes.org, Amsterdam, Netherlands). Ly6G-expressing neutrophils and Ly6C-expressing inflammatory monocytes were depleted by injecting mice with 2× 300 μg intraperitoneal doses of anti-Gr-1 monoclonal antibody (clone RB6-8C5) or a Rat IgG isotype control antibody (clone RTK2071) (Biolegend) on days −1 pre-infection and day 1 post-infection.
Assessment of liver fibrosis
Livers were removed from euthanized mice, embedded in OCT compound and immediately snap-frozen in liquid nitrogen. Sections 5μm thick were stained with hematoxylin-eosin to visualize infiltration of leukocytes or picrosirius red (Sigma Aldrich) to visualize hepatic collagen deposition. Imaging was performed on a widefield Zeiss Axioplan 2 microscope in at least 10 low-power non-overlapping random fields (magnification 20×) per mouse using a polarized light filter and quantified using ImageJ software (NIH, Bethesda, MD).
Immunohistochemistry
Sections of cryopreserved liver 5μm thick were stained with the following antibodies: anti- α-SMA-Cy3™ clone 1A4 dilution 1:500 (Sigma), anti-mouse EphB2 (clone 512001 or clone 512013 at 2μg/ml (R&D systems) and NIMP-14 clone ab2557 at dilution 1:200 (Abcam) using standard methodology. Sections were then stained with secondary rat or rabbit antibodies labelled with NorthernLights 493® or NorthernLights 577® (R&D Systems) and nuclei counterstained with mounting medium containing DAPI (VectorShield). Images were captured using a Carl Zeiss confocal microscope and analyzed using Image J software.
RNA extraction and cDNA synthesis
Cells or tissue were homogenized in RNA Stat60® and total RNA extracted using standard phenol-chloroform protocols followed by DNase treatment of RNA extracted using RNA-II purification kit (Nachery-Nagel). A total of 100ng of RNA per sample was converted into cDNA using Superscript II (Life Technologies) at 42°C for 50min, 70°C 15min, in the presence of 5uM oligo (dT)16–18, 5mM Dithiothreitol (DTT), 0.5mM dNTPs (all Life Technologies), 8U RNAsin (Promega), 50mM Tris-HCl pH8.3, 75mM KCl and 3mM MgCl2. The cDNA was treated with 2.5U RNAse H (Affymetrix) at 37°C for 20min to remove any remaining RNA residues.
Quantitative PCR
Real-time qPCR reactions were performed using Quantitect SYBR Green PCR reagent (Qiagen). PCR amplification was performed with 5μl cDNA sample (diluted 1:10), 2μM of each primer, 7μl of QPCR SYBR green mix and plates run using Applied BioSystems FAST 7000 Sequence detection system (ABI Prism FAST 7000). Primer sequences are shown in Supporting Table S1. Transcripts were normalized to two different housekeeping genes (Ubiquitin and β-actin) and expression levels calculated using the 2−ΔΔCt method.
Tissue processing for flow cytometry
Livers and brains were pressed through a 40μm cell strainer, suspended in Iscove's Modified Dulbecco's Medium (IMDM) containing 100units / ml penicillin, 100μg / ml streptomycin, 1μM of L-glutamine, 12mM HEPES, 0.5mM sodium pyruvate, 5 × 10−5M mercaptoethanol (all Gibco) and 10% heat inactivated fetal calf serum (FCS) (PAA Laboratories) containing Liberase-TL synthetic collagenase (Roche) at a final concentration of 0.3mg/ml and dispase (Invitrogen) at final concentration 2mg/ml and further incubated for 45 min at 37°C. The suspension was overlaid on a 30% Percoll gradient and centrifuged at 1800g for 10min. The pellet was collected and supernatant discarded. Red blood cells (RBC) were removed from aseptic single cell suspensions of splenocytes, liver and brain single cell suspensions by incubation in an NH4Cl-based RBC lyzing solution (eBioscience). Detailed procedure for flow cytometry and splenocytes FACS cell sorting are described in the supplementary methods.
A Complete description of the procedures used in this study is available in the Supporting Materials and Methods.
Statistical analysis
Differences between groups of animals were assessed using the non-parametric Mann Whitney-U test or parametric Student's t-test or General Linear Modelling (GLM), a variant of analysis of variance (ANOVA) including all 1st order interactions in Minitab software (Minitab, Inc.). Data or residual variation was assessed for normality using Anderson-Darling test and heterogeneity of variance using the F-test or Bartlett's test. Data that did not meet these requirements for parametric testing was logarithmically or square root transformed. For ANOVA, F values quoted are from the minimal model of the data with all insignificant terms removed. Values of p<0.05 were considered statistically significant.
Results
Rodent malaria infection increased EphB receptors mRNA in the liver
To ascertain whether transcripts of EphB receptors or ephrin B ligands were modulated by blood-stage malaria infection, the mRNA level for each member of the EphB/EphrinB molecules was monitored by qPCR in the spleen, brain, lung and liver of malaria-infected mice. Mice infected with PbA died between days 7–8 post-infection from ECM (Fig.1A) whilst PccAS circulating parasitemia peaked around day 9 post-infection (Fig. 1B). We noted that EphB receptors mRNA were mostly highly upregulated in the liver during Plasmodium infection (Fig. 1C,D). In the liver, PbA-infected mice upregulate EphB2 and EphB3 mRNA ~6-fold at day 4 PI and EphB6 mRNA increases ~4 fold at day 6 PI compared to naive mice (Fig. 1C). PccAS-infected mice also upregulate EphB2 and EphB3 mRNA ~10–15 fold in the liver at day 12 PI compared to naive mice (Fig. 1D). EphrinB1 and EphrinB2 mRNA were not altered in the liver of PbA and PccAS-infected mice (Supporting Fig. 1 A–D) while EphrinB3 mRNA appeared to be marginally upregulated in both infections (Fig. 1 C–D). The up-regulation of EphB2 mRNA was accompanied by increase EphB2 protein in the liver of PccAS and PbA infected mice (Supporting Fig. 1E).
Figure 1. Transcription of EphB receptors occurs predominantly in the liver of malaria infected mice.
