

Abstract
p53 and Notch-1 play important roles in breast cancer biology. Notch-1 inhibits p53 activity in cervical and breast cancer cells. Conversely, p53 inhibits Notch activity in T-cells but stimulates it in human keratinocytes. Notch co-activator MAML1 binds p53 and functions as a p53 co-activator. We studied the regulation of Notch signaling by p53 in MCF-7 cells and normal human mammary epithelial cells (HMEC). Results show that overexpression of p53 or activation of endogenous p53 with Nutlin-3 inhibits Notch-dependent transcriptional activity and Notch target expression in a dose-dependent manner. This effect could be partially rescued by transfection of MAML1 but not p300. Standard and quantitative co-immunoprecipitation experiments readily detected a complex containing p53 and Notch-1 in MCF-7 cells. Formation of this complex was inhibited by dominant negative MAML1 (DN-MAML1) and stimulated by wild-type MAML1. Standard and quantitative far-Western experiments showed a complex including p53, Notch-1 and MAML1. Chromatin immunoprecipitation (ChIP) experiments showed that p53 can associate with Notch-dependent HEY1 promoter and this association is inhibited by DN-MAML1 and stimulated by wild-type MAML1. Our data support a model in which p53 associates with the Notch transcriptional complex (NTC) in a MAML1-dependent fashion, most likely through a p53-MAML1 interaction. In our cellular models, the effect of this association is to inhibit Notch-dependent transcription. Our data suggest that p53-null breast cancers may lack this Notch-modulatory mechanism, and that therapeutic strategies that activate wild-type p53 can indirectly cause inhibition of Notch transcriptional activity.
Keywords: P53, Notch, MAML1, breast cancer
Introduction
The Notch signaling pathway participates in the development and progression of several malignancies, and is a potential therapeutic target (Espinoza and Miele, 2013a; Espinoza and Miele, 2013b; Espinoza et al., 2013; Pannuti et al., 2010). There is evidence that Notch plays a critical pro-oncogenic role in breast cancer and cancer stem cells (Hao et al., 2010; Harrison et al., 2010; Meurette et al., 2009; Rizzo et al., 2008; Stylianou et al., 2006). Notch-1 can transform human mammary cells in vitro and inhibits expression of p53-induced pro-apoptotic protein NOXA (Rizzo et al., 2008; Stylianou et al., 2006). High-level expression of Notch-1 and Notch ligand Jagged-1 are predictors of poor prognosis in breast cancer (Dickson et al., 2007). Enhanced Jagged-induced Notch signaling caused by loss of function of Lunatic Fringe (LFng) leads to mammary tumorigenesis (Xu et al., 2012). The p53 pathway is widely thought to play a key role in breast cancer biology (Agrawal et al., 2006; Borresen-Dale, 2003; Lacroix et al., 2006). Overall, inactivating mutations of the TP53 gene are found in 20–40% of breast cancers, and numerous studies suggest that such mutations are associated with poor prognosis and/or drug resistance (Borresen-Dale, 2003). Gene expression profiling revealed that inactivating mutations of p53 are far more common in the poor prognosis “basal-like” breast cancers (82%) and in ErbB2 overexpressing breast cancers (71%) compared to the more differentiated “luminal A” type breast cancers (17% of luminal A, 41% of luminal B). (Bertheau et al., 2013; Sorlie, 2004; Sorlie et al., 2001).
Notch proteins are transmembrane receptors that are activated upon interaction with ligands from the Delta or Jagged/Serrate families. Ligand binding triggers two successive cleavages: an extracellular cut by an ADAM metalloprotease followed by a γ-secretase-catalyzed transmembrane cut that releases the intracellular domain (NotchIC). NotchIC localizes to the nucleus where it displaces a corepressor complex associated with ubiquitous transcription factor CSL (Suppressor of Hairless in Drosophila, Lag-2 in C. elegans and CBF1/RBPJ-Jκ in mammals) and recruits a coactivator complex. The latter includes SKIP, MAML1 (Mastermind in Drosophila) and histone acetyltransferases PCAF, GCN5 or p300 (Artavanis-Tsakonas et al., 1999; Espinoza and Miele, 2013b; Kopan and Ilagan, 2009; Pannuti et al., 2010). The result is transcriptional activation of target genes. The ternary complex Notch-CSL-MAML1 complex functions as a basic platform for the recruitment of coactivators including p300 (Borggrefe and Oswald, 2009; Nam et al., 2007; Nam et al., 2006; Wilson and Kovall, 2006). However, relatively little is known on how the Notch transcriptional complex is directly regulated by other pathways in breast cancer cells.
Considerable evidence supports a context-dependent functional relationship between Notch and p53. In murine thymoma cell lines, p53 suppresses Notch activation by downregulating expression of presenilin-1 (Laws and Osborne, 2004). Ubiquitin ligase MDM2, which mediates constitutive degradation of p53, also induces degradation of Numb, an endocytic mediator that inhibits Notch signaling (Yogosawa et al., 2003). Notch has been reported to inhibit p53 activity in cervical cancer cells (Nair et al., 2003), in a transgenic model of Notch-1 induced lymphomagenesis (Beverly et al., 2005) and in colon carcinoma cells HCT116 (Kim et al., 2007). In the latter model, Kim et al. report that Notch-1IC interacts with p53, inhibiting its transcriptional activity. In cell lines of hepatoma, colon, prostate and breast origin, Notch-1 inhibits p53 via the mTOR pathway (Mungamuri et al., 2006). Suppression of Notch-1 increases p53 stability and induces p53-dependent apoptosis in K-ras-mutated lung adenocarcinoma cells (Licciulli et al., 2013). Conversely, in primary keratinocytes, where Notch-1 functions as a tumor suppressor, p53 induces Notch-1 expression, and Notch-1 inhibits ROCK1/2 and MRCKalpha kinases, two key effectors of small Rho GTPases (Lefort et al., 2007). However, in keratinocyte stem cells, the p53 homolog p63 has the opposite effect, inhibiting Notch activity (Okuyama et al., 2007). Zhao et al. reported that MAML1 can function as p53 coactivator independently of its Notch-related functions (Zhao et al., 2007). This raises the possibility that Notch and p53 may compete for limiting amounts of MAML1 under some conditions. In the context of mammary tumorigenesis, p53 has been reported to inhibit Notch4 via MDM2, through the formation of a ternary complex including p53, MDM2 and Notch4 (Sun et al., 2011). Here, we describe our study of transcriptional p53-Notch crosstalk in MCF-7 breast cancer cells and HMEC. Our data support the hypothesis that in these cells p53 modulates Notch-1 signaling at least in part by a novel mechanism involving MAML1-mediated association of p53 to the Notch transcriptional complex (NTC).
Material and Methods
Cell Culture
The MCF-7 cell lines [(MCF-7, MCF-7-p53i (p53−/−), MCF-7-Nci (p53+/+)] were generated by Drs. Vimla and Hamid Band (University of Nebraska). The cells were grown in DMEM supplemented with 10% FBS. MCF-7-p53−/− and p53+/+ were maintained in 100 μg/ml G418. The MCF-7-p53−/− cell line was generated by transfecting MCF-7 with p53 siRNA in the pSUPER vector and is referred to as p53siRNA in this manuscript. MCF-7-p53+/+ are control cells stably transfected with pSUPER-scrambled vector and are referred to as p53wt in this manuscript. HMEC were purchased from Lonza-Clonetics (Allendale, New Jersey) and cultured according to the manufacturer’s instructions.
