

Abstract
In equids, susceptibility to disease caused by Rhodococcus equi occurs almost exclusively in foals. This distribution might be attributable to the age-dependent maturation of immunity following birth undergone by mammalian neonates that renders them especially susceptible to infectious diseases. Expansion and diversification of the neonatal microbiome contribute to development of immunity in the gut. Moreover, diminished diversity of the gastrointestinal microbiome has been associated with risk of infections and immune dysregulation. We thus hypothesized that varying composition or reduced diversity of the intestinal microbiome of neonatal foals would contribute to increased susceptibility of their developing R. equi pneumonia. The composition and diversity indices of the fecal microbiota at 3 and 5 weeks of age were compared among 3 groups of foals: 1) foals that subsequently developed R. equi pneumonia after sampling; 2) foals that subsequently developed ultrasonographic evidence of pulmonary abscess formation or consolidation but not clinical signs (subclinical group); and, 3) foals that developed neither clinical signs nor ultrasonographic evidence of pulmonary abscess formation or consolidation. No significant differences were found among groups at either sampling time, indicating absence of evidence of an influence of composition or diversity of the fecal microbiome, or predicted fecal metagenome, on susceptibility to subsequent R. equi pneumonia. A marked and significant difference identified between a relatively short interval of time appeared to reflect ongoing adaptation to transition from a milk diet to a diet including available forage (including hay) and access to concentrate fed to the mare.
Introduction
Pneumonia is a major cause of disease and death in foals, and Rhodococcus equi is considered the most important cause of pneumonia among foals between approximately 1 and 6 months of age [1–3]. Susceptibility to R. equi pneumonia in horses appears to be age-related: the disease occurs almost exclusively among foals, whereas adult horses or other species of animals generally do not develop signs of infection unless they are immunocompromised [1–3]. One explanation for the age-restricted distribution of this disease is that mammalian neonates undergo an age-dependent maturation of their immune system following birth that renders newborns particularly vulnerable to infection [4–9]. Interaction with environmentally-derived microbes is considered to be a driving force for neonatal immune development [6, 7]. The intestinal microbiome of neonates is known to develop with age in various species, including foals [10–13]. In other species, expansion and diversification of the neonatal microbiome have been linked to development of intestinal function, including gut immunity [14, 15]. Furthermore, numerous studies have demonstrated that diminished diversity of the gastrointestinal microbiome leads to increased risk and severity of gastrointestinal diseases, systemic infections, and immune dysregulation [16]. Thus, we hypothesized that varying composition or reduced diversity of the neonatal foal intestinal microbiome would contribute to susceptibility of developing clinical signs of R. equi pneumonia. To test this hypothesis, we compared the composition and diversity indices of fecal samples collected at approximately 3 weeks and 5 weeks of age from 3 groups of foals: 1) foals that subsequently developed R. equi pneumonia after sampling; 2) foals that subsequently developed ultrasonographic evidence of pulmonary abscess formation or consolidation but not clinical signs; and, 3) foals that developed neither clinical signs nor ultrasonographic evidence of pulmonary abscess formation or consolidation.
Temporal changes in the foal microbiome remain ill-defined. To our knowledge, there are only 2 reports characterizing the microbiome of foals during the first month of life [13, 17]. While the fecal microbiome of foals changes rapidly and greatly during the first month of life [13, 17], current knowledge indicates that the fecal microbiome is relatively stable by 1 month of age. Thus, a second objective of our study was to better characterize the stability of the fecal microbiome in foals using sequential fecal samples collected over a 2-week interval spanning approximately 1 month of age.
Materials and Methods
Ethics statement
All protocols for this study were reviewed and approved by the Clinical Research Review Committee (CRRC Protocol 10–12) of the College of Veterinary Medicine & Biomedical Sciences, Texas A&M University. At the time this study was conducted, research involving client-owned animals at Texas A&M University was not subject to review by the Institutional Animal Care and Use Committee. All animals were monitored a minimum of twice daily by both veterinary staff and ranch personnel. All animals showing signs of pneumonia were examined by veterinary staff and treated as deemed appropriate by attending veterinarians, including the use of flunixin meglumine for analgesia. This study was conducted on private land (33°37′14″N 100°19′22″W) and specific permissions for use were granted by GPB. Written informed consent for participation was obtained for all foals included in the study, and the 6666 Ranch provided access to the foals included in this project. This study did not involve any endangered or protected species.