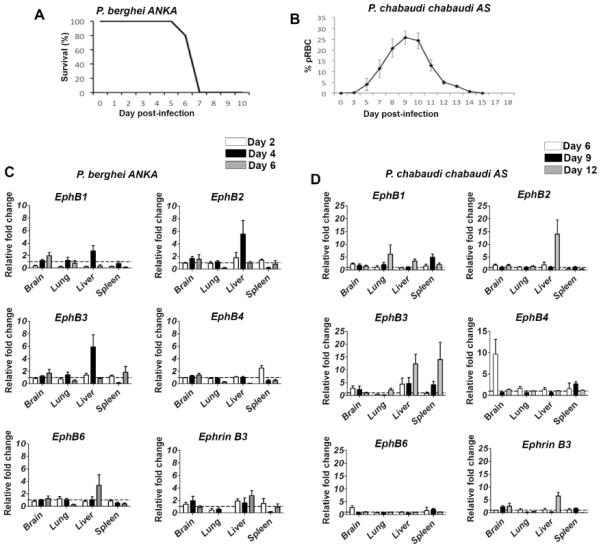
(A–B) C57BL/6 mice were infected with 106 PbA iRBCs or 105 PccAS iRBCs. As controls for infection, 2 mice were injected with Kreb's saline solution. (A) The survival of the mice infected with PbA is shown; (B) the % parasitemia curve for mice infected with PccAS is shown as a reference for each infection profile. (C–D) EphB1, EphB2, EphB3, EphB4, EphB6 and ephrin B3 mRNA from brain, lung, spleen and liver samples measured by qPCR is shown at days 2 (white bars), 4 (black bars) and 6 (grey bars) post PbA infection (C) or days 6 (white bars), 9 (black bars) and 12 (grey bars) post PccAS infection (D). RNA expression is quantified as fold change relative to the average transcripts measured in the naïve control mice at each time point. The bars represent the mean of data pooled from 2 identical experiments in total of n=8 mice. The error bars represent ±SEM from the cumulative data from both experiments.
Immune cells in the liver upregulate EphB receptors in PbA-infected mice
To determine which cells in the liver increased expression of EphB receptors in response to blood-stage malaria infection, flow cytometry was used to determine the % of HSCs or lymphocyte subsets (Fig. 2A) expressing EphB receptors on their surface at day 5 post-infection with PbA (Fig. 2B). There was an increase in surface expression of EphB receptors on HSCs as well as Kupffer cells, inflammatory monocytes and neutrophils (Fig. 2C). All cell types incubated with isotype control antibody, apart from HSCs, had significantly less staining than for samples incubated with recombinant mouse ephrin-B2-Fc chimeric protein (all Mann Whitney-U test p<0.05; HSCs p>0.05). These data suggest that several lymphocyte subsets in the liver upregulate EphB receptors in response to blood-stage malaria infection.
Figure 2. CD11b+ cells upregulate expression of EphB receptors in the liver during PbA infection.
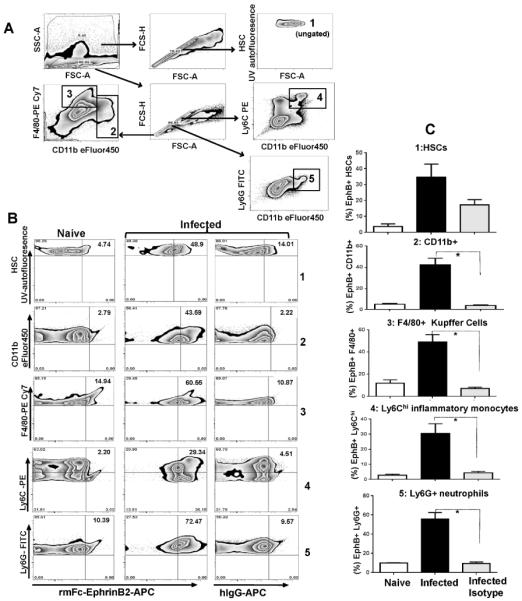
(A) C57BL/6 mice were infected with 106 P. berghei ANKA iRBCs with five mice per group. As controls for infection, 2 mice were injected with Kreb's saline solution. Leukocytes and stellate cells were isolated from livers at day 5 post-infection and gated according to the scheme shown in (A). Populations examined were hepatic stellate cells (1), CD11b+ cells (2), F4/80+ cells (3), inflammatory monocytes (4) and neutrophils (5). (B) Sample staining for each population from naïve and infected mice are shown with the inclusion of infected samples incubated with isotype control antibody for EphB receptor expression. (C) The average percent expression of EphB receptors on each cell population from naïve or infected animals was calculated from the percentage of cell in the upper-right quadrant. The bars represent the cumulative mean values and the error bars represent ±SEM. * p<0.05
EphB2−/− attenuates liver fibrosis in PbA and PccAS-infected mice
To probe the impact of EphB2 expression during malaria infection, we infected EphB2−/− mice or littermate control mice with malaria and assessed liver injury and fibrosis. We noted that malaria infection increases serum liver enzymes AST and ALT in both EphB2−/− and EphB2+/+ mice relative to naïve animals with no significant difference between the two groups of mice (Supporting Fig. 2 A–D). We observed that mice infected with PbA or PccAS had increased collagen deposition in the liver compared with naïve mice (p<0.05). Interestingly, EphB2−/− malaria-infected mice (day 6 in PbA and day 12 in PccAS) have reduced collagen deposition in the liver compared with infected littermate mice (Fig. 3A: PbA GLM F1,15=19.98 and p=0.001, day 6 PI; PccAS GLM F1,14=24.48 and p=0.001, day 12 PI). As further evidence to support the decrease in fibrosis in malaria infected EphB2−/− mice, there was a decreased in HSCs expressing α-SMA in PbA-infected EphB2−/− mice compared with HSCs from infected littermate control mice (Mann Whitney-U test p=0.03) (Fig. 3B). While the profibrotic marker COL1α1 mRNA was significantly down-regulated (Mann Whitney-U test p<0.05), a trend toward a decreased in TGF-β1 and α-SMA mRNA was observed in EphB2−/− infected mice compared with intact littermate mice (Fig. 3C and 3D). Interestingly, similar results were observed in the CCL4 model of liver fibrosis (Supporting Fig. 3A–C).
Figure 3. EphB2−/− mice are protected from liver fibrosis in rodent malaria infection.