Plasmids and Reagents
Dominant negative MAML1 (DN MAML1) in a migR1 vector was provided by Dr. John Aster, Harvard Medical School, and MAML1 cloned into pEF6-His vector was from Dr. James Griffin (Harvard Medical School). p53 siRNA was generated in Dr. Vimla Band’s laboratory, as was wt p53 cloned into pCMV. Notch-1 siRNA and pSUPER scrambled were from Dr. Debra Tonetti, University of Illinois at Chicago and CBF1-luciferase reporter construct (Ordentlich et al., 1998) from the late Dr. Thomas Kadesch (University of Pennsylvania). The plasmid coding for mouse full length Notch-1 tagged at the C-terminus with Renilla luciferase was a gift of Dr. Raphael Kopan’s lab (present address: Cincinnati Children’s Hospital Medical Center). Cdc2-Renilla Luciferase and GAPDH-Renilla luciferase were generated as follows: Plasmid PCS2+mN1FL6MT (Vooijs et al., 2004) was digested with restriction enzymes EcoRI and Mlu I. The plasmid backbone was gel-purified and used to ligate (in frame, upstream from the Renilla luciferase gene) PCR products coding for human Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or Cdc2 (from the first ATG to the last codon preceding the termination codon). Such PCR fragments were obtained using a lung cDNA library as the template and the following primers: GAPDH, 5′-GGGGAATTCATGGGGAAGGTGAAGGTC-3′ and 5′-GGGACGCGTCTCCTTGGAGGCCATGTG-3′; Cdc2, 5′-GGGGAATTCATGGAAGATTAT ACCAAA-3′ and 5′-GGGACGCGTCATCTTCTTAATCTGATT-3′ (underlined sequences represent initiation codons). Both recombinant plasmids pGAPDH-RL and pCDC2-RL were sequenced, and the expression of recombinant proteins was verified by Western blotting with Renilla antibodies. Renilla activity was assayed in transient transfection experiments followed by determination of Renilla luciferase activity. Nutlin-3 was purchased from EMD Millipore-Calbiochem (Billerica, MA).
Growth inhibition assays and isobologram
A 2×104 cells/ml cell suspension (200 μl) was seeded in 96 well plates. After 24 hours at 37°C, the medium was removed and fresh medium with indicated treatments was added to the plates. Plates were incubated for 2 days at 37°C, and 5% CO2. On the day of the assay, the medium in each well was discarded and 200 μl of a 0.2% crystal violet solution in 2% ethanol was added to each well and incubated at room temperature for 10 minutes. Plates were washed with deionized water and dried. Solvent solution (200 μl of 0.5% SDS in 50% ethanol) was added to each well and plates were incubated for one hour at room temperature to dissolve the crystals. Optical density at 562 nm was then measured using a microplate reader (Molecular Devices, CA). Isobolograms were constructed according to Chou and Talalay (Chou and Talalay, 1984). Briefly, the IC50 (inhibitory concentration at 50%) values of each drug alone are determined and plotted as axial points. A straight line connecting the two IC50s constitutes a line of additivity (dosages that produce growth inhibition in a simply additive combination). Dose-response curves of drug 1 in the presence of several concentrations of drug 2 are constructed. The IC50 values of drug 1 at each of the concentrations of drug 2 are plotted versus the concentration of drug 2. If the plot lies below the line of additivity, the effects of the drugs are synergistic.
Western Blot and co-immunoprecipitation
Cells were lysed in RIPA buffer (50mM Tris-Cl, pH 8.0, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1% Nonidet P-40) supplemented with fresh deoxycholic acid (50 μg/ml) and a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). The protein concentration of each lysate was determined using the BCA assay (Thermo-Pierce, Rockford, IL). Equal amounts of protein were loaded onto polyacrylamide gels (7% Tris-Acetate, NuPAGE, Thermo-Life Technologies, Rockford, IL). After electrophoresis, protein bands were transferred to PVDF membranes (Bio-Rad, Hercules, CA). Blots were probed with antibodies to Notch-1 (C-20), p53 (FL-393), Jagged-1 (C20), RBP-Jκ (CBF1, D-20), CDC2 (H297), p21 (C19) from Santa Cruz Biotechnologies (Dallas, TX), and Notch-1IC (Val1744, Cell Signaling, Danvers, MA). Antibodies to NOXA (OP180), MAML1 (AB5975), and Renilla Luciferase (MAB4410) were from EMD Millipore-Chemicon (Billerica, MA). Blocking and washes were performed in 2% blocking solution (Roche Diagnostics, Indianapolis, IN) in TBS. All blots were detected by chemiluminescence using BM Detection Solution (Roche Diagnostics, Indianapolis, IN). For co-immunoprecipitation, 500 μg of total cell lysate were pre-cleared with protein G Sepharose beads (GE Healthcare) for 1 hour at 4°C. Pre-cleared supernatants were incubated with 2 μg of primary antibodies or correspondent control IgG for 2 hours at 4°C. Antibody-protein complexes were then immunoprecipitated with protein G Sepharose beads for 1 hour with gentle rocking at 4°C. Beads-bound proteins were washed and separated by centrifugation. Immune complexes were eluted with Laemmli sample buffer (Bio-Rad) and resolved on 7% SDS gels and analyzed with Western blot analysis.
Luciferase Assays
Reporter assays were performed in 24-well plates using a Dual-Luciferase/Renilla Reporter Assay System (Promega, Madison, WI) as described by the manufacturer. pRL-TK (Renilla luciferase) was used as an internal control. Transient transfections were performed using Fugene 6.0 (Roche Diagnostics, Indianapolis, IN). A total of 0.2 μg of DNA was used throughout.
Quantitative Renilla immunoprecipitation
The quantitative Renilla immunoprecipitation assay was performed as previously described (Vooijs et al., 2004). MCF-7 cells were transfected with a total of 2.0 μg of DNA containing 1.0 μg of Renilla luciferase-tagged Notch-1 construct (Notch-1RL) and 1.0 μg of carrier DNA construct. Forty-eight hours after transfection, cells were washed and lysed in co-IP buffer (0.2M KCl, 25mM HEPES, pH 7.4, 1% Nonidet P-40, and 0.2 mM EGTA) supplemented with a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Cells lysates in 500 μl of co-IP buffer were immunoprecipitated with 2 μg of antibodies or correspondent control IgG for 2 hours at 4°C. The antibody was recovered after 1 hour of incubation with protein G Sepharose (GE Healthcare) at 4°C. Beads were washed and Renilla luciferase activity was assayed as described above using a kit from Promega (Madison, WI).
Extraction of nuclear proteins
Nuclear protein was extracted as previously described (Schreiber et al., 1989). Cell pellets were resuspended in 400 μl of ice-cold buffer 1 (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF). Lysates were pelleted and resuspended in 50 μl of ice-cold buffer 2 (20 mM HEPES, pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF).
Quantitative ChIP analysis
Quantitative ChIP analysis was carried out as described (Hao et al., 2010; Wu et al., 2005). Briefly, MCF-7 cells were crosslinked with 1% formaldehyde for 10 min at room temperature. Harvested cells were lysed in nucleus lysis buffer (1% SDS, 10 mM EDTA, 50mM Tris-HCl, pH 8.1). The lysate was diluted with IP dilution buffer (10 mM Tris-HCl, pH 8.1, 150 mM NaCl, 1 mM EDTA, 0.01% SDS, and 1% TritonX-100) and sonicated. Precleared chromatin was incubated with the indicated antibodies overnight. Immune complexes were collected by incubation with protein G Sepharose for 2 hours with gentle rocking at 4°C.
After phenol-chloroform extraction and ethanol precipitation, DNA was resuspended in 30 μl of TE. Aliquots (2 μl) were analyzed by quantitative real-time PCR with the indicated primer pairs. PCR products were quantified relative to a standard curve generated from a titration of input chromatin. Forward and reverse primers (5′ to 3′) designed to amplify the CBF1-binding site in the HEYL gene were as follows: 5′-AGCGTGGGAAAGGATGGT TG-3′ and 5′-CTCGCTTCATGCTGGCTCCC-3′. PCR was also performed with primers for the HBB (β-globin) gene (forward: 5′-AGTGCCAGAAGAGCCAAGGA-3′, reverse: 5′-CAGGGTGAGGTCTAAGTGATGACA-3′) as a negative control and for the RNA28S5 (28S rRNA) gene (forward: 5′-ATTAGTCAGCGGAGGAGAAGAAAC-3′, reverse: 5′-TCGCCGTTACTGAGGGAATC-3′) as an internal control.