Study population
The study was conducted at the 6666 Ranch because it agreed to provide access to foals, had history of R. equi pneumonia among foals with a cumulative incidence of ≥ 15% for the preceding 3 years, and because the farm’s veterinarian and general manager (GPB) had participated in a separate study during 2011 evaluating screening tests for R. equi pneumonia in foals; fecal samples for the study reported here were collected from the foals participating in the screening study [18]. For the study evaluating performance of screening tests, treatment for pneumonia was not initiated for any foal on the basis of screening test results alone, and the veterinarians making decisions about diagnosis and treatment of R. equi pneumonia were not informed of the results of screening tests. Bilateral thoracic ultrasonographic examination was performed on each foal at the farm at 2-week intervals, beginning at 3 weeks of age until either 19 weeks of age or until the foal developed clinical signs of pneumonia (as described below). The veterinarian performing the ultrasonographic examinations did not participate in either diagnosis or treatment of R. equi pneumonia. Ultrasonographic parameters recorded included: 1) the anatomic location (left versus right hemithorax; intercostal space; and, dorsal, middle, or ventral region); 2) maximal diameter of any areas of pulmonary abscesses or consolidation; and, 3) the total number of discrete lesions observed.
At the time of each sequential ultrasonographic examination, fecal samples were collected from foals, either by collecting freshly voided feces or by digital retrieval. When fresh fecal samples were collected, effort was made to obtain the central portion of the fecal matter (to limit environmental contamination). Fecal samples were double-bagged in sterile plastic sacks, frozen at -20°C for no more than 1 week, then shipped frozen and on ice to the Equine Infectious Disease Laboratory at Texas A&M University. Upon arrival, samples were evaluated for identifiers and date of collection (the 6666 Ranch was contacted to resolve any data that were missing or unclear), labelled, and stored in a freezer at -80°C. Data from samples (mare and foal identifiers, date collected, date received, weight of sample, and freezer location) were logged into a computerized spreadsheet for data management.
All foals [N = 270] born during 2011 at the ranch were eligible to be included in the study. Farm personnel monitored the foals daily for clinical signs of pneumonia until 20 weeks of age. Clinical signs indicative of pneumonia included fever, lethargy, signs of depressed attitude, cough, nasal discharge, polysynovitis, tachypnea, increased respiratory effort, respiratory distress, and detection of a tracheal rattle or pulmonary crackles or wheezes during thoracic auscultation. Thoracic ultrasonography and collection of a trans-endoscopic tracheobronchial aspirate (TBA) sample with a commercially available triple-guarded catheter (Triple stage tracheal wash catheter, MILA International Inc., Erlanger, KY) were performed for each foal that developed clinical signs of pneumonia. The endoscope was disinfected between procedures with a 3.4% glutaraldehyde solution (CIDEX-PLUS, Advanced Sterilization Products, Irvine, CA), following a standard protocol used in our laboratory and known to be microbiocidal againstR. equi. Each sample of TBA fluid was submitted for microbiologic culture and cytologic evaluation to the Texas Veterinary Medical Diagnostic Laboratory in College Station, Texas (culture) and Veterinary Clinical Pathology Laboratory, College of Veterinary Medicine and Biomedical Sciences, in College Station, Texas (cytology).
Foals with signs of pneumonia at 3 to 20 weeks of age, ultrasonographic evidence of peripheral pulmonary consolidation or abscesses at the time of examination for clinical signs of pneumonia, and R. equi isolated from TBA fluid via microbiologic culture, and cytological evidence of gram-positive intracellular coccobacilli in the TBA sample were defined as having R. equi pneumonia (clinical group; N = 43 [17%]). Foals having ultrasonographic evidence of peripheral pulmonary consolidation or abscesses, but lacking clinical signs of pneumonia were defined as being subclinical pneumonia foals (subclinical group; N = 156 [63%]) [19]. Foals having no clinical signs of pneumonia and no ultrasonographic evidence of pulmonary consolidation or abscessation were considered unaffected foals (healthy group; N = 49 [20%]). For purposes of this study, 25 clinical group foals were randomly selected from the 43 foals in the clinical group. These 25 foals were birthdate-matched both to 25 foals from the subclinical group and to 25 foals from the healthy group. Fecal samples from 2 time-points for each foal were selected for sequencing. The first sample for all foals (time 1) was the first fecal sample collected (3 weeks of age), and the second fecal sample was collected at 5 weeks of age. For foals that developed R. equi pneumonia, the second sample always preceded the onset of clinical signs of disease (i.e., before any treatment was instituted and before inappetance or other clinical signs of disease were observed).
