

Abstract
Occupational and environmental exposure to arsenic (III) and chromium VI (Cr(VI)) have been confirmed to cause lung cancer. Mechanisms of these metals-induced carcinogenesis are still under investigation. Selection of cell lines to be used is essential for the mechanistic studies. Human bronchial epithelial BEAS-2B cells are the cells to be utilized by most of scientists. However, due to p53 missense mutation (CCG → TCG) at codon 47 and the codon 72 polymorphism (CGC → CCC) in BEAS-2B cells, its usage has frequently been questioned. The present study has examined activity and expression of 53 and its downstream target protein p21 upon acute or chronic exposure of BEAS-2B cells to arsenic and Cr(VI). The results show that short-term exposure of BEAS-2B cells to arsenic or Cr(VI) was able to activate both p53 and p21. Chronic exposure of BEAS-2B cells to these two metals caused malignant cell transformation and tumorigenesis. In arsenic-transformed BEAS-2B cells reductions in p53 promoter activity, mRNA expression, and phosphorylation of p53 at Ser392 were observed, while the total p53 protein level remained the same compared to those in passage-matched parent ones. p21 promoter activity and expression were decreased in arsenic-transformed cells. Cr(VI)-transformed cells exhibit elevated p53 promoter activity, mRNA expression, and phosphorylation at Ser15, but reduced phosphorylation at Ser392 and total p53 protein level compared to passage-matched parent ones. p21 promoter activity and expression were elevated in Cr(VI)-transformed cells. These results demonstrate that p53 is able to respond to exposure of arsenic or Cr(VI), suggesting that BEAS-2B cells are an appropriate in vitro model to investigate arsenic or Cr(VI) induced lung cancer.
Keywords: Arsenic, Chromium(VI), BEAS-2B cells, p53, Cell transformation, Tumorigenesis
Introduction
Heavy metals, such as arsenic and Cr(VI) are confirmed human lung carcinogens by International Agency for Research on Cancer (IARC) (IARC, 2012; Martinez et al. 2011; Morales et al. 2000; Machle et al. 1948; Sorahan et al. 1998). While short-term study of those metals’ toxicity is initial step and necessary, chronic exposure at a low dose which simulates human environmental exposure is mostly adopted by researchers. Therefore, instead of primary cells, immortalized cell lines from normal human bronchial epithelium are essential for those studies of long-term exposure to metals. Among all available immortalized human bronchial/lung cell lines, BEAS-2B cells and human bronchial epithelial cells (HBECs) are most frequently used. BEAS-2B cells, immortalized human bronchial epithelial cells, were originally generated by infection with Ad12-SV40 virus (Lechner et al. 1982; Lechner et al. 1984). Wild type p53 was found in BEAS-2B cells with missense mutation (CCG → TCG) at codon 47 which causes a pro to ser substitution and the codon 72 polymorphism (CGC → CCC) which alters an arg residue to a pro residue (Matlashewski et al. 1987). p53 in BEA-2B cells binds to SV40 T antigen protein but does not bind to the heat shock 70 protein (Reddl, et al. 1988). Transfection of p53 plasmid with codon 47 mutation into human lung carcinoma cell line inhibited cells proliferation, indicating that p53 in BEAS-2B cells has normal wild type properties (Lehman et al. 1993). It has been reported that BEAS-2B cells are non-malignant as evidences by in vitro cell transformation assay and in vivo xenograft tumor model (Ramirez et al 2004). The cell line provider ATCC has described that BEAS-2B cells are p53 wild type (http://www.atcc.org/Products/All/CRL-9609.aspx#documentation). However, the missense mutation at codon 47 of p53 remains to be a concern for the scientists who utilize BEAS-2B cells as an in vitro model in the various research areas including respiratory cytotoxicity of chemicals/agents and malignant cell transformation of carcinogens and related cancer prevention.
The p53 signaling pathway is activated in response to a variety of stress signals, allowing p53 to coordinate transcription programs that ultimately contribute to tumor suppression (Vousden and Prives 2009). Loss of p53 function, through mutations in p53 itself or perturbations in pathways signaling to p53, is a common feature in the majority of human cancers. More than 75% of the mutations result in the expression of a p53 protein that has in most cases lost wild-type functions and may exert a dominant-negative regulation over any remaining wild-type p53 (Petitjean 2007). To evaluate whether exposure of BEAS-2B cells to the heavy metals is able to activate p53, in the present study BEAS-2B cells were exposed to arsenic or Cr(VI) in a short term, activities and expressions at mRNA and protein level of p53 were examined. p21, downstream target protein of p53, its activity and expressions were also evaluated. To best simulate the human environmental exposure, BEAS-2B cells were also chronically exposed to low dose of arsenic or Cr(VI) to generate malignant transformed cells. Expressions of p53 and p21 were examined in arsenic- or Cr(VI)-transformed cells. Tumorigenicity of arsenic- or Cr(VI)-transformed cells was also conducted in vivo.