Real Time qRT-PCR
Real-time qRT–PCR was performed as previously described (Hao et al., 2010). One μg of total RNA were reverse transcribed in a volume of 25 μl using 250 units of SuperScript III reverse transcriptase and 50 ng of random hexamers. Reaction conditions were as suggested by the manufacturer. Two μl of the cDNA mixture were used for real-time PCR experiments to measure the amount of HES1 and HEYL genes. Real-time PCR reactions were conducted on an Applied Biosystems 7500 Fast Real-Time PCR System using SYBR Green SuperMix with ROX kit (Quanta Biosciences, Gaithersburg, MD) according to the manufacturer’s protocol in a final volume of 25 μl. Primers concentration was 500 nM. The following primers were used: HES1: forward 5′-GCTGATAACAGCGGAATCCC-3′, reverse 5′-TGTGGCTACTTGGTGATCAGT-3′; HEYL: forward 5′-CGAGGCACCTTGGCCCTTCAAG-3′, reverse 5′-CTCCGCTGTTCTGCTGGGAACG-3′; Changes in gene expression were calculated by the 2−ΔΔCt formula using RPL13A as a reference gene.
Statistical Analysis
For pairwise comparisons, two-tailed unpaired Student’s t-tests were used with α=0.05. When more than two samples were compared, one-way analysis of variance was used with α=0.05. SPSS software (version 12.0 for Windows; SPSS Inc., Chicago, IL, USA) was used.
Results
p53 activation causes accumulation of Jagged-1 in MCF-7 cells but inhibits Notch transcriptional activity in a dose-dependent manner
We activated endogenous, wild-type p53 in MCF-7 cells by using an MDM2/HDM2 inhibitor, Nutlin-3 (Vassilev et al., 2004) and examined the expression of Jagged-1 and Notch-1 by Western blotting as compared to vehicle-treated cells. Nutlin-3 mediated p53 activation upregulated Jagged-1 and more modestly, the precursor and transmembrane forms of Notch-1 (Fig. 1A). In time-course experiments (Fig. 1B), Jagged-1 accumulation was observed after 4h, while Notch-1 accumulation was more modest and began after 6h, continuing up to 48h. It is unclear whether these are direct transcriptional effects of p53 or a response to cell stress subsequent to p53 accumulation. We examined the functional significance of this effect using CBF-1 luciferase reporter assays. Transfection of wild-type p53 into either MCF-7 cells or untransformed “normal” human mammary epithelial cells (HMEC) drastically inhibited CBF-1 reporter activity (Figure 1C). Nutlin-3 inhibited CBF-1 reporter activity in MCF-7 cells transfected with empty vector or Notch-1IC (Figure 1D). Transfection of wild-type p53 had identical effects (Figure 1E). Conversely, a stable MCF-7 cell line expressing p53 siRNA (Figure 2D insert (i)) had significantly higher CBF-1 transcriptional activity than the vector-transfected control line (Figure 1F). Importantly, transfection of wild-type p53 rescued the effect of p53 siRNA in a dose-dependent fashion (Supplemental Figure 1A, B). To determinate the effects of p53 on endogenous Notch-1 target genes, we measured the expression of HES1 and HEYL after p53 transfection. Real-time RT–PCR confirmed that transfection of p53 (≥ 0.5 μg DNA, causing detectable protein accumulation compared to control) inhibited the expression of both Notch target genes (Figure 1G, H). These experiments suggested that, despite upregulating expression of at least one Notch ligand (Jagged-1), p53 inhibits Notch-1 transcriptional activity in these cells when it reaches sufficient intracellular concentrations.
Figure 1. p53 increases Jagged-1 and Notch-1, in a time-dependent manner, but inhibits Notch activity.
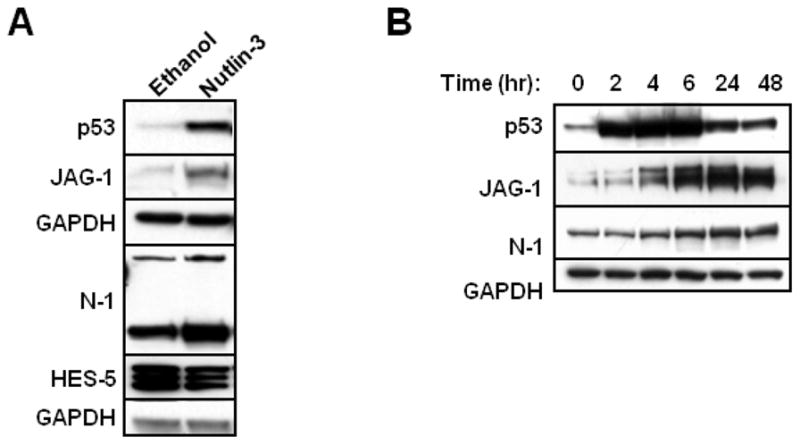


A, B: Western blots. MCF-7 cells were treated with Nutlin-3 (10 μM) for 24h in (A) and for 0–48h in (B). Whole cell lysates (20 μg) were immunoblotted for p53, Jagged-1, and Notch-1. GAPDH was used as the loading control.
C: MCF-7 or HMEC cells were transfected with empty pCMV vector or pCMV-p53 plasmid (100 ng) plus CBF-1 luciferase reporter and pRL-TK (Renilla luciferase) as an internal control and reporter activity was measured 48 h later; D: MCF-7 cells were transfected with empty vector (EC) or Notch-1IC (ICN) plus CBF-1 luciferase reporter and pRL-TK (Renilla luciferase) as an internal control. Twenty-four h after transfection, cells were treated with vehicle or 10 μM Nutlin-3 for 24 h and reporter activity was measured by the Dual-Luciferase/Renilla Reporter Assay System (Promega); E: experiments were performed as described above, but instead of treatment with vehicle or Nutlin-3, cells were transfected with empty pCMV or p53 48 h before luciferase activity was measured. Note that both Nutlin-3 and p53 expression suppress reporter activity in the presence or absence of Notch-1IC; F: MCF-7 cells stably expressing p53 siRNA (p53siRNA) have higher basal CBF-1 reporter activity than empty vector-transfected controls (p53wt). In all cases, ** indicates p<0.005; F insert (i): Western blot demonstrating p53 knockdown in p53siRNA cells. G: MCF-7 cells were transfected with the empty vector (EV) or different concentrations of p53 plasmid (0.15, 0.25, 0.5μg) and analyzed by Western blotting to confirm p53 expression; H: RNA was extracted from the cells shown in G, and RT-PCR was performed for Notch target genes HES1 and HEYL.
Figure 2. p53 inhibits Notch transcriptional activity in a dose-dependent fashion.