DNA extraction
The fecal sample was removed from storage at -80°C and 200 mg of feces was harvested from the frozen sample. Genomic DNA was extracted from using a commercially available fecal DNA extraction kit (QIAamp Fast DNA Stool Mini Kit, Qiagen) according to manufacturer’s protocol with slight modifications. Briefly, 200 mg of frozen feces was placed in a 2-ml tube that contained 1 ml Inhibitex buffer and 50 mg each of sterile/DNAase free 0.1- and 0.5-mm silica zirconium beads. This was then homogenized for 90 seconds at 6.5 m/sec with FastPrep FP120 cell disrupter (Qbiogene, Carlsbad,CA). The sample was then heated at 70°C for 10 minutes prior to following the manufacturer’s protocol for DNA extraction. DNA was suspended in tris-EDTA buffer (Integrated DNA Technologies, Coralville, Iowa) and stored at -80°C.
Sequencing of 16S rRNA gene
Amplification and sequencing of the V4 variable region 16S rRNA gene was performed at MR DNA (www.mrdnalab.com, Shallowater, TX, USA) [20]. Briefly samples were barcoded and PCR primers 515F/806R were used in a 28 cycle PCR using the HotStarTaq Plus Master Mix Kit (Qiagen, USA) under the following conditions: 94°C for 3 minutes, followed by 28 cycles of 94°C for 30 seconds, 53°C for 40 seconds and 72°C for 1 minute, with a final elongation step at 72°C for 5 minutes. A DNA library was prepared according to Illumina TruSeq DNA library preparation protocol. Sequencing was performed on a MiSeq (Illumina) following the manufacturer’s guidelines. Sequence data was uploaded into NCBI GenBank database submission number SUB995994.
Analysis of sequencing data
The software Quantitative Insights Into Microbial Ecology (QIIME v1.9 (http://qiime.sourceforge.net) was used for data processing and analysis [21]. The raw sequence data were de-multiplexed, and low quality reads were filtered using database’s default parameters. Chimeric sequences were detected using Uchime and removed prior to further analysis [22]. Sequences were then assigned to operational taxonomic units (OTUs) using an open-reference OTU picking protocol in QIIME [23] against the Greengenes database filtered at 97% identity [24, 25]. To adjust for uneven sequencing depth among the samples, each sample was rarefied to an even sequencing depth per sample prior to further analysis.
Alpha rarefaction, beta diversity measures, richness, taxonomic summaries, and tests for significance were calculated and plotted using QIIME. Diversity indices were compared using a generalized linear model with a given index as the outcome variable and the 3 health groups as independent variables, with post hoc testing for pairwise differences among groups using the method of Sidak. The same approach was used to compare differences between times. Differences in microbial communities among the groups were investigated by visual assessment of clustering on principal coordinates analysis (PCoA) plots, and by analysis of similarity (ANOSIM) [26] calculated on unweighted and weighted UniFrac distance metrics [27] using available QIIME scripts. When ANOSIM identified significant differences among groups, then similarity percentage (SIMPER) [26] was used with Bray Curtis dissimilarity metric [28] to examine which features contributed to the differences among groups. SIMPER was performed with PAST v3.05 [29]. Differences in the proportions of bacteria making up the composition of the microbiota in different groups was also determined by the Mann-Whitney U test when only 2 comparisons were made (i.e., time differences) and the Kruskal-Wallis test when multiple comparisons were made (e.g., differences between the 3 phenotypes of disease). For all analyses, significance was set at P ≤ 0.05 after correction using the Benjamini-Hochberg FDR procedure. The groups compared were: 1) healthy, subclinical, and clinical foals at time 1 (age 3 weeks); 2) healthy, subclinical, and clinical foals at time 2 (age 5 weeks); and, 3) all foals at time 1 compared with all foals at time 2, regardless of health status.