Materials and Methods
Chemicals and reagents
Sodium arsenite (NaAsO2) and sodium dichromate dehydrate (Na2Cr2O7) were from Sigma (St Louis, MO). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), gentamicin, and L-glutamine were from Invitrogen (Carlsbad, CA). RNeasy mini kit and plasmid prep kit were from Qiagen (Valencia, CA, USA). M-MLV reverse transcriptase was from Promega (Madison, WI). Oligo (dT)20, AccuPrime Taq DNA polymerase high fidelity and pGEM-T easy cloning vector were from Invitrogen (Carlsbad, CA). Luciferase assay system was from Promega (Fitchburg, WI). Antibodies against p53 and phospho-p53 were from Cell Signaling Tech (Danvers, MA). Antibody against p21 antibody was from Santa Cruz (Santa Cruz, CA).
Immunoblot analysis
Cell lysates were prepared in RIPA buffer. Protein concentration was measured using Bradford protein assay reagent (Bio-Rad) and 30 μg of protein was separated by SDS-PAGE, and incubated with primary antibodies. The blots were then re-probed with second antibody conjugated to horseradish peroxidase. Immunoreactive bands were detected by the enhanced chemiluminescence reagent (Amersham).
Luciferase Assay
p53 and p21 promoter activity were measured using luciferase reporter assay. BEAS-2B cells were seeded on 12-well plates (5×105 cells/well). The cells were transfected with 2 μg of plasmid using Lipofectamine 2000. After 24 hours of transfection, the cells were treated for various doses of arsenic or Cr(VI) for 24 hours. The cells were then harvested for luciferase assay in a Glomax luminometer (Promega). Transfections were performed in triplicate and each experiment was repeated at least three times. Data were normalized to total protein determined by Bradford assay.
Real time PCR
RNA was extracted and purified using the RNeasy mini kit. 0.5 μg of RNA was reverse transcribed using qScript cDNA synthesis kit (Quanta Biosciences). Connexin primers were designed using Primer-Blast yielding the following sense and anti-sense sequences: p53, forward-GTTCCGAGAGCTGAATGAGG, reverse-TTATGGCGGGAGGTAGACTG; p21, forward-GGAAGACCATGTGGACCTGT, reverse-GGCGTTTGGAGTGGTAGAAA. Values were normalized by β-actin, forward-TCACCCACACTGTGCCCATCTACGA, reverse-CAGCGGAACCGCTCATTGCCAATGG. All primers were tested using standard curves with 10-fold serial dilutions. The qPCR was performed in the CFX96 Real-Time PCR Detection System (Bio-Rad) using Perfecta Sybr Green Fastmix (Quanta Biosciences), and the data analyzed with CFX Manager software (Bio-Rad).
Cell transformation Assay
BEAS-2B cells were from the American Type Culture Collection (Rockville, MD). The cells were maintained in DMEM supplemented with 10% heat-inactivated FBS and 1% penicillin–streptomycin in 10-cm dishes. After 90% of confluence, cells were treated with 0.1 μM arsenic or 0.25 μM Cr(VI). The fresh medium was added for every 3 days. After 24 weeks, 1×104 cells were suspended in 2 mL culture medium containing 0.35% agar and seeded into 6-well plates with 0.5 % agar base layer, and maintained in an incubator for 4 weeks. The cells were stained with 1 mg/mL iodonitrotetrazolium violet, and colonies greater than 0.1 mm in diameter were scored by microscope examination.
The arsenic- or Cr(VI)-transformed cells from anchorage-independent colonies were picked up and continued to grow in DMEM. Passage-matched cells without Cr(VI) treatment were used as control.
Xenograft tumorigenesis
6-week-old female athymic nude mice were purchased from The Jackson Laboratory (Bar Harbor, ME). The mice were housed in sterilized filter-topped cages and maintained in a pathogen-free animal facility at the Chandler Medical Center, University of Kentucky. All animals were handled according to the Institutional Animal Care and Use Committee (IACUC) guidelines. BEAS-2B, BEAS-2B-As, and BEAS-2B-Cr (1×106 cells) in 100 μL of a mixture of 1xDMEM and Matrigel (BD Biosciences, CA) were subcutaneously (s.b.) injected on the left flank of each mouse. After 21 days, mice were euthanized. Tumors were measured using an external caliper and volume was calculated using the formula: (length × width2)/2.
Statistical analysis
Data were expressed as the mean ± standard deviation (SD). Statistical significance of differences among treatment groups were determined by Student’s t-test. A p< 0.05 was considered as statistically significance.