A: MCF-7 cells were treated with vehicle or with the indicated concentrations of Nutlin-3 for 48 h and CBF-1 reporter activity was measured as described in Figure 1. Parallel experiments conducted simultaneously were analyzed by Western blotting for p53, p21waf1/cip1, NOXA and GAPDH; B: MCF-7 cells were transfected with empty pCMV vector or with the indicated amounts of pCMV-p53. DNA amount was kept constant at 100 ng. CBF-1 reporter activity was measured after 48 h. Parallel experiments conducted simultaneously were analyzed by Western blotting for p53, p21waf1/cip1, NOXA and GAPDH. Data are representative of 3 independent experiments. *p<0.05, **p<0.005; C: dose-dependent inhibition of CBF-1 reporter activity by GSI MRK-003 in MCF-7 cells. **p<0.005; D, isobologram showing synergism between Nutlin-3 and MRK-003 in growth inhibition assays in MCF-7.
p53 inhibits Notch transcriptional activity in a dose-dependent manner
The effects of p53 on cell proliferation and apoptosis are well known to be dose-dependent (Riley et al., 2008). Thus, we examined the dose-dependence of p53-mediated Notch inhibition in MCF-7 cells. Both with Nutlin-3 treatment and p53 transfection, the strongest inhibition of CBF-1 reporter activity was seen at high levels of p53 (Figure 2A–B; upper panels). Western blot analysis showed that inhibition of CBF-1 by Nutlin3 or high concentrations of p53 is accompanied by induction of pro-apoptotic mediator NOXA, while cell cycle modulator p21waf1/cip1 was induced at lower concentrations (Figure 2A, B; lower panels). These results suggest that inhibition of Notch activity by p53 may be part of a pro-apoptotic signal. Since Notch-1IC inhibits induction of NOXA and apoptosis by p53 in MCF-7 cells (Stylianou et al., 2006), it is reasonable to postulate that execution of an apoptotic program by p53 in these cells may include inhibition of Notch signaling. Consistent with this model, a GSI (MRK-003) which prevents the cleavage and activation of Notch-1, exhibits dramatic synergism with Nutlin-3. In the presence of Nutlin-3, the IC50 of MRK-003 decreased by one order of magnitude (Figures 2C, D).
MAML1 but not p300 partially reverses Notch inhibition by p53
Based on the observation that Notch-CBF-1 coactivator Mastermind-like 1 (MAML1) also binds p53 and can function as a p53 coactivator (Zhao et al., 2007), we tested whether MAML1 participates in cross-talk between Notch and p53. Our initial hypothesis was that there may be competition between p53 and Notch-1 for limiting amounts of MAML1. Since there are previous reports of competition of p53 and Notch for p300 (Oswald et al., 2001) and MAML1 functions at least in part by recruiting p300 to the Notch transcriptional complex (NTC), we also determined whether p300 may relieve p53-mediated inhibition of Notch-1 transcriptional activity. In reporter experiments, co-transfection of a fixed amount of MAML1 with p53 partially rescued p53-mediated inhibition of CBF-1 luciferase activity. The addition of increasing amounts of p300 to a fixed amount of MAML1 did not further increase the rescue effect (Figure 3A). When both MAML1 and p300 were concurrently increased, they had dose-dependent and statistically significant rescue effects on p53-mediated CBF-1 inhibition (Figure 3B). Transfection of increasing amounts of MAML1 (5, 25 100 ng) in the presence of a fixed amount of p300 also had dose-dependent and statistically significant rescue effect on p53-mediated CBF-1 inhibition, even though baseline activity was not recovered (Figure 3C). These results suggest that MAML1 may be the rate-limiting factor in rescuing the inhibitory effect of p53. In control experiments, neither MAML1 alone nor p300 alone stimulated baseline CBF-1 transcriptional activity in amounts up to 100 ng. Even higher amounts (300 ng) suppressed transcriptional activity (Supplemental Figure 2A. B).
Figure 3. MAML1, but not p300 rescues p53 inhibition of Notch transcriptional activity.
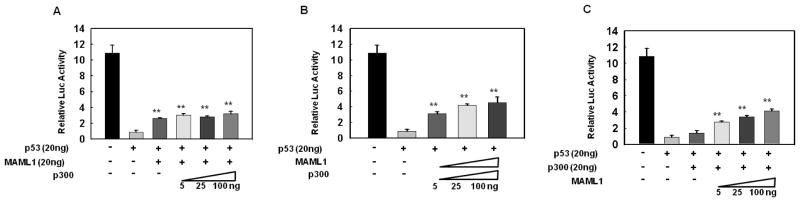
MCF-7 cells were transfected with CBF1-luc and a constant amount of p53 (20 ng) plus (A) a constant amount of p300 (20 ng) and increasing amounts of MAML1; (B), a constant amount of MAML1 (20 ng) and increasing amounts of p300; or (C), increasing amounts of both p300 and MAML1. **p<0.005
p53 co-immunoprecipitates with Notch-1 in MCF-7 cells in a MAML-1 dependent fashion
We then conducted a series of immunoprecipitation experiments to test the hypothesis that p53 and Notch compete for MAML1. Based on a report showing a direct interaction between Notch-1IC and p53 (Kim et al., 2007), we tested whether MAML1 may participate in this interaction. Consistent with the results of Kim et al. (Kim et al., 2007), our experiments revealed that p53 and Notch-1 can be readily co-immunoprecipitated from MCF-7 cells by either p53 or Notch-1 antibodies. This was observed with transfected p53 (Figure 4A) and endogenous p53 after Nutlin-3 treatment (Figure 5B). This putative complex was detectable in small amounts in untransfected cells with Notch-1 but not p53 immunoprecipitation, and was nearly undetectable in a MCF-7 cells stably expressing p53 siRNA (Figure 4A, B). To confirm the existence of a p53 and Notch-1-containing complex, we used a quantitative immunoprecipitation assay based on a full-length Notch-1 chimeric construct labeled at the C-terminus with Renilla luciferase (Vooijs et al., 2004). We first used this assay in nuclear lysates of MCF-7 cells transfected with p53 (Figure 4C–F). Results showed a specific interaction between Notch-1-Renilla and p53, as well as known Notch-1 binding partners CBF-1 and MAML1 that we used as positive controls (Figure 4C). Negative controls GAPDH-Renilla and Cdc2-Renilla did not show any specific interactions with p53, MAML1 or CBF-1 (Figure 4C–D). No specific interactions between p53 and Notch-1 could be detected by this assay in untransfected cells, or in MCF-7 cells stably expressing p53 siRNA, which we used as negative controls (Figure 4D–F). The vast majority of the Notch-1-Renilla specifically immunoprecipitable with p53 antibodies was in the nuclei, with cytoplasmic extracts showing approximately 10-fold lower values (data not shown). When cells were treated with Nutlin-3, a strong Notch-1-Renilla luciferase immunoprecipitation signal was observed that was abrogated in cells expressing p53 siRNA (Figure 4F). These data are consistent with the hypothesis that above a threshold of p53 accumulation, nuclear complexes containing p53 and Notch-1 are formed.
Figure 4. p53 and Notch can be co-immunoprecipitated.
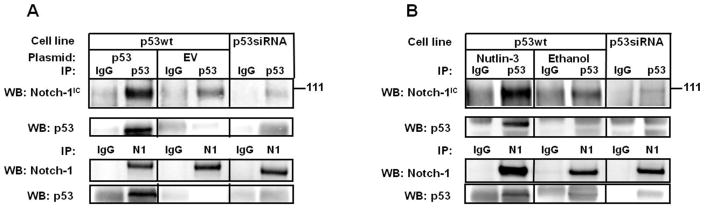

A: MCF-7 cells stably transfected with control pSUPER scrambled vector (wt) were transiently transfected with p53 or empty vector control. As an additional control, MCF-7 cells stably expressing p53 siRNA (see Figure 1) were used. Whole cell lysates were immunoprecipitated with p53 antibody or Notch-1 antibody and Western blot analysis was performed for Notch-1IC or p53 (upper panel: Notch-1IC (Cell signaling); lower panel: Notch (C20; Santa Cruz). B: Parental MCF-7 cells were treated antibody or Notch-1 antibody, and Western blot analysis was performed for Notch-1 or p53 respectively. C: Notch-1-Renilla luciferase is specifically immunoprecipitated by antibodies to p53, CBF-1 while negative controls GAPDH-RL and Cdc2-RL are not; D: Western blotting with Renilla luciferase antibodies confirming expression of Renilla-tagged proteins in these assays. “NT” indicates “Not Transfected”; E: MCF-7 cells were transfected with Notch-1-Renilla luciferase expression vector (1 μg) along with p53 or empty vector control. As an additional negative control, MCF-7 cells stably expressing p53 siRNA were processed in parallel. Nuclear extracts were immunoprecipitated with p53 antibody or control IgG, and Renilla luciferase activity analysis was performed in triplicate. Data are representative of 3 independent experiments; F: MCF-7 cells were transfected with Notch-Renilla luciferase expression vector (1 μg) and treated with Nutlin-3 (10 μM) or vehicle (ethanol). Nuclear extracts were immunoprecipitated with p53 antibody or control IgG, and Renilla luciferase activity analysis was performed in triplicate. Data are representative of three independent experiments.