The software Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) was used to predict the metagenome [30]. Sequencing data were prepared as described above, but sequences were then clustered into OTUs using a closed-reference OTU picking protocol at the 97% sequencing identity level. The resulting OTU table was normalized by the expected copy number(s) of the 16s rRNA gene in each OTU. PICRUSt was then used to predict the metagenome. Each sample was rarefied to an even sequencing depth to adjust for uneven sequencing depth prior to further analysis. Differences in the metagenomes among the groups were investigated by visual assessment of clustering on principal coordinates analysis (PCoA) plots, and by analysis of similarity (ANOSIM) calculated on Bray Curtis dissimilarity metric using the available QIIME scripts. The KEGG orthologies and pathways identified [31] via PICRUSt were analyzed and compared as described above.
Results
Data
A total of 16,386,847 sequences were obtained from the sequencing facility. After quality filtering, 9,471,315 reads remained with a median of 65,108 sequences per sample (range, 3 to 136,547 sequences per sample). The Usearch61 method of chimera detection was used to identify 820,087 chimeric sequences, and these were removed prior to OTU picking. Open reference OTU picking was used to cluster sequences into 86,866 unique OTUs with a median of 57,740 counts/sample (range, 9 to 121,213 counts/sample) with a total count of 8,532,117. Very low frequency OTUs (i.e., OTUs represented fewer than 4 times) or OTUs present in fewer than 4 animals were removed. This reduced the number of OTUs into which sequences were clustered to 37,184 with a total count of 8,389,450. Although this filtering reduced the number of OTUs by 58% it resulted in only a 2% decrease in the number of counts (i.e., 98% of the counts were retained). Finally, an even sequencing depth of 10,800 reads per sample was selected. Although alpha rarefaction curves had not plateaued at this sampling depth, Good’s coverage index estimates indicated that over 90% of the species were represented at both time points (median, 91%; Fig 1A and 1B). Following quality filtering, OTU picking, removal of ultra-low abundance OTUs, and selecting an even sampling depth of 10,800 reads/sample, 139 samples remained for analysis.
Fig 1. Data filtering and even sampling depth suggest adequate coverage for subsequent analysis.

A) Bar charts of estimated goods coverage at an even sampling depth of 10,800 reads/sample at time 1 (dotted pattern) and time 2 (checkerboard pattern). Error bars represent the standard deviation. B) Alpha rarefaction curves for all samples showing numbers of observed species at each sampling depth on the y-axis and sequences/sample on the x-axis up to a sampling depth of 10,800 sequences/sample. C) Bar charts of estimated goods coverage at an even sampling depth of 3,000,000 predicted KEGG orthologies/sample at time 1 (dotted pattern) and time 2 (checkerboard pattern). Error bars represent the standard deviation. D) Alpha rarefaction curves for all samples showing numbers of observed species (KEGG orthologies) at each sampling depth on the y-axis and sequences/sample on the x-axis up to a sampling depth of 3,000,000 sequences/sample.
Closed reference OTU picking was used to identify OTUs for use in PICRUSt to predict the metagenome. Closed reference OTU picking resulted in sequences being clustered into 11,300 unique OTUs with a total count of 7,026,925 (median, 47,894 counts/sample). These were filtered as above, such that removal of OTUs identified fewer than 4 times reduced the number of unique OTUs to 6,923 and reduced the total OTU count to 7,020,091; removal of OTUs identified in fewer than 4 animals further reduced the number of unique OTUs to 6,393 and total count to 7,016,295. The resulting OTU table was normalized by the expected copy number(s) of the 16S rRNA gene for each OTU prior to metagenome predictions. Metagenome predictions resulted in OTUs being clustered into 6,909 unique KEGG orthologies. An even sampling depth of 3,000,000 reads per sample was chosen. Alpha rarefaction curves and Good’s coverage index suggest 3,000,000 reads/sample adequately covered the predicted metagenome (Fig 1C and 1D).
Differences in fecal microbiota among the health groups at time 1
There were no significant differences in the diversity indices or richness calculated for the health groups at time 1 ( Fig 2A–2C). Moreover principal coordinate analysis (PCoA) plots based on the unweighted UniFrac distance metric revealed no apparent clustering of the health groups at time 1 (Fig 2D). ANOSIM analysis of both the weighted (R -0.0023, P-value 0.502) and unweighted (R = 0.0029; P = 0.379) Unifrac distance metric revealed no significant difference among the health groups at time 1, and Kruskal-Wallis analysis revealed no significant differences in abundances of any OTUs among the health groups at time 1(data not shown).
Fig 2. No differences in the fecal microbiota among the health groups at time 1.