Results
Activation of p53 upon short-term exposure of BEAS-2B cells to arsenic or Cr(VI) exposure
To investigate whether short term exposure of BEAS-2B cells to arsenic or Cr(VI) is able to activate p53, promoter activity, phosphorylation, and expression of p53 at both transcription and translation level were measured. The results show that both arsenic and Cr(VI) treatment for 24 hours increased p53 activity in a dose-dependent manner (Figs. 1A and 3A). Similarly, mRNA level of p53 was also elevated upon arsenic or Cr(VI) treatment (Figs. 1B and 3B). To confirm that exposure of these two metals are able to activate p53 at translation level, immunoblotting analysis was employed. The results show that arsenic treatment caused phosphorylation of p53 at Ser392, but not at Ser15, while the total p53 level remained unchanged (Fig. 1C). In contrast, Cr(VI) treatment at 5 μM and 10 μM markedly increased phosphorylation of p53 at Ser15 (Fig. 3C), but not at Ser392 (data not shown). Cr(VI) treatment did not alter total protein level of p53.
Fig. 1.
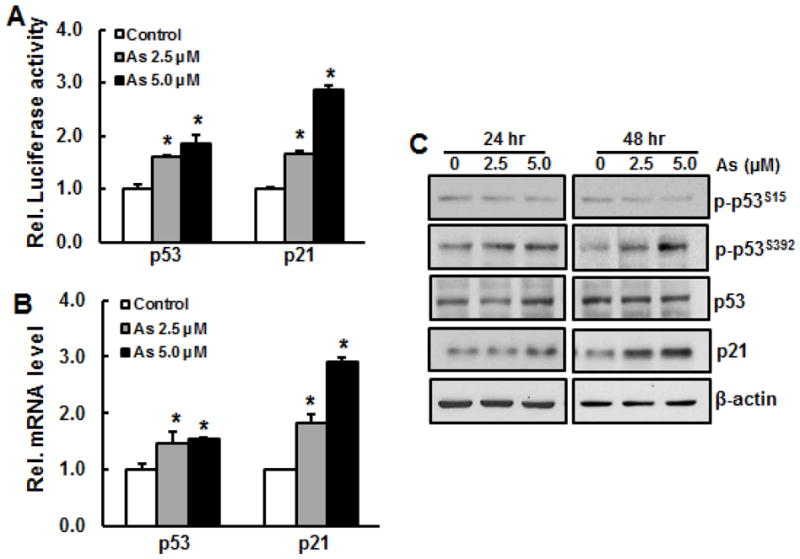
Activations of p53 and p21 upon acute exposure of BEAS-2B cells to arsenic. A, p53 and p21 activity. BEAS-2B cells were transfected with either p53 or p21 luciferase report for overnight, followed by treatment with arsenic at doses of 2.5 μM and 5.0 μM for 24 hr. The cells were harvested for luciferase assay. B, BEAS-2B cells were treated with 2.5 μM and 5.0 μM of arsenic for 24 hr. RNA was isolated from the cells and subjected for RT-PCR analysis. C, BEAS-2B cells were treated with 2.5 μM and 5.0 μM of arsenic for 24 hr and 48 hr. Whole cell lysate was prepared for immunoblotting. *, p<0.05 compared to control group without arsenic treatment.
Fig. 3.

Activations of p53 and p21 upon acute exposure of BEAS-2B cells to Cr(VI). A, p53 and p21 activity. BEAS-2B cells were transfected with either p53 or p21 luciferase report for overnight, followed by treatment with Cr(VI) at doses of 5 μM and 10 μM for 24 hr. The cells were harvested for luciferase assay. B, BEAS-2B cells were treated with 5 μM and 10 μM of Cr(VI) for 24 hr. RNA was isolated from the cells and subjected for RT-PCR analysis. C, BEAS-2B cells were treated with Cr(VI) for 24 hr and 48 hr. Whole cell lysate was prepared for immunoblotting. *, p<0.05 compared to control group without Cr(VI) treatment.
Increased expression of p21 upon short-term exposure of BEAS-2B cells to arsenic or Cr(VI)
p21, a downstream protein of p53, plays an important role in cell cycle and cell proliferation. To investigate whether activation of p53 by arsenic or Cr(VI) exposure causes p21 activation, p21 promoter activity and its expression at transcription and translation level were measured. The results show that both arsenic and Cr(VI) were able to increase promoter activity and mRNA level of p21 (Figs. 1A, 1B, 3A, and 2B). Consequently, p21 protein level was elevated upon arsenic exposure (Fig. 1C). p21 protein level was slightly increased upon 24 hr of Cr(VI) exposure, but it was reduced at 48 hr (Fig. 3C).
Fig. 2.