Figure 5. Dominant negative MAML1 inhibits and wt-MAML1 stimulates formation of p53 and Notch-1-containing complexes.

(A–C): MCF-7 cells were transfected with Notch-RL vector (1μg) and p53 vector (100 ng). Quantitative immunoprecipitation was performed as described for Figure 6; A: Dominant negative MAML1 (DN MAML1) inhibits complex formation in a dose-dependent manner; B: wild type MAML1 (WT MAML1) increases complex formation in a dose-dependent manner; C: WT MAML1 rescues the inhibitory effect of DN MAML1 (100 ng). (*p<0.05, **p<0.005). Data are representative of three independent experiments; (D–F): MCF-7 cells were transfected with Notch-RL vector (1 μg) and treated with Nutlin-3 (10 μM). D: dominant negative MAML1 (DN MAML1) inhibits complex formation in a dose-dependent manner; E: wild type MAML1 (WT MAML1) increases complex formation in a dose-dependent manner; F: WT MAML1 rescues the inhibitory effect of DN MAML1 (10 ng). (*p<0.05, **p<0.005). In each panel, “C” represents “Control” and “EV” represents “Empty Vector”. Data are representative of three independent experiments.
Given the fact that Notch coactivator MAML1 interacts with p53, we asked whether the formation of the Notch-1/p53 complexes was affected by MAML1. Transfection of a dominant negative MAML1 (DN-MAML1), consisting of the N-terminal 70 amino acids linked to GFP (Maillard et al., 2004) together with p53, inhibited co-immunoprecipitation of Notch-1-Renilla with p53 in a dose-dependent fashion (Figure 5A). Conversely, transfection of wild-type MAML1 enhanced complex formation in a dose-dependent fashion (Figure 5B) and abrogated the inhibitory effect of DN-MAML1 (Figure 5C). Similarly, in Nutlin-3 stimulated cells (Figure 5D–F), the formation of complexes of endogenous p53 with Notch-1-Renilla was virtually abrogated by DN-MAML-1 in a dose-dependent fashion (Figure 5D), while wild-type MAML1 stimulated complex formation (Figure 5E) and rescued the inhibitory effect of DN-MAML1 (Figure 5F). These data suggested that the putative Notch-1 and p53-containing complex requires MAML1.
MAML1 stabilizes the p53/Notch-1 complex
Based on the observation that p53 can bind MAML1 (Zhao et al., 2007), while Notch-1 and CSL cooperatively bind the N-terminal helix of MAML1 (Nam et al., 2006; Wilson and Kovall, 2006), we hypothesized that p53 may bind to MAML1 in Notch-1/CSL transcriptional complexes (NTC). We first explored this hypothesis by double-immunoprecipitation using the Notch-1-Renilla construct (Figure 6). Results showed that when we co-transfected p53 and wild-type MAML1 into MCF-7 cells, either p53 antibodies or MAML1 antibodies specifically immunoprecipitated Notch-1-Renilla (Figure 6A). Beads from the primary immunoprecipitation were eluted and re-precipitated with the opposite antibody Figure 6B) (i.e., MAML1 immunoprecipitates were re-precipitated with p53 antibody and p53 immunoprecipitates were re-precipitated with MAML1 antibody). Figure 6B shows that Notch-1-Renilla could be specifically precipitated by MAML1 or p53 antibodies in p53 immunoprecipitates or in MAML1 immunoprecipitates, but not in IgG immunoprecipitates. When the same experiment was performed using Western blotting to detect endogenous Notch-1 as opposed to transfected Notch-1-Renilla, identical results were obtained. Moreover, we could verify by Western blotting that the complexes containing p53, MAML1 and Notch-1 also contained CBF-1 (Figure 6C). This is consistent with structural data indicating that MAML1 binds to a composite surface formed by the ankyrin region of Notch-1 and the β-trefoil domain of CBF-1 (Nam et al., 2006; Wilson and Kovall, 2006). Additional quantitative co-immunoprecipitation experiments using transient transfection of p53 or Nutlin-3 treatment (Figures 7A and B, respectively) confirmed that MAML1 stabilizes, and DN-MAML1 destabilizes the specific immunoprecipitation of Notch-1-Renilla with antibodies to p53, MAML1 and CBF-1. Taken together, these data indicate that p53 can bind to the NTC in a MAML1-dependent fashion, possibly by binding MAML1 at a site different from the one used by Notch-1.
Figure 6. p53-Notch complexes contain MAML1.

A: MCF-7 cells were transfected with Notch-RL plasmid. Cells were co-transfected with either empty vectors to balance DNA amounts (EV) or with p53 plus WT MAML1 (p53 + MAML1). Whole cell lysates were immunoprecipitated with IgG, p53 antibody or MAML1 antibody (1st IP). Renilla luciferase activity analysis was performed to detect Notch-1-RL; B: IP beads from each of the 1st IPs shown in the right panel (cells co-transfected with Notch-1RL, MAML1 and p53) were eluted and recovered proteins were re-immunoprecipitated (2nd IP) with either IgG, p53 or MAML1 antibodies. Renilla luciferase activity was measured again to detect Notch-1-RL. (**p<0.005). Data are representative of three independent experiments. C: An identical experiment was performed as shown in (A) and (B) without transfection of Notch-RL, and the second immunoprecipitates were analyzed by Western blotting for endogenous Notch-1.
Figure 7. MAML1 stabilizes p53–Notch-1 complexes.

A: MCF-7 cells were transfected with Notch-RL construct plus, as indicated at the bottom, p53 empty vector, p53 alone, WT MAML1 alone, p53 plus WT MAML1, p53 plus DN MAML1 and p53 plus both WT and DN MAML1. Nuclear extracts were immunoprecipitated with p53, MAML1, or CBF1 antibodies respectively. Renilla luciferase activity was measured. Data are representative of three independent experiments. B: MCF-7 cells were transfected with Notch-RL construct plus, as indicated at the bottom, cells were treated with vehicle (ethanol), Nutlin-3 (10 μM), transfected with WT MAML1, treated with Nutlin-3 and transfected with WT MAML1, treated with Nutlin-3 and transfected with DN MAML1 and treated with Nutlin-3 and transfected with both WT and DN MAML1. Nuclear extracts were immunoprecipitated with p53, MAML1, or CBF1 antibodies respectively. Renilla luciferase activity was measured. Data are representative of three independent experiments.
p53 binds to a Notch-responsive promoter in a MAML1-dependent fashion
The Notch transcriptional complex is physiologically found at many Notch-dependent promoters in genomic chromatin (Wang et al., 2011). To further test the hypothesis that p53 can bind to the Notch transcriptional complex, we used a quantitative ChIP-real time PCR approach. We examined the HEY1 promoter, which we have previously shown is occupied by Notch-1 under basal conditions in MCF-7 cells (Hao et al., 2010). Figure 8A shows that in cells transfected with p53, both p53 and Notch-1 were detectable above background at the HEY1 promoter. In cells transfected with MAML1, Notch-1 and MAML1 were detectable at the HEY1 promoter. In cells co-transfected with MAML1 and p53, detection of Notch-1, p53 and MAML1 at the HEY1 promoter was significantly enhanced. Conversely, transfection of DN-MAML1 abolished the specific detection of Notch-1, p53 or MAML1 by ChIP. This effect was completely reversed by co-transfection of wild-type MAML1 to compete with DN-MAML1. In untransfected cells, Notch-1 was detectable on the HEY1 promoter under basal conditions, but p53 and MAML1 were below the limits of detection (data not shown). Figure 8B shows that treatment with Nutlin-3 caused p53 to become detectably associated with the HEY1 promoter. Transfection of MAML1 in vehicle treated-cells caused Notch-1 and MAML1 to be strongly associated to the HEY1 promoter, while p53, as expected, was undetectable. Treatment with Nutlin-3 in MAML1 transfected cells caused strong association of p53 with the HEY1 promoter, in the co-presence of Notch-1 and MAML1. Conversely, transfection with DN-MAML1 drastically inhibited the association of p53 and MAML1 to the promoter induced by Nutlin-3. Finally, co-transfection of wild-type MAML1 completely reversed the effect of DN-MAML1 (Figure 8B). Taken together, these data support a model in which, when cellular levels of p53 reach a certain threshold, p53 associates with Notch transcriptional complexes in a MAML1-dependent fashion, inhibiting Notch-dependent transcription.