A) Scatter dot plot of Shannon diversity index for all health groups; healthy (dots), subclinical (squares), and clinical (triangles) at time 1. Horizontal lines represent the mean for each group and error bars represent the standard deviation. There were no statistical differences among the groups. B) Scatter dot plot of Simpson diversity index for all health groups; healthy (dots), subclinical (squares), and clinical (triangles) at time 1. Horizontal lines represent the mean for each group and error bars represent the standard deviation. There were no statistical differences among the groups. C) Alpha rarefication curves for each of the health groups; healthy (red), clinical (blue), subclinical (orange) at time 1 showing numbers of observed species at each sampling depth on the y-axis and sequences/sample on the x-axis up to 10,800 sequences/sample. Error bars represent standard deviations of each group at the specified sampling depth. D) Principal coordinate analysis of unweighted Unifrac distance metric for all heath groups; healthy (red), clinical (blue), subclinical (orange) at time 1. There were no differences among the groups.
Differences in predicted metagenome among health groups at time 1
Principal coordinate analysis plots based on the Bray Curtis dissimilarity metric revealed no graphical evidence of clustering of the predicted metagenome of the health groups at time 1 (Fig 3). ANOSIM analysis of the dissimilarity metric revealed no significant difference in the predicted metagenome among the health groups at time 1 (R = 0.0016; P = 0.370). In addition, Kruskal-Wallis testing revealed no significant differences in the abundance of any KEGG pathways among the health groups at time 1 (data not shown).
Fig 3. Principal coordinate analysis plots of the predicted metagenome for health groups at time 1 based on the Bray Curtis dissimilarity metric.
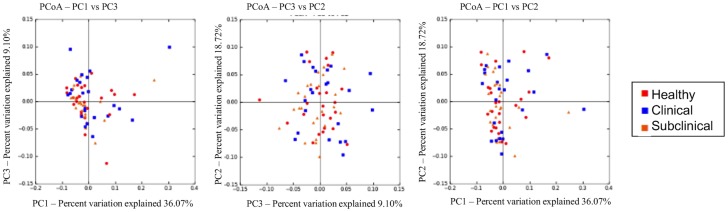
Principal coordinate analysis of the Bray Curtis dissimilarity metric of the predicted metagenome for all heath groups; healthy (red), clinical (blue), subclinical (orange) at time 1. There were no differences among the groups.
Differences in fecal microbiota among the health groups at time 2
There were no differences in the diversity indices or richness calculated for the health groups at time 2 (Fig 4A–4C). Moreover, principal coordinate analysis (PCoA) plots based on the unweighted UniFrac distance metric revealed no apparent clustering of the health groups at time 2 (Fig 4D). The unweighted Unifrac distance metric was small but significantly different than 0 using ANOSIM analysis of the distance metric at time 2 (R = 0.072; P = 0.005). When using the weighted distance metric, however, the distance metric was small and there was no significant difference among the groups at time 2 (R = 0.0303; P = 0.083). Kruskal-Wallis tests revealed no significant differences in abundances of any OTUs among the health groups at time 2 (data not shown).
Fig 4. No differences in the fecal microbiota among the health groups at time 2.

A) Scatter dot plot of Shannon diversity index for all health groups; healthy (dots), subclinical (squares), and clinical (triangles) at time 2. Horizontal lines represent the mean for each group and error bars represent the standard deviation. There were no statistical differences among the groups. B) Scatter dot plot of Simpson diversity index for all health groups; healthy (dots), subclinical (squares), and clinical (triangles) at time 2. Horizontal lines represent the mean for each group and error bars represent the standard deviation. There were no statistical differences among the groups. C) Alpha rarefication curves for each of the health groups; healthy (red), clinical (blue), subclinical (orange) at time 2 showing numbers of observed species at each sampling depth on the y-axis and sequences/sample on the x-axis up to 10,800 sequences/sample. Error bars represent standard deviations of each group at the specified sampling depth. D) Principal coordinate analysis of unweighted Unifrac distance metric for all heath groups; healthy (red), clinical (blue), subclinical (orange) at time 2. There were no differences among the groups.
Differences in predicted metagenome among health groups at time 2
Principal coordinate analysis plots based on the Bray Curtis dissimilarity metric revealed no visual clustering of the predicted metagenome of the health groups at time 2 (Fig 5). ANOSIM analysis of the distance metrics revealed no significant difference in the predicted metagenome among the health groups at time 1 (R = 0.0149; P = 0.212). In addition, Kruskal-Wallis test revealed no significant differences in the abundance of any KEGG pathways among the health groups at time 2 (data not shown).