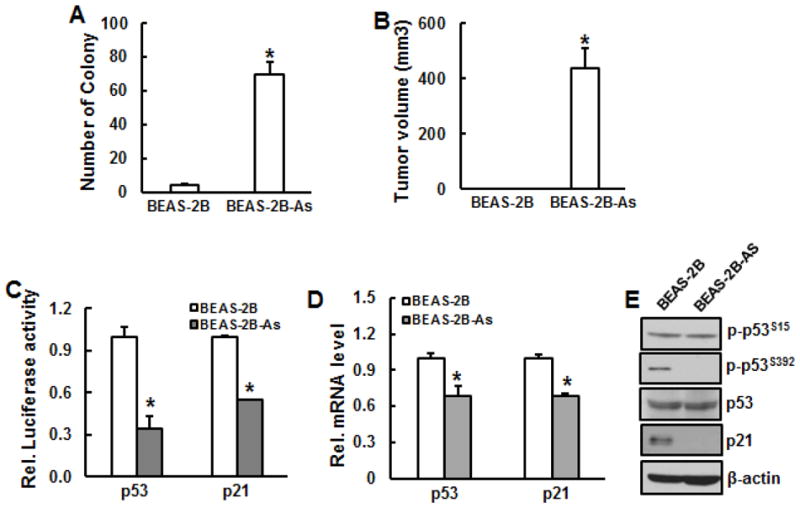
Tumorigeneicity and inactivation of p53 in arsenic-transformed BEAS-2B cells. BEAS-2B cells were chronically exposed to 0.1 μM arsenic for 24 weeks. A, Cell transformation assay. Chronically arsenic-exposed BEAS-2B cells and their passage-matched parent cells were used to examine cell transformation. B, C, and D, The transformed cells from single colony were picked and continued to grow. Arsenic-transformed cells (BEAS-2B-As) and passage-matched non-transformed ones (BEAS-2B) were prepared for tumorigenecity assay (B), luciferase assay (C), RT-PCR (D), and immunoblotting (E). *, p<0.05 compared to passage-matched parent BEAS-2B cells.
Cell transformation induced by chronic exposure to arsenic or Cr(VI)
BEAS-2B cells were chronically exposed to low dose of arsenic (0.1 μM) or Cr(VI) (0.25 μM) for 24 weeks. Soft agar assay was used to examine cell transformation. The results show that the number of colony in the cells exposed to arsenic or Cr(VI) is dramatically increased compared to the passage-matched BEAS-2B cells without treatment, indicating that chronic exposure of BEAS-2B cells to arsenic or Cr(VI) is able to cause malignant cell transformation, but not in the passage-match parent BEAS-2B cells.
Tumorigenecity of arsenic- or Cr(VI)-induced transformed cells
To examine whether arsenic- or Cr(VI)-transformed cells exhibit tumorigenecity, immunodeficient nude mice were s.b. injected with transformed cells and passage-matched parent cells. The volume of the tumor was calculated after 4 weeks of injection. The results show that tumors were only observed in arsenic- or Cr(VI)-transformed cells, but not in the passage-matched parent cells (Figs. 2B and 4B), indicating that arsenic- or Cr(VI)-transformed cells are tumorigenic and normal BEAS-2B cells are not tumorigenic.
Fig. 4.
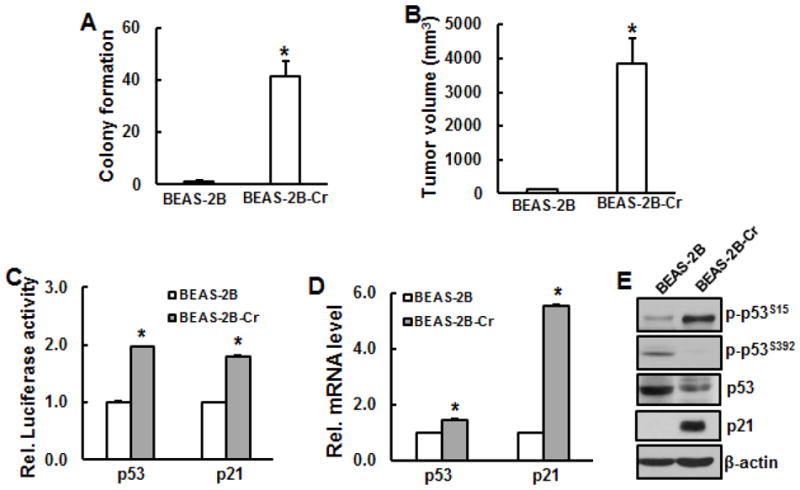
Tumorigenecity and inactivation of p53 in Cr(VI)-transformed cells. BEAS-2B cells were chronically exposed to 0.25 μM Cr(VI) for 24 weeks. A, Cell transformation assay. Chronically Cr(VI)-exposed BEAS-2B cells and their passage-matched parent cells were used to examine cell transformation. B, C, and D, Transformed cells from single colony were picked and continued to grow. Cr(VI)-transformed cells (BEAS-2B-Cr) and passage-matched non-transformed ones (BEAS-2B) were prepared for tumorigenecity assay (B), luciferase assay (C), RT-PCR (D), and immunoblottinh (E). *, p<0.05 compared to passage-matched parent BEAS-2B cells.