C57BL/6 mice were infected with 106 PbA iRBCs or 105 Pcc iRBCs with 5 mice per group. As controls for infection, 2 mice were injected with Kreb's saline solution. (A) Picrosirius staining of the liver sections was used to detect collagen deposition in the liver at day 6 post PbA infection or day 12 post PccAS infection. (B) HSCs were identified according to the gating strategy outlined in Figure 6A and the production of α-SMA evaluated by flow cytometry. (C–D) RNA was extracted from the livers of PbA day 6 post-infection (C) or PccAS day 12 post-infection (D) and the level of profibrotic markers COL1a1, TGF-β1 and α-SMA mRNA measured by qPCR. (E) For liver parasites burden mRNA level of PbA 18s rRNA and PccAS MSP-1 measured by qPCR is shown as -ΔΔCt values. The % circulating parasitemia for PbA at day 6 post-infection is shown for comparison. The bars represent the cumulative data from 2 identical experiments and in total n=10 mice. The error bars represent ±SEM from the cumulative data of both experiments. * p<0.05; +/+=EphB2 WT; −/−=EphB2 KO.
It has been suggested that liver damage/fibrosis is a function of parasite burden in the liver (4), yet assessment of parasite loads by qPCR in the liver indicated no significant difference in parasite burden between infected EphB2−/− mice and littermate controls (Fig. 3E Mann Whitney-U test PbA: p=0.780; PccAS: p=0.560); additionally peripheral parasitemia in EphB2+/+ and EphB2−/− PbA-infected mice was not different (Fig. 3E Mann Whitney-U test p=0.742).
EphB2 deficiency attenuates inflammatory responses in the liver
The release of hepatic pro-inflammatory cytokines is usually linked with the development of liver fibrosis. We next evaluated whether the decrease in collagen deposition in EphB2−/− mice was correlated with a decrease in the transcription of pro-inflammatory cytokines. We observed a drastic reduction of TNFα (mRNA and proteins levels), IL-6 and iNOS mRNA in the liver of PbA-infected EphB2−/− mice compared to intact littermate control mice (Fig. 4A Mann Whitney-U test: TNF-α p=0.002; IL-6 p=0.028; iNOS p=0.028). This decrease was unlikely to be related to an increase in the immunoregulatory cytokine IL-10, the transcription of which did not differ between the two groups of mice examined (Fig. 4A Mann Whitney-U test p=0.885). Similar results were observed in the CCL4 model of liver fibrosis (Supporting Fig. 4) suggesting that the link between EphB2 and fibrogenesis is not restricted to malaria infection.
Figure 4. EphB2−/− hepatocytes have defective inflammatory potential.
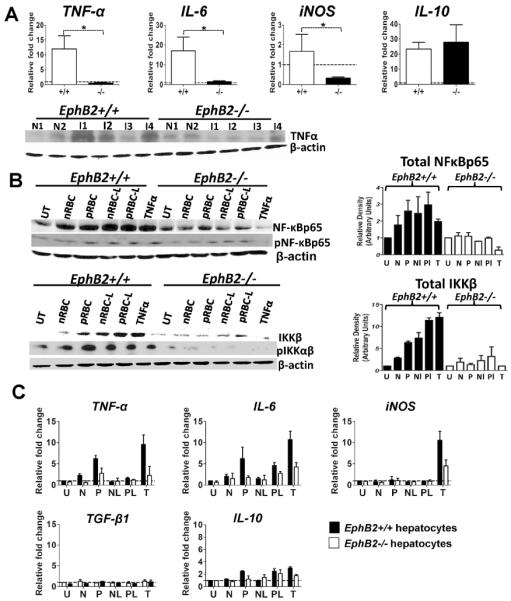
(A) C57BL/6 mice were infected with 106 PbA iRBCs with 5 mice per group. As controls for infection, 2 mice were injected with Kreb's saline solution. At day 6 post-infection RNA was extracted from the livers and the level of TNFα, IL-6, iNOS and IL-10 mRNA measured by qPCR. Immunoblots of liver lysates stained with TNFα and β-actin antibodies. The graphs represent the mean ± SEM from data pooled from 2 separate experiments. (B) Primary hepatocytes were cultured from EphB2−/− or littermate control animals and pulsed for 30 minutes with red blood cells (RBCs) obtained from naïve animals (nRBC), PbA-infected animals (pRBC) or lysates of these red blood preparations (Nl and Pl respectively). As control, hepatocyte cultures were left unstimulated (UT) or pulsed with recombinant mouse TNF-α (T). Densitometry was performed on the bands and is representative of three separate experiments. (C) Hepatocytes were stimulated as described above for 3 hours and RNA extracted for cytokine analysis by qPCR. The bars represent the mean ± SEM of two representative experiments. N=Naïve; I=Infected mice * p<0.05; +/+=EphB2 WT; −/−EphB2 KO.
We next explored whether the apparent reduced liver inflammation in PbA-infected EphB2−/− animals may have been due to differences in the activation of hepatocytes in response to iRBCs. Primary mouse hepatocytes derived from liver of naïve EphB2−/− mice exposed to intact and lyzed iRBCs had reduced activation of the NFκB pathway when compared with those delivered from livers of naïve littermate control mice (Fig. 4B). Since activation of NFκB leads to the transcription of inflammatory genes, we also observed reduced levels of transcription of the TNF-α and IL-6 genes in iRBC-stimulated EphB2−/− hepatocytes compared to those derived from littermate control mice (Fig. 4C). This same trend was observed in hepatocytes stimulated through the TNF receptor (Figs. 4B,C). This effect was unlikely to be mediated by the action of immunoregulatory cytokines on hepatocytes because we observed very little up-regulation in mRNA levels encoding TGF-β1 or IL-10 in stimulated hepatocytes (Fig. 4C).
EphB2 deficiency impairs infiltration of leukocytes into the liver
During fibrogenesis, the inflammatory response leads to recruitment of leukocytes into the liver (22). Interestingly, the Eph/ephrin system has been implicated in the regulation of cell trafficking (23). Given that liver inflammation during malaria infection and CCL4-induced fibrosis was reduced in the absence of EphB2 we asked whether EphB2−/− mice might have reduced trafficking of leukocytes to the liver. We found that with PbA infection this was indeed the case in EphB2−/− mice in comparison to infected littermate control mice (Fig. 5A,B GLM F1,23=9.26 p=0.006). Similar results were observed in the CCL4 model of liver fibrosis (Supporting Fig. 5). There was no reduction in the cellularity of the spleen, in the number of cells trafficking to the brain in response to sequestered PbA iRBC or in the numbers of IFN-γ-producing splenic T cells in PbA-infected EphB2−/− mice compared to littermate controls (Supporting Fig. 6A–D). These data indicate that EphB2 likely modulates leukocyte trafficking to the liver and that EphB2 does not affect the activation of splenic T cells in malaria infected mice.