Fig 5. Principal coordinate analysis plots of the predicted metagenome for health groups at time 2 based on the Bray Curtis dissimilarity metric.
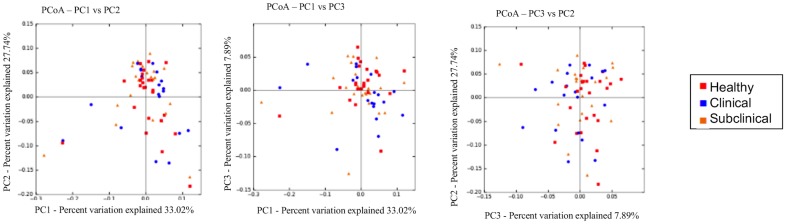
Principal coordinate analysis of the Bray Curtis dissimilarity metric of the predicted metagenome for all heath groups; healthy (red), clinical (blue), subclinical (orange) at time 2. There were no differences among the groups.
Differences in fecal microbiota between time 1 and time 2
The distribution of the major (frequency > 1%) families of microbes present at time 1 and time 2 were plotted as pie charts (Fig 6). There were no differences in the diversity indices or richness calculated for horses at times 1 or 2 irrespective of health status (Fig 7A–7C). The PCoA plots of the first 3 principal coordinates based on the unweighted UniFrac distance metric revealed obvious visual clustering of the health groups by time (Fig 7D). ANOSIM revealed significant differences of both the unweighted Unifrac distance metric (R = 0.6945; P = 0.001) and weighted Unifrac distance metric (R = 0.2566; P = 0.001). In order to identify bacterial taxa that contributed to the difference between times, we performed SIMPER analysis at the levels of both the phyla and family. Mean change in abundance with time and percent contribution to differences in the Bray Curtis dissimilarity metric were tabulated (Table 1 for Phyla and S1 Table for Family). Mann-Whitney U tests on the proportion of OTUs per sample revealed many differences in the abundance of OTUs between time 1 and time 2 samples (S2 Table).
Fig 6. Major bacterial families present at (A) time 1 and (B) time 2.

A) Pie charts showing the distribution of the major (>1% mean abundance) of the families of bacteria and archea present at time 1. Each Family is colored by a different color and the color for each Family and the Phylum to which it belongs is shown in the figure legend accompanying the pie chart. B) Pie charts showing the distribution of the major (>1% mean abundance) of the families of bacteria and archea present at time 2. Each Family is colored by a different color and the color for each family and the phylum to which it belongs is shown in the figure legend accompanying the pie chart.
Fig 7. No differences in the diversity or richness of the fecal microbiota between time 1 and time 2 but the composition of the fecal microbiota is different between time 1 and time 2.

A) Scatter dot plot of Shannon diversity index for time 1 (dots) and time 2 (squares). Horizontal line represents mean and error bars represent the standard deviation. There were no statistical differences between the groups. B) Scatter dot plot of Simpson diversity index for time 1 (dots) and time 2 (squares). Horizontal line represents mean and error bars represent the standard deviation. There were no statistical differences between the groups. C) Alpha rarefication curves for time 1 (red) and time 2 (blue) showing numbers of observed species at each sampling depth on the y-axis and sequences/sample on the x-axis up to 10,800 sequences/sample. Error bars represent standard deviations of each group at the specified sampling depth. D) Principal coordinate analysis of unweighted Unifrac distance metric for time 1 (red) and time 2 (blue). There was obvious clustering of the time 1 samples and the time 2 samples.
Table 1. SIMPER analysis identifying the % contribution of each phylum to the Bray Curtis dissimilarity metric between time 1 and time 2.