Inactivation of p53 and p21 expression in arsenic-transformed cells but increased activities of p53 and p21 in Cr(VI)-transformed cells
It has been reported that p53 expression is inhibited in various cancer cells and tumor tissues. To explore whether function of p53 is lost in arsenic- or Cr(VI)-transformed cells, p53 promoter activity and its expressions at both transcription and translation levels were examined in those transformed cells and their passage-matched parent ones. The results show that p53 promoter activity was markedly decreased in arsenic-transformed cells compared to passage-matched ones (Fig. 2C). Similarly, p53 mRNA level and phosphorylation at Ser392 were also reduced in arsenic-transformed cells compared to passage-matched parent cells (Figs. 2D and 2E) while p53 total protein level and phosphorylation at Ser15 remained the same (Fig. 2E). Difference from arsenic-transformed cells, Cr(VI)-transformed cells display increased p53 promoter activity and mRNA expression (Figs. 4C and 4D). An enhanced phosphorylation of p53 at Ser15, a loss of phosphorylation of p53 at Ser392, and remarkable reduction of total p53 protein level were observed in Cr(VI)-transformed cells compared to normal BEAS-2B cells (Fig. 4E). p21 promoter activity and expressions at both transcription and translation level in arsenic-transformed cells were dramatically reduced compared to their passage-matched parent cells (Figs. 2C–2E). In contrast, p21 was markedly increased in Cr(VI)-transformed cells at promoter activity and transcription and translation level (Figs. 4C–4E).
Discussion
The results from present study show that short-term exposure of BEAS-2B cells to arsenic or Cr(VI) increased promoter activity and mRNA expression of p53. While Cr(VI) exposure increased phosphorylation of p53 at Ser15, but not at Ser392, arsenic exposure increased phosphorylation of p53 at Ser392. Total p53 protein level remained the same upon Cr(VI) or arsenic treatment compared to control without treatment. p53, a well-known tumor suppressor, functions as a node for organizing whether the cell responds to various types and levels of stress with apoptosis, cell cycle arrest, senescence, DNA repair, cell metabolism, or autophagy (Kruse 2009). p53-controlled transactivation of target genes is an essential feature of each stress response pathway, although some effects of p53 may be independent of transcription (Vogelstein et al. 2000; Vousden and Lane 2007; Marchenko and Moll 2007). Phosphorylation of serine residues within the N-terminal p53 transactivation domain was among the first posttranslational modifications of p53 identified. Phosphorylation of p53 is regarded as the first crucial step of p53 stabilization (Kruse 2009). The highly conserved residue, Ser392 is a major phosphorylation site in p53. The induction of Ser392 phosphorylation by diverse stimuli can be explained by a common mechanism in which its phosphorylation at a low rate, coupled with the rapid turnover of p53, limits the accumulation of phosphorylated molecules until a stimulus stabilizes p53 and allows the Ser392-phosphorylated p53 to accumulate (COX and Meek 2010). It has been reported that arsenic treatment up to 48 hours caused increases of total p53 level and p21 expression in BEAS-2B cells (Liao et al. 2007; Liao et al. 2010). Activation of p53 has been believed to play a centric role in mediating stress and DNA damage in response to arsenic exposure (Sandoval 2007). It has also reported that treatment of rat proximal tubular cells with 20 μM of sodium arsenic for 6 hours increased p53 expression at protein level, but not at transcription level, indicating that intracellular accumulation of 53 induced by arsenic is controlled by ubiquitin-conjugating system (Tokumoto 2013). However, a genome-wide analysis showed that p53 expression decreased 1.63-fold in the BEAS-2B cells exposed to high dose (15 μM) of sodium arsenite compared to control without arsenic treatment (Chilakapati 2010). It appears that p53 response upon arsenic exposure depends on cell type, previous p53 stage, and timing.
N-terminal phosphorylation at Ser15 and Ser20 have been generally thought to stabilize p53 by inhibiting the interaction between p53 and Mdm2. Ser15 and Ser20 are phosphorylated after DNA damage and other types of stress by ATM, ATR, DNA-PK, Chk1, and Chk2 (Appella and Anderson 2001; Shieh et al. 1997; Shieh et al. 2000). The results from present study show that Cr(VI) treatment increased phosphorylation of p53 at Ser15. Similarly, our previous publication has demonstrated that ROS-mediated phosphorylation of p53 at Ser15 is essential for Cr(VI)-induced cell death in JB6 cells (Son et al. 2010). It has been reported that 10 μM of Cr(VI) treatment induced phosphorylation of p53 at Ser15 as early as 1 hour of post-treatment in BEAS-2B cells while the level of phosphorylation of p53 at Ser392 and total p53 protein level remained the same upon Cr(VI) treatment compared to control without treatment (Lu et al. 2013). The same results were observed when BEAS-2B cells were treated with doxorubicin, a genotoxic agent which induces double-strand DNA breaks (Lu et al. 2013). Hydrogen peroxide (H2O2) treatment elevated phosphorylation of p53 at Ser15 and Ser392 in BEAS-2B cells (Lu et al. 2013). Cr(VI) generates reactive oxygen species (ROS) that induce DNA damage, which is thought to trigger DNA damage responses in somatic cells (Shi et al. 1994). Phosphorylation of p53 at Ser15 is believed to be an integral part of DNA damage response in vivo (Toledo and Wahl 2006; Kruse and Gu, 2009). Therefore, it is very possible that increased ROS generation is responsible for p53 activation upon Cr(VI) exposure.