Figure 5. Immune cells infiltration into the liver of malaria-infected mice is impaired in the absence of EphB2.
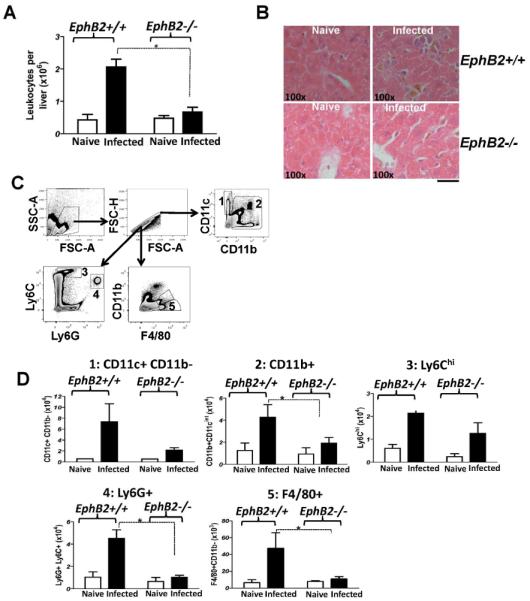
(A) EphB2−/− and littermate control mice were infected with 106 PbA iRBCs with 5 mice per group. As controls for infection, 2 mice were injected with Kreb's saline solution. The numbers of leukocytes at day 6 post-infection with PbA were enumerated. (B) Representative H&E stained sections of liver from animals that were infected with PbA for 6 days are shown. (C–D) A breakdown of the immune cell types infiltrating the liver was identified by flow cytometry (C) and the numbers of CD11c+, CD11b+, Ly6Chi, Ly6G+ and F4/80+cells recruited in the liver of PbA-infected mice at day 6 post infection are shown in (D). The bars represent the cumulative data from 2 identical experiments and in total n=10 mice. Graphs represent the mean ± SEM. * p<0.05
By undertaking FACS analysis of the immune cells isolated from the livers of day 6 PbA-infected mice (Fig. 5C) we found that there was a general trend towards decreased numbers of all cell types analyzed in EphB2−/− mice compared with littermates controls with statistically significant reductions in CD11b+, F4/80+ cells and Ly6G+ neutrophils (Mann Whitney-U test p<0.05) (Fig. 5D). The trends towards a reduction in CD11c+ dendritic cells and Ly6Chi inflammatory monocytes in EphB2−/− mice compared with intact littermate controls did not reach statistical significance. CD4+ and CD8+ T cells were also significantly reduced in the livers of both PbA (day 6) and PccAS (day12) EphB2−/− infected mice as well as INF-γ mRNA (Supporting Fig. 7A–D).
Transcription of adhesion molecules and chemokines is reduced in the livers of EphB2−/− PbA-infected mice
Chemokines and integrins are essential mediators for recruiting immune cells and for activating non-parenchymal liver cells (24). Our data show that PbA infection upregulates the transcription of genes encoding the adhesion molecules VCAM1, ICAM1 and P-selectin (Fig. 6A) as well as chemokines MIP-2, CXCL10, CCL2 and the chemokine receptor CCR2 (Fig. 6B). However, as expected with reduced inflammation, transcription for the majority of these molecules was significantly reduced in EphB2−/− PbA-infected mice compared with infected littermate control animals (Mann Whitney-U test p<0.05 for VCAM1, ICAM-1, CCR2, CXCL10, CCL2, P-selectin). The trend towards decreased transcription of MIP2 in EphB2−/− mice did not reach statistical significance (Mann Whitney-U test p=0.114). Similar results were noted in the CCL4 model of liver fibrosis (Supporting Fig. 8). Thus, reduced chemokines in the liver of EphB2−/− mice may in turn reduce the ability of immune cells to traffic to the liver in response to stimuli.
Figure 6. Livers of EphB2ȡ/− mice have defective transcription of chemokines and adhesion molecules during PbA infection.

(A–B) C57BL/6 mice were infected with 106 PbA iRBCs with 5 mice per group. As controls for infection, 2 mice were injected with Kreb's saline solution. At day 6 post infection RNA was extracted from liver and mRNA level of adhesion molecules VCAM-1, ICAM-1, P-selectin (A) or the chemokines MIP2, CCR2, CXCL10 and CCL2 (B) measured by qPCR. Transcription is shown as fold change relative to the average transcripts measured in the naïve control mice. The bars represent the cumulative data from 2 identical experiments and in total n=10 mice. The error bars represent ±SEM from the cumulative data of both experiments. *p<0.05
Kupffer cells are required for up-regulation of EphB2 expression in PbA-infected liver
EphB2 deficiency resulted in a significant decreased of CD11b+ cells, Ly6G+ neutrophils and F4/80+ cells in the liver in response to malaria infection (Fig. 4D) and these immune cells types are usually involved in the progression of fibrosis. To confirm that accumulation of these cells types in the liver led to the initial observation of increased EphB2 expression in infected mice as suggested in Figs. 1 and 2, we depleted macrophages using clodronate-loaded liposomes and Ly6G+/Ly6C+ neutrophils/inflammatory monocytes using anti-Gr-1 monoclonal antibody administration and infecting with PbA. Flow cytometry analysis revealed that F4/80+/CD11b− Kupffer cells were successfully depleted from the liver using this approach (Fig. 7A). The depletion of neutrophils/monocytes was confirmed by IHC staining of liver sections (Fig. 7B). Only macrophage depletion but not neutrophil/monocyte depletion abrogated the EphB2 mRNA or protein up-regulation observed in PbA-infected mice (Mann Whitney-U test p=0.003, Fig. 7C and Supporting Fig. 9A). These data indicate that Kupffer cells are the main liver cells responsible for the up-regulation of EphB2 in response to rodent malaria infection.
Figure 7. Macrophages/Kupffer cells but not Ly6G+ and Ly6C+ cells are required for EphB2 upregulation in the livers of P. berghei ANKA-infected mice.