| Phyla | % Contribution | Cumulative % | Mean abundance time 1 | Mean abundance time 2 | Change in abundance with time |
|---|---|---|---|---|---|
| k__Bacteria;p__Firmicutes | 31.71 | 31.71 | 77.00% | 70.10% | -6.90% |
| k__Bacteria;p__Bacteroidetes | 18.23 | 49.94 | 8.27% | 10.80% | 2.53% |
| k__Bacteria;p__Proteobacteria | 14.89 | 64.82 | 5.32% | 3.02% | -2.30% |
| k__Archaea;p__Euryarchaeota | 13.84 | 78.66 | 2.82% | 7.14% | 4.32% |
| k__Bacteria;p__Verrucomicrobia | 7.509 | 86.17 | 2.35% | 3.65% | 1.30% |
| k__Bacteria;p__Spirochaetes | 4.411 | 90.58 | 0.49% | 2.11% | 1.62% |
| k__Bacteria;p__Actinobacteria | 2.868 | 93.45 | 1.88% | 1.20% | -0.68% |
| k__Bacteria;p__Chlamydiae | 1.484 | 94.93 | 0.21% | 0.51% | 0.30% |
| k__Bacteria;p__WPS-2 | 1.142 | 96.07 | 0.43% | 0.09% | -0.34% |
| k__Bacteria;p__Fusobacteria | 0.8746 | 96.95 | 0.31% | 0.19% | -0.13% |
| k__Bacteria;p__Tenericutes | 0.5851 | 97.53 | 0.32% | 0.17% | -0.15% |
| Unassigned;Other | 0.5642 | 98.1 | 0.23% | 0.34% | 0.10% |
| k__Bacteria;p__Fibrobacteres | 0.4846 | 98.58 | 0.02% | 0.21% | 0.19% |
Similarity percentage analysis of the phyla differences between time 1 and time 2. The first column identifies the phylum explained by that row, the second column shows the % dissimilarity explained by that phylum, the third column tallies the cumulative Bray Curtis dissimilarity metric for the phyla thus far represented in the table, and the last three columns show mean abundance at time 1, mean abundance at time 2, and change in mean abundance respectively
Differences in predicted metagenome between time 1 and time 2
As expected from the observed time-dependent fecal microbiota differences, there also were time-dependent differences in the predicted metagenome revealed by PCoA of the Bray Curtis dissimilarity metric (Fig 8A). ANOSIM results calculated on the Bray Curtis dissimilarity metric revealed significant differences of the predicted metagenome at time 1 and time 2 (R = 0.2376; P = 0.001). There were 367 KEGG orthologies that were significantly (adjusted P < 0.05) decreased more than 2-fold between time 1 and time 2, and 486 KEGG orthologs were significantly (adjusted P < 0.05) increased by more than 2-fold between time 1 and time 2 (S3 Table). The different pathways to which these KEGG orthologies belong are represented as pie charts (Fig 8B and 8C). Approximately half (13) of the top 25 predicted functional pathways that were decreased by 2-fold or more between 3 and 5 weeks were related to metabolism. Moreover, the majority of these decreased metabolic pathways were related to metabolism of simple sugars (sucrose, glucose, fructose, and mannose) and amino acids (tyrosine, arginine, and proline). Predicted functional pathways that were increased more than 2-fold over the same time period were evenly distributed among metabolic pathways, transcriptional pathways, and a myriad of unclassified or unknown pathways.
Fig 8. The composition of the predicted metagenome changes between time 1 and time 2.

(A) Principal coordinate analysis of the Bray Curtis dissimilarity metric of the predicted metagenome for time 1 (red) and time 2 (blue). There was obvious visual clustering of the predicted metagenome at time 1 and time 2. B) Pie chart showing the predicted functional pathways that were significantly (Mann Whitney U test FDR P value < 0.05) decreased >2 fold between time 1 and time 2. The figure legend accompanying the figure explains which pathways are represented by which colors. C) Pie chart showing the predicted functional pathways that were significantly (Mann Whitney U test FDR P value < 0.05) increased >2 fold between time 1 and time 2. The figure legend accompanying the figure explains which pathways are represented by which colors.