p53 mutation and dysfunction have been found in over 50% of all types of human cancers, resulting in inactivation, silence, or even dominant-negative inhibition of wild-type p53 (Muller and Vousden 2013). Many of these mutant p53 proteins acquire oncogenic properties that enable them to promote invasion, metastasis, proliferation, and cell survival. The tumor suppressor p53 is involved in the maintenance of genome stability, and its inactivation is a common characteristic of tumors (Aylon and Oren 2007; Kastan 2007). Even though p53 can also function in a transcription-independent manner, the best understood functions of p53 have been attributed to its transcriptional activity. In fact, approximately half of all cancers bear p53 gene mutations, the vast majority of which impair the ability of p53 to act as a sequence-specific transcriptional activator (Aylon and Oren, 2007). Loss of p53 function probably contributes to tumor progression through a combination of increased genetic instability, loss of growth-arresting signals, and inappropriate cell survival. If the primary reason that p53 loss contributes to tumor development is the generation of increased genetic instability, then p53 dysfunction might act as a “hit-and-run” mutation. In other words, p53 dysfunction would contribute to tumor development because it increases the rate of genetic mutations, but p53 dysfunction may not be required for tumor maintenance once the genetic changes that create the transformed phenotype are in place (Kastan 2007). It has been shown that chronic exposure of BEAS-2B cells to low dose of arsenic alleviated phosphorylation of p53 (Xu et al. 2012). It was also reported that suppression of p53 function by its inhibitor pifithrin-α in BEAS-2B cells promoted cell malignant cell transformation induced by chronic arsenic exposure (Liao et al. 2010). The present study shows that in arsenic-transformed cells promoter activity and mRNA expression of p53 were markedly reduced compared to passage-matched parent cells. Phosphorylation of p53 at Ser392 was also markedly reduced in arsenic-transformed cells, while the total p53 protein level and phosphorylation of p53 at Ser15 remained the same, indicating dysfunction of p53 in arsenic-transformed cells. Differently and more complicatedly, Cr(VI)-transformed cells exhibit a very high expression of phosphorylation of p53 at Ser15, a diminished phosphorylation at Ser392, and a reduced p53 expression, indicating that that Cr(VI) is a strong DNA damage agent which causes constitutive activation of p53. Its mechanism needs to be further investigation.
Activation of the cyclin-dependent kinase inhibitor p21 can be induced by both p53-dependent and p53-independent mechanisms following DNA damage and oxidative stress (Gartel and Tyner 2002). Activated p53 acts as a transcription factor for numerous specific target genes (Smeenk et al. 2008; Millau et al. 2009a, b). Among these genes, p21 mediates p53-depedent G1 growth arrest (Abbas and Dutta 2009). p21 is believed to function as both a sensor and an effector of multiple anti-proliferative signals. The present study has found that p21 was activation at multiple levels upon arsenic or Cr(VI) exposure at short term. This activation of p21 allows cell growth arrest and repairs damaged cells. Rapid proliferation is characteristic of cancer cells (Hanahan and Weinberg, 2000). Earlier studies support the view that p21 suppresses tumors by promoting cell cycle arrest in response to various stimuli. Additionally, substantial evidence from biochemical and genetic studies indicates that p21 acts as a master effector of multiple tumor suppressor pathways for promoting anti-proliferative activities. However, p21 has also be found to be oncogenic due to its overexpression in a variety of human cancer including prostate, cervical, breast, and squamous cell carcinomas. In many cases, p21 up-regulation correlates positively with tumor grade, invasiveness, and aggressiveness and is s poor prognostic indicator (Abbas and Dutta 2009). A microarray analysis has indicated that p21 mRNA was increased in arsenic-transformed BEAS-2B cells with down-regulation of CKS1B, suggesting cell cycle arrest at G1 phase, a common mechanism in aggressive cancer (Stueckle et al. 2012). In contrast, another study showed that knock down of oncogene K-Ras in arsenic-transformed human prostate epithelial cells reduced cell proliferation and anchorage-independent cell growth concurrent with elevated p21 expression (Ngalame et al. 2014).
In summary, results from present study demonstrate that p53 missense mutation (CCG → TCG) at codon 47 and the codon 72 polymorphism (CGC → CCC) in BEAS-2B cells do not cause dysfunction of p53 in response to arsenic or Cr(VI) treatment, indicating that BEAS-2B cells are an appropriate in vitro model to investigate environmental agents induced pulmonary diseases, such as cancer.
Highlights.
Short-term exposure of BEAS-2B cells to arsenic or Cr(VI) activates p53 and p21.