(A) macrophages/Kupffer cells were depleted from mice using clodronate-loaded liposomes the day before infection with 106 PbA with eight mice per group. As controls for liposome injection, mice were injected with PBS-loaded liposomes. As controls for infection, 2 mice each were injected clodronate-loaded or PBS-loaded liposomes and injected with Kreb's saline solution. Clodronate treatment successfully removed F4/80+ cells from the liver as identified by flow cytometry (gate 1) in (A). (B) C57BL/6 mice were depleted of neutrophils (Ly6G+) and inflammatory monocytes (Ly6C+) by injection of anti-Gr-1 monoclonal antibody the day before infection with 106 PbA iRBCs and again on day 1 post-infection. Ly6G+ and Ly6C+ cells depletion were successful as demonstrated by immunohistochemical analysis of liver sections stained with NIMP-R14 antibody shown in (B). (C–D) At day 4 post-infection, RNA was extracted from the livers of Kupffer-cell depleted (C) or neutrophil/inflammatory monocyte-depleted (D) PbA-infected mice and mRNA level of EphB2 analyzed by qPCR. For visual clarity two mice were removed from the group of infected mice injected with anti-Gr-1 that had a relative fold change in EphB2 transcription of 111 and 100 in (D). (E) CD11b+, CD11c+, CD8+ and CD4+ cells were sorted from splenocytes and RNA was extracted for analysis of mRNA level of EphB2 by qPCR. Transcription for EphB2 shown as fold change relative to the average transcripts measured in the naïve control mice at each time point. Each point represents 1 mouse and 1 outlier was removed from day 2 CD11b+ with a fold up-regulation of 152 fold relative to the average of naïve animals. ND: not done.
Kupffer cells that participate in inflammatory immune responses are thought to arise from infiltrating monocytes and CD11b+F4/80− monocytes lose CD11b expression upon entering the liver (25). We screened several cell types that were FACS sorted from the spleen, a key site for the generation of immunity against blood-stage malaria (26) at different time points post-PbA infection and found a strong up-regulation of EphB2 transcription at day 2 PI in CD11b+ cells that contain Kupffer cell precursors (Fig. 7E). Altogether these data support a model by which EphB2-expressing resident F4/80+ and infiltrating CD11b+ macrophages accumulate in the liver in response to malaria infection leading to an increase in liver EphB2 expression.
Kupffer cells mediate liver fibrosis in malaria-infected mice
Having established that macrophages upregulate EphB2 and accumulate in the liver, and that depletion of F4/80+ Kupffer cells abrogates EphB2 expression increased during PbA infection, we next sought to investigate whether this depletion of Kupffer cells would phenocopy the reduction in liver fibrosis observed in EphB2−/− PbA-infected mice. We reasoned that during malaria infection, Kupffer cell activation is the critical factor providing the pro-inflammatory microenvironment necessary for HSC transformation into myofibroblast collagen-producing cells. At day 6 PI, we observed that collagen deposition was reduced in mice treated with clodronate-loaded liposomes when compared to control animals injected with PBS-liposomes (Fig. 8A,B Mann Whitney-U test p=0.01) along with reduced levels of Col1a1 and TGF-β1 mRNA (Fig. 8C Mann Whitney-U test p<0.05). This reduction again appeared to be independent of parasite burden which was higher in both the peripheral blood circulation (Mann Whitney-U test p=0.0001) and in the liver of EphB2−/− mice, although the latter was not statistically significant (Fig. 8D Mann Whitney-U test p=0.055). Reduced liver fibrosis was accompanied with reduced accumulation of leukocytes and F4/80+ cells in the liver (Mann Whitney-U test p<0.05) (Fig. 8E,F and Supporting Fig. 9B). We also noted a reduction in inflammatory cytokines/chemokines mRNA and αSMA protein in the liver of PbA-infected clodronate-treated mice (Supporting Fig. 9C,D). Altogether these data suggest a critical implication of Kupffer cells in the up-regulation EphB2 and the modulation of pro-inflammatory response required for induction of fibrogenesis during blood-stage malaria infection.
Figure 8. Collagen deposition in the liver of PbA-infected animals dependent on macrophages/Kupffer cells.

(A–B) Collagen deposition was measured by picrosirius staining of the liver sections at day 6 post PbA infection. Representative sections are shown in (A) and quantified using Image J software across 4 (naïve) or 8 (infected) individual animals per group pooled from 2 experiments (B). The bars represent the mean ±SEM. (C) At day 6 post-infection, RNA was extracted from the livers of PbA-infected mice and mRNA level of Col1a1 and TGF-β1 analyzed by qPCR. (D) For liver parasites burden, mRNA level of PbA 18s rRNA was measured by qPCR and is shown as -ΔΔCt values with the % circulating parasitemia at day 6 post-infection shown for comparison. Graphs represent the mean ± SEM from the cumulative data from 2 experiments. (E–F) The total number of leukocytes and Kupffer cells (gate 1, Figure 7) recruited in the liver on day 6 post-infection with PbA are shown. * p<0.05.
Discussion
Malaria infection significantly contributes to chronic hepatomegaly and hepatic fibrosis among children living in rural areas of Sub-Saharan Africa (27, 28). Rodent models of Plasmodium infection support a model whereby liver injury/fibrosis occur as a result of localized inflammation related to sequestration of iRBCs and accumulation of haemozoin (4) as well as exposure to free heme (29) from hemolysis of infected and uninfected RBCs in the liver. Fibrogenesis is a complex process that involves the activation of quiescent HSCs into myofibroblasts-producing collagen. Based on a growing body of experimental evidence, hepatic inflammation, recruitment of Kupffer cells and HSCs activation are critical events for induction of liver fibrosis (30–32) making them an attractive target for anti-fibrotic strategies (33). Our study demonstrates that EphB2 deficiency lessens liver fibrosis in malaria by dampening inflammation, immune cells infiltration in the liver (Figs. 3–6) and that Kupffer cells are required for EphB2 up-regulation, inflammation, leukocyte infiltration and HSC-mediated collagen deposition (Figs. 7 and 8 and Supporting Fig. 9). In the CCL4 model of liver fibrosis EphB2 deficiency also drastically reduced hepatic fibrosis (Supporting Figs. 3–5 and 8). Altogether, these data highlight EphB2 RTKs as a new player in liver fibrosis in general. We propose a dual mechanism of EphB2 in liver fibrosis during malaria infection or CCL4 exposure in which EphB2 initially influences the inflammatory potential of hepatocytes and subsequently the accumulation of immune cells in the inflamed liver as separate, but related events (Supporting Fig. 10). The relationship between hepatocyte inflammation and macrophage-mediated fibrosis is consistent with results reported by Sunami et al (8).