Figure 8. Quantitative ChIP analysis of Notch-1, p53 and MAML1 at the HEY1 promoter.

A: MCF-7 cells were transfected as indicated at the bottom with p53 vector, p53 alone, WT MAML1 alone, p53 plus WT MAML1, p53 plus DN MAML1 and p53 plus both WT and DN MAML1. Anti-Notch-1, anti-p53 and anti-MAML1 were used for ChIP and specific PCR products were quantified by real-time PCR. No complexes were found at the β-globin promoter analyzed as negative control from each of these IPs (data not shown). Data are representative of three independent experiments; B: MCF-7 cells were transfected with WT MAML1 or/and DN MAML1 and treated as indicated at the bottom with ethanol (V), Nutlin-3 (10 μM), ethanol plus transfection with MAML1, Nutlin-3 plus transfection with MAML1, Nutlin-3 plus transfection with DN MAML1 and Nutlin-3 plus transfection with both DN MAML1 and WT MAML1. Notch-1, p53 and MAML1 antibodies were used for ChIP and specific PCR products were quantified by real-time PCR. No complexes were found at the β-globin promoter analyzed as negative control from each of these IPs (data not shown). Data are representative of three independent experiments.
Discussion
Our data describe a novel mechanism whereby p53 inhibits Notch-1 signaling by directly associating with the NTC in a MAML1-dependent fashion. Our data do not indicate whether the association of p53 with the NTCs is mediated by direct association with MAML1 or through additional proteins. However, the observations of Zhao et al. suggest that MAML1 may interact directly with Notch-1 and p53 (Zhao et al., 2007). These authors mapped the interaction between p53 and MAML1 to the N-terminal region of MAML1 and the DNA-binding domain of p53 respectively. That interaction was stable in the absence of CBF-1 in GST-pulldown assays. The N-terminal region of MAML1 is also involved in the interaction with Notch-1 and CBF-1. However, in that case the interaction requires the presence of both Notch-1 and CBF-1, which form a composite binding surface for MAML1 (Nam et al., 2006; Wilson and Kovall, 2006). This surface binds the N-terminal 70 residues of MAML1, which form a kinked α-helix (Nam et al., 2006; Wilson and Kovall, 2006). The helix does not appear completely buried by Notch and MAML1, and thus may potentially provide a binding site for p53. Whether p53 binds exclusively to MAML1, or to a surface including regions of Notch-1 and CBF-1 cannot be determined from our data. Our observations do not rule out that additional protein(s) may be required for the formation of this complex. Additional mapping of the interaction surfaces will be required. An interesting question that requires further exploration is whether p53 inhibits the activity of only Notch-1 containing NTCs or of NTCs formed by Notch-2, -3 or -4. Another open question is whether p53 “loss of function” mutations found in tumors interfere with the ability of p53 to inhibit Notch.
The mechanism we have uncovered is likely to be one of multiple avenues of cross-talk between these two key cell fate regulatory pathways. Overall, our data suggest that in luminal A breast cancer cells, and possibly in mammary epithelial cells, as in T-cells, p53 functions as a negative regulator of Notch signaling. In both mammary epithelial cells and T-cells, Notch-1 is oncogenic and has transforming activity in vitro and in vivo. (see (Espinoza and Miele, 2013a; Espinoza and Miele, 2013b; Pannuti et al., 2010) for reviews). Conversely, in keratinocytes, where Notch-1 functions as a tumor suppressor, Notch-1 is a p53-target gene and mediates some of the effects of p53. This is not necessarily surprising, in light of the fact that both Notch and p53 are well known to have effects that are exquisitely dose- and context-dependent. Thus, it is not unreasonable that p53 may stimulate Notch activity by directly increasing Notch-1 levels in a cell type in which Notch-1 suppresses growth and promotes differentiation, while it inhibits Notch activity in other cell types in which Notch functions as an oncogene. While we did observe accumulation of Notch-1, and more prominently, Jagged-1, upon activation of p53 in MCF-7 cells with Nutlin-3, the net effect of p53 on Notch activity in our experimental system was inhibitory. This was not limited to MCF-7 cells, because inhibition of Notch activity by p53 was observed in untransformed HMEC as well. Whether this inhibitory effect is simply not operative in normal keratinocytes, or perhaps requires higher or lower amounts of HDM2-free p53 than can be attained in those cells under most circumstance remains to be established.
The mechanisms controlling the activity of the Notch pathway in the different subtypes of breast cancer remain incompletely understood. The Notch pathway is controlled with exquisite precision at multiple levels. (see (Espinoza and Miele, 2013a; Espinoza and Miele, 2013b; Pannuti et al., 2010) for reviews). Among these, an intricate cross-talk with p53 signaling is emerging. Loss of endocytic mediator Numb has been suggested to cause increased Notch activity in a fraction of breast cancers (Pece et al., 2004). Numb is also a substrate of MDM2. (Sczaniecka et al., 2012). Thus, Numb can compete with MDM2 for p53, stabilizing p53 levels. On the other hand, non-degradative ubiquitination of Notch-1 receptor by MDM2 has been suggested to activate Notch signaling (Pettersson et al., 2013). This may be a mechanism for the modest Notch-1 protein accumulation we observed upon p53 transduction (Figure 1). Our data indicate that the level of p53 is one of the mechanisms controlling Notch-1 activity in p53-wild-type breast cancer cells. Both inactivating p53 mutations and high levels of Notch-1 are associated with poor prognosis in breast cancer. Clinical studies are necessary to determine whether the presence of p53 inactivating mutations correlates with high levels of Notch targets in breast cancer.
Our observation may have significant clinical implications. Chemotherapy or radiation therapy in p53-wild-type cells cause activation of p53, which participates in apoptosis induction when cellular stress exceeds levels compatible with survival. Our observations imply that one of the effects of p53 activation is suppression of Notch signaling. This may contribute to the induction of apoptosis. Notch-1 has been shown to prevent p53-mediated induction of pro-apoptotic, BH3-only protein NOXA in MCF-7 breast cancer cells (Stylianou et al., 2006). Moreover, knockdown of Notch-1 or inhibition of γ-secretase in T47D (Luminal B, ERα+, p53 mutant) and MDA-MB231 (claudin-low, ERα−, p53 mutant) cells leads to upregulation of NOXA regardless of ER or p53 status (Rizzo et al., 2008). NOXA upregulation was observed in early stage ERα+ breast cancers in a pilot clinical trial of GSI MK-0752 plus endocrine therapy (Albain et al., 2010). Thus, suppression of NOXA expression may be a key mechanism whereby Notch-1 transmits a survival signal. It is reasonable to postulate that at least in some cells, suppression of Notch-1 activity may be part of the apoptotic program triggered by p53 (Figure 9). This mechanism may be defective in p53-mutant breast cancers. Whether Notch signaling contributes to the notorious resistance to apoptosis of p53-mutant breast cancers remains to be determined. However, our data suggest that p53-wild-type and p53-mutant breast cancer cells may respond differently to DNA damage or other chemotherapy-induced cell stress in terms of Notch activation. Further experiments will establish whether chemotherapeutic combinations including Notch inhibitors have different effects on breast cancer cells depending on their p53 status.