Chronic exposure of BEAS-2B cells to arsenic or Cr(VI) causes cell transformation and tumorigenesis.
Arsenic-transformed cells exhibit reduced activities of p53 and p21.
Cr(VI)-transformed cells exhibit increased activities of p53 and p21.
Acknowledgments
This research was supported by National Institutes of Health/National Institutes of Environmental Health Sciences (R01ES018883 and R01ES021771).
Footnotes
Conflict of interest
The authors declare that there is no conflict interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–14. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Type Culture Collection (ATCC) http://www.atcc.org/Products/All/CRL-9609.aspx#documentation.
- Appella E, Anderson CW. Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem. 2001;268:2764–72. doi: 10.1046/j.1432-1327.2001.02225.x. [DOI] [PubMed] [Google Scholar]
- Aylon Y, Oren M. Living with p53, dying of p53. Cell. 2007;130:597–600. doi: 10.1016/j.cell.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Chilakapati J, Wallace K, Ren H, Fricke M, Bailey K, Ward W, Creed J, Kitchin K. Genome-wide analysis of BEAS-2B cells exposed to trivalent arsenicals and dimethylthioarsinic acid. Toxicology. 2010;268:31–39. doi: 10.1016/j.tox.2009.11.018. [DOI] [PubMed] [Google Scholar]
- Cox ML, Meek DW. Phosphorylation of serine 392 in p53 is a common and integral event during p53 induction by diverse stimuli. Cell Signal. 2010;22:564–71. doi: 10.1016/j.cellsig.2009.11.014. [DOI] [PubMed] [Google Scholar]
- Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21in apoptosis. Mol Cancer Ther. 2002;1:639–649. [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- International Agency for Research on Cancer (IARC) Arsenic and arsenic compounds. IARC Monogr Eval Carcinog Risks Hum. 2012;100C:1–54. [PMC free article] [PubMed] [Google Scholar]
- International Agency for Research on Cancer (IARC) Chromium (VI) compounds. IARC Monogr Eval Carcinog Risks Hum. 2012;100C:1–22. [PMC free article] [PubMed] [Google Scholar]
- Kastan MB. Wild-type p53: tumors can’t stand it. Cell. 2007;128:837–840. doi: 10.1016/j.cell.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–22. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner JF, Haugen A, McClendon IA, Pettis EW. Clonal growth of normal adult human bronchial epithelial cells in a serum-free medium. In Vitro. 1982;18:633–642. doi: 10.1007/BF02796396. [DOI] [PubMed] [Google Scholar]
- Lechner JF, Haugen A, McClendon IA, Shamsuddin AM. Induction of squamous differentiation of normal human bronchial epithelial cells by small amounts of serum. Differentiation. 1984;25:229–237. doi: 10.1111/j.1432-0436.1984.tb01361.x. [DOI] [PubMed] [Google Scholar]
- Lehman TA, Modali R, Boukamp P, Stanek J, Bennett WP, Welsh JA, Metcalf RA, Stampfer MR, Fusenig N, Rogan EM. p53 mutations in human immortalized epithelial cell lines. Carcinogenesis. 1993;14:833–9. doi: 10.1093/carcin/14.5.833. [DOI] [PubMed] [Google Scholar]
- Liao WT, Lin P, Cheng TS, Yu HS, Chang LW. Arsenic promotes centrosome abnormalities and cell colony formation in p53 compromised human lung cells. Toxicol Appl Pharmacol. 2007;225:162–70. doi: 10.1016/j.taap.2007.07.017. [DOI] [PubMed] [Google Scholar]
- Liao WT, Yu HS, Lin P, Chang LW. Arsenite promotes centrosome abnormalities under a p53 compromised status induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) Toxicol Appl Pharmacol. 2010;243:55–62. doi: 10.1016/j.taap.2009.11.012. [DOI] [PubMed] [Google Scholar]
- Lu Y, Xu D, Zhou J, Ma Y, Jiang Y, Zeng W, Dai W. Differential responses to genotoxic agents between induced pluripotent stem cells and tumor cell lines. J Hematol Oncol. 2013;6:71. doi: 10.1186/1756-8722-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machle W, Gregorius F. Cancer of the respiratory system in the United States chromate producing industry. Public Health Rep. 1948;63:1114–1127. [PubMed] [Google Scholar]
- Marchenko ND, Moll UM. The role of ubiquitination in the direct mitochondrial death program of p53. Cell Cycle. 2007;6:1718–1723. doi: 10.4161/cc.6.14.4503. [DOI] [PubMed] [Google Scholar]