The Eph receptors have been linked with influencing inflammation via the NFκB signalling pathway (34). A connection between EphB2 ligation and NFκB activation has been suggested for diseases of the central nervous system (35). Our data showing less inflammation in the liver of malaria-infected, CCL4 injected EphB2−/− mice and reduced NF-κB activation in EphB2−/− primary hepatocytes stimulated with iRBC and TNF-α further strengthen the connection between EphB2 and NF-κB activation (Fig. 4 and Supporting Fig. 4). Although the molecular mechanisms leading to the modulation of NF-κB activity upon EphB2 activation is currently unclear we can speculate that EphB2 might be a hub connecting signalling induced by TNFR1 as well as other pattern recognition receptors (PRR) mediating recognition of iRBCs with activation of NF-κB in hepatocytes. Furthermore, in silico analysis revealed the presence of canonical NF-κB binding sites in the promoter of the mouse and human EphB2 gene (data not shown). Further work is necessary for a better understanding of the EphB2/NF-κB signalling pathway and its relationship with liver fibrosis.
Several studies have shown the importance of chemokine/chemokine receptors in the recruitment of immune cells, especially macrophages, in hepatic fibrosis (24) and their potential as targets for the treatment of fibrosis (36). Eph receptors and their ephrin ligands are known to play a role in regulating cell migration by modulating the activity of chemokines (37) and are able to regulate cell-cell adhesion and movement in combination with integrins expressed on the liver vasculature (38). The reason why migrating macrophages do not infiltrate the liver of EphB2−/− mice may be because sequestration of iRBCs does not provoke an inflammatory response of sufficient magnitude to upregulate chemokines and adhesion molecules. Indeed, we observed that EphB2−/m mice had greatly reduced levels of transcription of chemokines, chemokine receptors and adhesion molecules in the liver compared to littermate controls (Fig. 6) supporting this hypothesis.
Macrophages/Kupffer cells are a principal source of inflammatory cytokines and chemokines and, in addition to HSCs, constitute a pool of cells required for fibrogenesis. We showed that upon malaria infection, surface EphB receptors were significantly increased in various subsets of liver CD11b+macrophages (Ly6C+, Ly6G+ and F4/80+ population) and splenic CD11b+macrophages upregulate EphB2 mRNA at day 2 post-PbA infection (Fig. 7E). More importantly selective depletion of macrophages or neutrophils/monocytes followed by infection with PbA demonstrates that F4/80+ macrophages/Kuppfer cells were the main cells required for EphB2 up-regulation and development of malaria-associated hepatic fibrosis (Figs. 7 and 8). Although inflammatory monocytes (39) and neutrophils(29) have previously been reported to be implicated in liver fibrosis, here we show that Kupffer cells which are liver primary phagocytic cells are essentials for liver EphB2 up-regulation and initiate the cascade of inflammation, recruitment of immune cells and production of collagen by HSCs independently of liver parasite burden (Fig. 8). During arteriosclerosis, EphB2-expressing macrophages migrate to areas of arteriosclerotic sites via interaction with ephrinB2 expressed on the inflamed endothelium (40). Our data potentially support a similar role for EphB2 in mediating trafficking of macrophages to the liver in response to the inflammatory response triggered by sequestered iRBCs during blood stage malaria infection.
Activation of Eph receptors signalling can be contact-dependent (via interaction with Ephrin-expressing cells) or contact-independent. In the context of contact-dependent activation of EphB2 for liver fibrosis to occur, we are hypothesizing that EphB2- expressing Kupffer cells need to interact with cells expressing ephrinB ligands within the liver. As expression of Eph/ephrin molecules is ubiquitous, we can speculate that EphB2-expressing Kupffer cells interact with ephrinB-expressing HSCs and modulate collagen production and α-SMA via ephrinB-mediated control of the distribution, internalization and signalling of platelet-derived growth factor receptor-β (PDGFRβ). Ligation of PDGFRβ and the TGF-β receptor are important in the activation of HSCs (41). In fact, EphrinB2 a ligand for the EphB2 receptor, was found to control PDGFR-β distribution and internalization on vascular smooth muscle cells (42) and it is possible that ephrinB ligands perform a similar function on HSC leading to liver fibrosis. To support this plausible model, hepatic macrophages isolated from fibrotic livers have been shown to promote the survival of activated HSCs and this correlated with up-regulation of ephrinB ligand transcripts (6). Whether an actual physical interaction between Kupffer cells and HSCs is required or not for fibrosis to occur, the inflammatory microenvironment induced by Kupffer cells is critical for the transdifferentiation of HSC into myofibroblast collagen producing cells. Further mechanistic work is needed to demonstrate how EphB2 promotes liver fibrosis in mice and humans. However, our data from mouse malaria models and CCL4 fibrosis has uncovered EphB2 as a potential profibrotic molecule that could be targeted for the development of anti-fibrotic therapies.
Supplementary Material
Acknowledgements
We thank Professor Jonathan Gibbins, University of Reading, UK for providing EphB2−/− animals and the staff of the Division of Animal Resources for excellent animal husbandry. We also thank Mr Aaron Rae of the Emory Pediatric Flow Cytometry Core for cell sorting, Miss Katie Casper in the Emory Pediatric Immunology Core for assistance with qPCR analysis and primers designed, Miss Deborah E. Martison and Mr Neil Anthony in the Emory University Integrated Cellular Imaging Core for assistance with microscopy.
Financial Support TJL is supported by National Institute for Neurological Disorders and Strokes (1R21NS085382-01A1), Children's Healthcare of Atlanta, the Royal Society and Emory Egleston Children's Research Centre. MT is supported by the NRSA training grant T32 (2T32AI70081-06AI, FY2013-2014) and F32 Fellowship grant (1F32DK101163-01, FY2014-2015). AG is supported in part by EVC/CFAR Immunology Core P30 AI050409, Yerkes Research Center Base Grant RR-00165, and Public Health Service AI070101.