Figure 9. Working model of the interaction of p53 with the Notch Transcriptional Complex.

Under normal conditions, p53 induces MDM2 expression, limiting its own protein levels. A multitude of cellular stresses, including DNA damage, metabolic stress and other pro-apoptotic signals stabilize p53 and increase its intracellular levels, activating its transcriptional activity. MAML1 has been shown to function as a transcriptional co-activator for p53 as well as intracellular Notch1 (NIC). At low levels, p53is known to induces gene that cause cell cycle arrest, allowing DNA repair and/or recovery from cell stress. At higher levels, p53 induces a group of pro-apoptotic genes that trigger programmed cell death. We hypothesize that under these conditions, p53 interacts with the Notch transcriptional complex (NTC) through MAML1 (and/or possibly other MAML isoforms). This prevents the recruitment of p300 and other coactivators to the NTC, decreasing Notch1 transcriptional activity. This prevents NIC from triggering anti-apoptotic, pro-survival pathways (e.g., suppression of NOXA expression, induction of NF-κB etc.), thereby contributing to the pro-apoptotic function of p53. This model predicts that MDM2 inhibitors by inducing accumulation of p53 would synergize with Notch inhibitors, as we have observed. Abbreviations: CoA, co-activator; NIC, Notch1 intracellular fragment; CSL, CBF-1, Suppressor of Hairless, LAG1; MDM-2Is, MDM2 inhibitors.
Supplementary Material
Acknowledgments
This work was supported in part by grant PO1CA166009 (L.M, B.A.O, T.E.G). Drs. Yun, Kim and Park were supported in part by a grant (KBS4151022) of the Korean Health Technology R&D Project, Ministry for Health, Welfare & Family Affairs, Republic of Korea.
Abbreviations
- ADAM
A Disintegrin and metalloproteinase domain-containing protein
- CSL
CBF-1, Suppressor of Hairless/LAG1DN-MAML1
- ErbB2
V-Erb-B2 Avian Erythroblastic Leukemia Viral Oncogene Homolog 2
- GST
Glutathione S-transferase
- HMEC
Human Mammary Epithelial Cells
- HES1
Hairy/Enhancer of Split family1
- HEY1
Hairy/enhancer-of-split related with YRPW motif protein 1
- HEYL
Hairy/enhancer-of-split related with YRPW motif-like protein
- MAML1
Mastermind-like protein 1
- MDM2
Mouse double minute 2 homolog
- Notch IC
Notch Intracellular Domain
- NTC
Notch Transcriptional Complex
- PCAF
P300/CBP-associated factor
- SKIP
Ski-interacting protein
Footnotes
This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: [10.1002/jcp.25052]
The authors have no conflicts of interest pertaining to the contents of this manuscript
Additional Supporting Information may be found in the online version of this article.
References
- Agrawal A, Yang J, Murphy RF, Agrawal DK. Regulation of the p14ARF-Mdm2-p53 pathway: an overview in breast cancer. Exp Mol Pathol. 2006;81(2):115–122. doi: 10.1016/j.yexmp.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Albain K, Czerlanis C, Rajan P, Zlobin A, Godellas C, Bova D, Lo SS, Robinson P, Sarker S, Gaynor ER, Cooper R, Aranha G, Czaplicki K, Busby B, Rizzo P, Chisamore M, Demuth T, Blackman S, Waters J, Stiff P, Fuqua SA, Miele L. Combination of Notch inhibitor MK-0752 and endocrine therapy for early stage ERα+ breast cancer in a pre-surgical window study. Cancer Res. 2010;70(24 Suppl):113s–114s. [Google Scholar]
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284(5415):770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- Bertheau P, Lehmann-Che J, Varna M, Dumay A, Poirot B, Porcher R, Turpin E, Plassa LF, de Roquancourt A, Bourstyn E, de Cremoux P, Janin A, Giacchetti S, Espie M, de The H. p53 in breast cancer subtypes and new insights into response to chemotherapy. Breast. 2013;22(Suppl 2):S27–29. doi: 10.1016/j.breast.2013.07.005. [DOI] [PubMed] [Google Scholar]
- Beverly LJ, Felsher DW, Capobianco AJ. Suppression of p53 by Notch in lymphomagenesis: implications for initiation and regression. Cancer Research. 2005;65(16):7159–7168. doi: 10.1158/0008-5472.CAN-05-1664. [DOI] [PubMed] [Google Scholar]
- Borggrefe T, Oswald F. The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci. 2009;66(10):1631–1646. doi: 10.1007/s00018-009-8668-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borresen-Dale AL. TP53 and breast cancer. Hum Mutat. 2003;21(3):292–300. doi: 10.1002/humu.10174. [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Dickson BC, Mulligan AM, Zhang H, Lockwood G, O’Malley FP, Egan SE, Reedijk M. High-level JAG1 mRNA and protein predict poor outcome in breast cancer. Mod Pathol. 2007;20(6):685–693. doi: 10.1038/modpathol.3800785. [DOI] [PubMed] [Google Scholar]
- Espinoza I, Miele L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer letters. 2013a;341(1):41–45. doi: 10.1016/j.canlet.2013.08.027. [DOI] [PubMed] [Google Scholar]
- Espinoza I, Miele L. Notch inhibitors for cancer treatment. Pharmacol Ther. 2013b;(13):10. doi: 10.1016/j.pharmthera.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza I, Pochampally R, Xing F, Watabe K, Miele L. Notch signaling: targeting cancer stem cells and epithelial-to-mesenchymal transition. Onco Targets and therapy. 2013;6:1249–1259. doi: 10.2147/OTT.S36162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao L, Rizzo P, Osipo C, Pannuti A, Wyatt D, Cheung LW, Sonenshein G, Osborne BA, Miele L. Notch-1 activates estrogen receptor-alpha-dependent transcription via IKKalpha in breast cancer cells. Oncogene. 2010;29(2):201–213. doi: 10.1038/onc.2009.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, Bundred NJ, Clarke RB. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70(2):709–718. doi: 10.1158/0008-5472.CAN-09-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SB, Chae GW, Lee J, Park J, Tak H, Chung JH, Park TG, Ahn JK, Joe CO. Activated Notch1 interacts with p53 to inhibit its phosphorylation and transactivation. Cell Death Differ. 2007;14(5):982–991. doi: 10.1038/sj.cdd.4402083. [DOI] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix M, Toillon RA, Leclercq G. p53 and breast cancer, an update. Endocr Relat Cancer. 2006;13(2):293–325. doi: 10.1677/erc.1.01172. [DOI] [PubMed] [Google Scholar]
- Laws AM, Osborne BA. p53 regulates thymic Notch1 activation. European Journal of Immunology. 2004;34(3):726–734. doi: 10.1002/eji.200324772. [DOI] [PubMed] [Google Scholar]
- Lefort K, Mandinova A, Ostano P, Kolev V, Calpini V, Kolfschoten I, Devgan V, Lieb J, Raffoul W, Hohl D, Neel V, Garlick J, Chiorino G, Dotto GP. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 2007;21(5):562–577. doi: 10.1101/gad.1484707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licciulli S, Avila JL, Hanlon L, Troutman S, Cesaroni M, Kota S, Keith B, Simon MC, Pure E, Radtke F, Capobianco AJ, Kissil JL. Notch1 is required for Kras-induced lung adenocarcinoma and controls tumor cell survival via p53. Cancer Res. 2013;73(19):5974–5984. doi: 10.1158/0008-5472.CAN-13-1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillard I, Weng AP, Carpenter AC, Rodriguez CG, Sai H, Xu L, Allman D, Aster JC, Pear WS. Mastermind critically regulates Notch-mediated lymphoid cell fate decisions. Blood. 2004;104(6):1696–1702. doi: 10.1182/blood-2004-02-0514. [DOI] [PubMed] [Google Scholar]