- Martinez VD, Vucic EA, Becker-Santos DD, Gil L, Lam WL. Arsenic exposure and the induction of human cancers. J Toxicol. 2011:431287. doi: 10.1155/2011/431287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlashewski GJ, Tuck S, Pim D, Lamb P, Schneider J, Crawford LV. Primary structure polymorphism at amino acid residue 72 of human p53. Mol Cell Biol. 1987;7:961–3. doi: 10.1128/mcb.7.2.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millau JF, Bastien N, Bouchard EF, Drouin R. p53 pre- and post-binding event theories revisited: stresses reveal specific and dynamic p53-binding patterns on the p21 gene promoter. Cancer Res. 2009a;69:8463–71. doi: 10.1158/0008-5472.CAN-09-2036. [DOI] [PubMed] [Google Scholar]
- Millau JF, Bastien N, Drouin R. P53 transcriptional activities: a general overview and some thoughts. Mutat Res. 2009b;681:118–33. doi: 10.1016/j.mrrev.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Morales KH, Ryan L, Kuo TL, Wu MM, Chen CJ. Risk of internal cancers from arsenic in drinking water. Environ Health Persp. 2000;108:655–661. doi: 10.1289/ehp.00108655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller PAJ, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- Ngalame NN, Tokar EJ, Person RJ, Waalkes MP. Silencing KRAS overexpression in arsenic-transformed prostate epithelial and stem cells partially mitigates malignant phenotype. Toxico Sci. 2014;142:489–96. doi: 10.1093/toxsci/kfu201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, Peyton M, Zou Y, Kurie JM, Dimaio JM, Milchgrub S, Smith AL, Souza RF, Gilbey L, Zhang X, Gandia K, Vaughan MB, Wright WE, Gazdar AF, Shay JW, Minna JD. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64:9027–34. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- Reddel RR, Ke Y, Gerwin BI, McMenamin MG, Lechner JF, Su RT, Brash DE, Park JB, Rhim JS, Harris CC. Transformation of Human Bronchial Epithelial Cells by Infection with SV40 or Adenovirus-12 SV40 Hybrid Virus, or Transfection via Strontium Phosphate Coprecipitation with a Plasmid Containing SV40 Early Region Genes. Cancer Res. 1988;48:1904–1909. [PubMed] [Google Scholar]
- Sandoval M, Morales M, Tapia R, del Carmen Alarcón L, Sordo M, Ostrosky-Wegman P, Ortega A, López-Bayghen E. p53 response to arsenic exposure in epithelial cells: protein kinase B/Akt involvement. Toxicol Sci. 2007;99:126–40. doi: 10.1093/toxsci/kfm153. [DOI] [PubMed] [Google Scholar]
- Shi X, Mao Y, Knapton AD, Ding M, Rojanasakul Y, Gannett PM. Reaction of Cr(VI) with ascorbate and hydrogen peroxide generates hydroxyl radicals and causes DNA damage: role of a Cr(IV)-mediated Fenton-like reaction. Carcinogenesis. 1994;15:2475–2478. doi: 10.1093/carcin/15.11.2475. [DOI] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–34. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000;14:289–300. [PMC free article] [PubMed] [Google Scholar]
- Smeenk L, van Heeringen SJ, Koeppel M, van Driel MA, Bartels SJ, Akkers RC, Denissov S, Stunnenberg HG, Lohrum M. Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res. 2008;36:3639–54. doi: 10.1093/nar/gkn232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son YO, Hitron JA, Wang X, Chang Q, Pan J, Zhang Z, Liu J, Wang S, Lee JC, Shi X. Cr(VI) induces mitochondrial-mediated and caspase-dependent apoptosis through reactive oxygen species-mediated p53 activation in JB6 Cl41 cells. Toxicol Appl Pharmacol. 2010;245:226–35. doi: 10.1016/j.taap.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorahan T, Burges DC, Hamilton L, Harrington JM. Lung cancer mortality in nickel/chromium platers, 1946–1995. Occup Environ Med. 1998;55:236–242. doi: 10.1136/oem.55.4.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stueckle TA, Lu Y, Davis ME, Wang L, Jiang BH, Holaskova I, Schafer R, Barnett JB, Rojanasakul Y. Chronic occupational exposure to arsenic induces carcinogenic gene signaling networks and neoplastic transformation in human lung epithelial cells. Toxi Appl Pharm. 2012;261:204–216. doi: 10.1016/j.taap.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumoto M, Lee JY, Fujiwara Y, Uchiyama M, Satoh M. Inorganic arsenic induces apoptosis through downregulation of Ube2d genes and p53 accumulation in rat proximal tubular cells. J Toxicol Sci. 2013;38:815–20. doi: 10.2131/jts.38.815. [DOI] [PubMed] [Google Scholar]
- Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–283. doi: 10.1038/nrm2147. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Xu Y, Li Y, Pang Y, Ling M, Shen L, Jiang R, Zhao Y, Zhou J, Wang X, Liu Q. Blockade of p53 by HIF-2α, but not HIF-1α, is involved in arsenite-induced malignant transformation of human bronchial epithelial cells. Arch Toxicol. 2012;86:947–959. doi: 10.1007/s00204-012-0810-x. [DOI] [PubMed] [Google Scholar]