List of Abbreviations
- iRBCs
infected red blood cells
- CM
cerebral malaria
- BBB
blood brain barrier
- TGF-β1
transforming growth factor-β1
- HSC
hepatic stellate cell
- ExCM
extracellular matrix
- αSMA
α-smooth muscle actin
- IKK
IκB kinase
- NFκB
nuclear factor κ light chain enhancer of activated B cells
- ECM
experimental cerebral malaria
- IFN-γ
interferon-γ
- CD
cluster of differentiation
- PbA
Plasmodium berghei ANKA
- Eph
Erythropoietin producing hepatocellular
- Ephrin
Eph receptor interacting
- GPI
glycosylphosphatidyl inositol
- PccAS
Plasmodium chabaudi chabaudi AS
- WT
wild type
- MSP-1
merozoite protein-1
- LPS
lipopolysaccharide
- TNF-α
tumor necrosis factor-α
- IκBα
nuclear factor of κ light polypeptide gene enhancer in B cells inhibitor, α
- ANOVA
analysis of variance
- qPCR
quantitative polymerase chain reaction
- Col1a1
collagen type 1 α1
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- PRR
pattern recognition receptor
- VCAM
vascular adhesion molecule
- ICAM
intercellular adhesion molecule
- IHC
Immunohistochemistry
- PI
Post-infection
- CXCL10
C-X-C chemokine ligand 10
- MIP-2
macrophage inflammatory protein-2
- CCR2
C-C chemokine receptor 2
- CCL2
C-C chemokine ligand 2
- CCL4
Carbon tetrachloride
- PDGFRβ
platelet-derived growth factor receptor-β
- GLM
General Linear Modelling
Footnotes
Conflicts of interest: None to disclose
Author contributions PNM conceived the design of the experiments, performed the experiments, analyzed the data and wrote the manuscript.
LMB, CFB, MT, CDM, TPK, SM, KQ and CL performed the experiments
EM and AG contributed reagents and intellectual input
TJL conceived the design of the experiments, analyzed the data and wrote the manuscript.
Publisher's Disclaimer: This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1002/hep.27792
References
- 1.Viriyavejakul P, Khachonsaksumet V, Punsawad C. Liver changes in severe Plasmodium falciparum malaria: histopathology, apoptosis and nuclear factor kappa B expression. Malaria journal. 2014;13:106. doi: 10.1186/1475-2875-13-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dorovini-Zis K, Schmidt K, Huynh H, Fu W, Whitten RO, Milner D, Kamiza S, et al. The Neuropathology of Fatal Cerebral Malaria in Malawian Children. The American Journal of Pathology. 2011;178:2146–2158. doi: 10.1016/j.ajpath.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martins YC, Daniel-Ribeiro CT. A new hypothesis on the manifestation of cerebral malaria: The secret is in the liver. Medical Hypotheses. 2013;81:777–783. doi: 10.1016/j.mehy.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haque A, Best SE, Amante FH, Ammerdorffer A, de Labastida F, Pereira T, Ramm GA, et al. High parasite burdens cause liver damage in mice following Plasmodium berghei ANKA infection independently of CD8(+) T cell-mediated immune pathology. Infection and immunity. 2011;79:1882–1888. doi: 10.1128/IAI.01210-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, Pradere J-P, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4 doi: 10.1038/ncomms3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pradere J-P, Kluwe J, De Minicis S, Jiao J-J, Gwak G-Y, Dapito DH, Jang M-K, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58:1461–1473. doi: 10.1002/hep.26429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Helk E, Bernin H, Ernst T, Ittrich H, Jacobs T, Heeren J, Tacke F, et al. TNFα-Mediated Liver Destruction by Kupffer Cells and Ly6Chi Monocytes during Entamoeba histolytica Infection. PLoS Pathog. 2013;9:e1003096. doi: 10.1371/journal.ppat.1003096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sunami Y, Leithäuser F, Gul S, Fiedler K, Güldiken N, Espenlaub S, Holzmann K-H, et al. Hepatic activation of IKK/NFκB signaling induces liver fibrosis via macrophage-mediated chronic inflammation. Hepatology. 2012;56:1117–1128. doi: 10.1002/hep.25711. [DOI] [PubMed] [Google Scholar]
- 9.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rupani AB, Amarapurkar AD. Hepatic changes in fatal malaria: an emerging problem. Annals of tropical medicine and parasitology. 2009;103:119–127. doi: 10.1179/136485909X385054. [DOI] [PubMed] [Google Scholar]
- 11.Whitten R, Milner DA, Jr., Yeh MM, Kamiza S, Molyneux ME, Taylor TE. Liver pathology in Malawian children with fatal encephalopathy. Human pathology. 2011;42:1230–1239. doi: 10.1016/j.humpath.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villegas-Mendez A, Greig R, Shaw TN, de Souza JB, Gwyer Findlay E, Stumhofer JS, Hafalla JC, et al. IFN-gamma-producing CD4+ T cells promote experimental cerebral malaria by modulating CD8+ T cell accumulation within the brain. J Immunol. 2012;189:968–979. doi: 10.4049/jimmunol.1200688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nitcheu J, Bonduelle O, Combadiere C, Tefit M, Seilhean D, Mazier D, Combadiere B. Perforin-dependent brain-infiltrating cytotoxic CD8+ T lymphocytes mediate experimental cerebral malaria pathogenesis. J Immunol. 2003;170:2221–2228. doi: 10.4049/jimmunol.170.4.2221. [DOI] [PubMed] [Google Scholar]
- 14.Dey S, Bindu S, Goyal M, Pal C, Alam A, Iqbal MS, Kumar R, et al. Impact of Intravascular Hemolysis in Malaria on Liver Dysfunction. Journal of Biological Chemistry. 2012;287:26630–26646. doi: 10.1074/jbc.M112.341255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirai H, Maru Y, Hagiwara K, Nishida J, Takaku F. A novel putative tyrosine kinase receptor encoded by the eph gene. Science. 1987;238:1717–1720. doi: 10.1126/science.2825356. [DOI] [PubMed] [Google Scholar]
- 16.Pasquale EB. Eph-Ephrin Bidirectional Signaling in Physiology and Disease. Cell. 2008;133:38–52. doi: 10.1016/j.cell.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 17.Hjorthaug HS, Aasheim HC. Ephrin-A1 stimulates migration of CD8+CCR7+ T lymphocytes. European journal of immunology. 2007;37:2326–2336. doi: 10.1002/eji.200737111. [DOI] [PubMed] [Google Scholar]