- Meurette O, Stylianou S, Rock R, Collu GM, Gilmore AP, Brennan K. Notch activation induces Akt signaling via an autocrine loop to prevent apoptosis in breast epithelial cells. Cancer Research. 2009;69(12):5015–5022. doi: 10.1158/0008-5472.CAN-08-3478. [DOI] [PubMed] [Google Scholar]
- Mungamuri SK, Yang X, Thor AD, Somasundaram K. Survival signaling by Notch1: mammalian target of rapamycin (mTOR)-dependent inhibition of p53. Cancer Res. 2006;66(9):4715–4724. doi: 10.1158/0008-5472.CAN-05-3830. [DOI] [PubMed] [Google Scholar]
- Nair P, Somasundaram K, Krishna S. Activated Notch1 inhibits p53-induced apoptosis and sustains transformation by human papillomavirus type 16 E6 and E7 oncogenes through a PI3K-PKB/Akt-dependent pathway. J Virol. 2003;77(12):7106–7112. doi: 10.1128/JVI.77.12.7106-7112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam Y, Sliz P, Pear WS, Aster JC, Blacklow SC. Cooperative assembly of higher-order Notch complexes functions as a switch to induce transcription. Proc Natl Acad Sci USA. 2007;104(7):2103–2108. doi: 10.1073/pnas.0611092104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam Y, Sliz P, Song L, Aster JC, Blacklow SC. Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell. 2006;124(5):973–983. doi: 10.1016/j.cell.2005.12.037. [DOI] [PubMed] [Google Scholar]
- Okuyama R, Ogawa E, Nagoshi H, Yabuki M, Kurihara A, Terui T, Aiba S, Obinata M, Tagami H, Ikawa S. p53 homologue, p51/p63, maintains the immaturity of keratinocyte stem cells by inhibiting Notch1 activity. Oncogene. 2007 doi: 10.1038/sj.onc.1210235. [DOI] [PubMed]
- Ordentlich P, Lin A, Shen CP, Blaumueller C, Matsuno K, Artavanis-Tsakonas S, Kadesch T. Notch inhibition of E47 supports the existence of a novel signaling pathway. Mol Cell Biol. 1998;18(4):2230–2239. doi: 10.1128/mcb.18.4.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald F, Tauber B, Dobner T, Bourteele S, Kostezka U, Adler G, Liptay S, Schmid RM. p300 acts as a transcriptional coactivator for mammalian Notch-1. Mol Cell Biol. 2001;21(22):7761–7774. doi: 10.1128/MCB.21.22.7761-7774.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannuti A, Foreman KE, Rizzo P, Osipo C, Golde TE, Osborne BA, Miele L. Targeting Cancer Stem Cells through Notch Signaling. Clin Cancer Res. 2010;16 doi: 10.1158/1078-0432.CCR-09-2823. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pece S, Serresi M, Santolini E, Capra M, Hulleman E, Galimberti V, Zurrida S, Maisonneuve P, Viale G, Di Fiore PP. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol. 2004;167(2):215–221. doi: 10.1083/jcb.200406140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson S, Sczaniecka M, McLaren L, Russell F, Gladstone K, Hupp T, Wallace M. Non-degradative ubiquitination of the Notch1 receptor by the E3 ligase MDM2 activates the Notch signalling pathway. Biochem J. 2013;450(3):523–536. doi: 10.1042/BJ20121249. [DOI] [PubMed] [Google Scholar]
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9(5):402–412. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- Rizzo P, Miao H, D’Souza G, Osipo C, Song LL, Yun J, Zhao H, Mascarenhas J, Wyatt D, Antico G, Hao L, Yao K, Rajan P, Hicks C, Siziopikou K, Selvaggi S, Bashir A, Bhandari D, Marchese A, Lendahl U, Qin JZ, Tonetti DA, Albain K, Nickoloff BJ, Miele L. Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. 2008;68(13):5226–5235. doi: 10.1158/0008-5472.CAN-07-5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 1989;17(15):6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sczaniecka M, Gladstone K, Pettersson S, McLaren L, Huart AS, Wallace M. MDM2 protein-mediated ubiquitination of numb protein: identification of a second physiological substrate of MDM2 that employs a dual-site docking mechanism. The Journal of biological chemistry. 2012;287(17):14052–14068. doi: 10.1074/jbc.M111.303875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T. Molecular portraits of breast cancer: tumour subtypes as distinct disease entities. Eur J Cancer. 2004;40(18):2667–2675. doi: 10.1016/j.ejca.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de RM, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein LP, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98(19):10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stylianou S, Clarke RB, Brennan K. Aberrant activation of notch signaling in human breast cancer. Cancer Research. 2006;66(3):1517–1525. doi: 10.1158/0008-5472.CAN-05-3054. [DOI] [PubMed] [Google Scholar]
- Sun Y, Klauzinska M, Lake RJ, Lee JM, Santopietro S, Raafat A, Salomon D, Callahan R, Artavanis-Tsakonas S. Trp53 regulates Notch 4 signaling through Mdm2. Journal of cell science. 2011;124(Pt 7):1067–1076. doi: 10.1242/jcs.068965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Vooijs M, Schroeter EH, Pan Y, Blandford M, Kopan R. Ectodomain shedding and intramembrane cleavage of mammalian Notch proteins is not regulated through oligomerization. J Biol Chem. 2004;279(49):50864–50873. doi: 10.1074/jbc.M409430200. [DOI] [PubMed] [Google Scholar]
- Wang H, Zou J, Zhao B, Johannsen E, Ashworth T, Wong H, Pear WS, Schug J, Blacklow SC, Arnett KL, Bernstein BE, Kieff E, Aster JC. Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc Natl Acad Sci USA. 2011;108(36):14908–14913. doi: 10.1073/pnas.1109023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JJ, Kovall RA. Crystal structure of the CSL-Notch-Mastermind ternary complex bound to DNA. Cell. 2006;124(5):985–996. doi: 10.1016/j.cell.2006.01.035. [DOI] [PubMed] [Google Scholar]
- Wu J, Iwata F, Grass JA, Osborne CS, Elnitski L, Fraser P, Ohneda O, Yamamoto M, Bresnick EH. Molecular determinants of NOTCH4 transcription in vascular endothelium. Mol Cell Biol. 2005;25(4):1458–1474. doi: 10.1128/MCB.25.4.1458-1474.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Usary J, Kousis PC, Prat A, Wang DY, Adams JR, Wang W, Loch AJ, Deng T, Zhao W, Cardiff RD, Yoon K, Gaiano N, Ling V, Beyene J, Zacksenhaus E, Gridley T, Leong WL, Guidos CJ, Perou CM, Egan SE. Lunatic fringe deficiency cooperates with the Met/Caveolin gene amplicon to induce basal-like breast cancer. Cancer Cell. 2012;21(5):626–641. doi: 10.1016/j.ccr.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogosawa S, Miyauchi Y, Honda R, Tanaka H, Yasuda H. Mammalian Numb is a target protein of Mdm2, ubiquitin ligase. Biochem Biophys Res Commun. 2003;302(4):869–872. doi: 10.1016/s0006-291x(03)00282-1. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Katzman RB, Delmolino LM, Bhat I, Zhang Y, Gurumurthy CB, Germaniuk-Kurowska A, Reddi HV, Solomon A, Zeng MS, Kung A, Ma H, Gao Q, Dimri G, Stanculescu A, Miele L, Wu L, Griffin JD, Wazer DE, Band H, Band V. The notch regulator MAML1 interacts with p53 and functions as a coactivator. J Biol Chem. 2007;282(16):11969–11981. doi: 10.1074/jbc.M608974200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.