- 18.Ivanov AI, Steiner AA, Scheck AC, Romanovsky AA. Expression of Eph receptors and their ligands, ephrins, during lipopolysaccharide fever in rats. Physiological genomics. 2005;21:152–160. doi: 10.1152/physiolgenomics.00043.2004. [DOI] [PubMed] [Google Scholar]
- 19.Yu G, Luo H, Wu Y, Wu J. Mouse ephrinB3 augments T-cell signaling and responses to T-cell receptor ligation. J Biol Chem. 2003;278:47209–47216. doi: 10.1074/jbc.M306659200. [DOI] [PubMed] [Google Scholar]
- 20.Pfaff D, Héroult M, Riedel M, Reiss Y, Kirmse R, Ludwig T, Korff T, et al. Involvement of endothelial ephrin-B2 in adhesion and transmigration of EphB-receptor-expressing monocytes. Journal of Cell Science. 2008;121:3842–3850. doi: 10.1242/jcs.030627. [DOI] [PubMed] [Google Scholar]
- 21.Engwerda C, Belnoue E, Gruner AC, Renia L. Experimental models of cerebral malaria. Curr Top Microbiol Immunol. 2005;297:103–143. [PubMed] [Google Scholar]
- 22.Karlmark KR, Zimmermann HW, Roderburg C, Gassler N, Wasmuth HE, Luedde T, Trautwein C, et al. The fractalkine receptor CX3CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology. 2010;52:1769–1782. doi: 10.1002/hep.23894. [DOI] [PubMed] [Google Scholar]
- 23.Pfaff D, Fiedler U, Augustin HG. Emerging roles of the Angiopoietin-Tie and the ephrin-Eph systems as regulators of cell trafficking. Journal of Leukocyte Biology. 2006;80:719–726. doi: 10.1189/jlb.1105652. [DOI] [PubMed] [Google Scholar]
- 24.Karlmark KR, Wasmuth HE, Trautwein C, Tacke F. Chemokine-directed immune cell infiltration in acute and chronic liver disease. Expert Review of Gastroenterology & Hepatology. 2008;2:233–242. doi: 10.1586/17474124.2.2.233. [DOI] [PubMed] [Google Scholar]
- 25.Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, Pierce RH, et al. Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood. 2007;110:4077–4085. doi: 10.1182/blood-2007-02-073841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Engwerda CR, Beattie L, Amante FH. The importance of the spleen in malaria. Trends in Parasitology. 2005;21:75–80. doi: 10.1016/j.pt.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 27.Walters JH, Mc GI. The mechanism of malarial hepatomegaly and its relationship to hepatic fibrosis. Trans R Soc Trop Med Hyg. 1960;54:135–145. doi: 10.1016/0035-9203(60)90050-x. [DOI] [PubMed] [Google Scholar]
- 28.Wilson S, Vennervald BJ, Dunne DW. Chronic Hepatosplenomegaly in African School Children: A Common but Neglected Morbidity Associated with Schistosomiasis and Malaria. PLoS Negl Trop Dis. 2011;5:e1149. doi: 10.1371/journal.pntd.0001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dey S, Bindu S, Goyal M, Pal C, Alam A, Iqbal MS, Kumar R, et al. Impact of intravascular hemolysis in malaria on liver dysfunction: involvement of hepatic free heme overload, NF-kappaB activation, and neutrophil infiltration. The Journal of biological chemistry. 2012;287:26630–26646. doi: 10.1074/jbc.M112.341255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. The Journal of clinical investigation. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heymann F, Hammerich L, Storch D, Bartneck M, Huss S, Russeler V, Gassler N, et al. Hepatic macrophage migration and differentiation critical for liver fibrosis is mediated by the chemokine receptor C-C motif chemokine receptor 8 in mice. Hepatology. 2012;55:898–909. doi: 10.1002/hep.24764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, Hartland SN, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E3186–3195. doi: 10.1073/pnas.1119964109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baeck C, Wei X, Bartneck M, Fech V, Heymann F, Gassler N, Hittatiya K, et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C+ macrophage infiltration in mice. Hepatology. 2013 doi: 10.1002/hep.26783. n/a-n/a. [DOI] [PubMed] [Google Scholar]
- 34.Chan B, Sukhatme VP. Receptor tyrosine kinase EphA2 mediates thrombin-induced upregulation of ICAM-1 in endothelial cells in vitro. Thrombosis Research. 2009;123:745–752. doi: 10.1016/j.thromres.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pozniak P, White M, Khalili K. TNF-α/NF-κB Signaling in the CNS: Possible Connection to EPHB2. Journal of Neuroimmune Pharmacology. 2013:1–9. doi: 10.1007/s11481-013-9517-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mackay CR. Moving targets: cell migration inhibitors as new anti-inflammatory therapies. Nat Immunol. 2008;9:988–998. doi: 10.1038/ni.f.210. [DOI] [PubMed] [Google Scholar]
- 37.Sharfe N, Freywald A, Toro A, Dadi H, Roifman C. Ephrin stimulation modulates T cell chemotaxis. Eur J Immunol. 2002;32:3745–3755. doi: 10.1002/1521-4141(200212)32:12<3745::AID-IMMU3745>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 38.Sharfe N, Nikolic M, Cimpeon L, Van De Kratts A, Freywald A, Roifman CM. EphA and ephrin-A proteins regulate integrin-mediated T lymphocyte interactions. Molecular immunology. 2008;45:1208–1220. doi: 10.1016/j.molimm.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 39.Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, Merad M, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009;50:261–274. doi: 10.1002/hep.22950. [DOI] [PubMed] [Google Scholar]
- 40.Braun J, Hoffmann SC, Feldner A, Ludwig T, Henning R, Hecker M, Korff T. Endothelial cell ephrinB2-dependent activation of monocytes in arteriosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:297–305. doi: 10.1161/ATVBAHA.110.217646. [DOI] [PubMed] [Google Scholar]
- 41.Lopez-Sanchez I, Dunkel Y, Roh Y-S, Mittal Y, De Minicis S, Muranyi A, Singh S, et al. GIV/Girdin is a central hub for profibrogenic signalling networks during liver fibrosis. Nat Commun. 2014;5 doi: 10.1038/ncomms5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakayama A, Nakayama M, Turner CJ, Höing S, Lepore JJ, Adams RH. Ephrin-B2 controls PDGFRβ internalization and signaling. Genes & Development. 2013;27:2576–2589. doi: 10.1101/gad.224089.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.