Discussion
The distribution of virulent R.equi at equine breeding farms appears to be widespread, and foals are exposed to the bacterium from birth [1–3, 32–41]. Commensurate with this widespread exposure, many if not most foals at affected breeding farms appear to develop pulmonary abscess formation or consolidation attributed to R. equi without developing disease (i.e., subclinical pneumonia); however, most of these foals with subclinical pulmonary lesions do not develop clinical signs of disease [18, 42–45]. It is unclear why, among foals from the same environment with presumably similar exposure to virulent R. equi, some develop clinical signs of pneunonia while others either develop subclinical pneumonia or remain free of disease and pulmonary lesions. One possible explanation for this phenomenon is variation in immune function among foals. All foals are born with an immune system that is naïve, lacking circulating immunoglobulins, and with various diminutions in innate and adaptive immune responses [4, 5]. The intestinal microbiome in neonates (including foals) is known to expand and diversify following birth, and this expansion and diversification of the microbiome is known to influence various host developmental processes including intestinal and systemic immunity to infection [10–16]. Thus, we hypothesized that the difference in susceptibility to R. equi pneumonia among foals might be attributable to differences in the composition or diversity of the intestinal microbiome resulting in less developed immune responses in some foals that rendered these foals more susceptible to infection. To test this hypothesis, we compared the fecal microbiome using samples collected prior to the onset of clinical disease in 75 foals: 25 foals that developed R. equi pneumonia; 25 that developed subclinical pneumonia presumed to be attributed to R. equi infection; and, 25 healthy foals (i.e., foals free of clinical signs and ultrasonographic evidence of pulmonary lesions). In addition to being appropriate for predictive purposes, the strategy of collecting samples prior to the onset of disease also had the advantage of avoiding the influence of disease or any antimicrobial treatment for disease on the fecal microbiome; antimicrobial treatment, systemic inflammatory responses, inflammation, and reduced appetite can alter the intestinal environment and consequently the microbiota [46]. Thus, collecting samples prior to onset of disease and treatment obviated the confounding effects of these factors on the association of the fecal microbiome composition and diversity with R. equi pneumonia. Our goal was to examine changes in the fecal microbiome during a brief interval after the initial rapid colonization of the foal’s gastrointestinal tract (i.e., around age 1 month) when the foals’ microbiota were more stable but before any confounding effects of illness or treatment might have impacted the microbiota.
Our results did not provide strong evidence against the null hypothesis of no difference in the microbiota among the 3 clinical groups (R. equi pneumonia, subclinical pneumonia, or healthy). At either time-point, the diversity of organisms was similar among the 3 groups (Figs 3A–3B and 5A–5B) and there was no significant difference in any of the diversity indices evaluated among groups. At the first sample-time (3 weeks of age), neither PCoA, alpha diversity indices, nor measure of overall richness (Fig 3) revealed evidence of significant differences among groups. At time 2 (5 weeks), there was a weak but statistically significant difference detected when using ANOSIM to determine similarity of the Unifrac unweighted distance metric based on health status (Fig 5C). When OTUs were weighted for abundance, however, this association was no longer significant. In light of the graphical and statistical results, we interpret these findings to indicate that there were no clinically important differences in the fecal microbiome among the groups at either time-point.
A strong and significant difference in the 2 sample time-points was observed in both the composition of the fecal microbiota and the predicted metagenome. This finding was somewhat surprising to us considering that most (88%; 22/25) of these samples were collected 2 weeks apart. The observed changes were largely characterized by a decrease in members of the phylum Firmicutes and an increase in members of the phylum Bacteroidetes. In fact, 50% of the dissimilarity between the fecal microbiota from time 1 and time 2 was explained by changes in members of these 2 phyla. A recent study [47] identified the core microbiota of the large intestine and feces of adult horses. In that study, the relative abundance of Firmicutes and Bacteroidetes in feces was nearly equal at 46% and 43% respectively. Among our foals, the relative abundance of Firmicutes was 73% and the relative abundance of Bacteroidetes was 10%; therefore, the decreased abundance of Firmicutes and increased abundance of Bacteroidetes between the later and earlier samples in our study might have reflected continuation of a gradual transition to an adult horse-like composition of the fecal microbiota. These findings support the notion that the microbiota of foals is rapidly changing up to 7 weeks of age and thus may influence immune development. Foals gradually consume less milk and more solid feedstuffs between birth and ~21 weeks of age as they transition to becoming more reliant on hind-gut fermentation for meeting caloric requirements [48, 49]. Clearly, as a foal’s diet changes, their microbiota adapts to changes in dietary composition. Despite the fact that this physiological transition is occurring over a period of months, we noted large changes during a period as short as 2 weeks between 3 and 5 weeks of age. Other studies have demonstrated that the foals fecal microbiota community changes gradually with age but appears to climax around 30 days of age with no additional significant changes between 30 and 60 days of age [17]. Our second (latter) sample was collected from 5-week-old foals; therefore, it is possible that we have characterized the final stages of maturation of the fecal microbiota. Alternatively, our results indicate that the fecal microbiota of foals might be continuing to change significantly beyond the first month of life. Interestingly, these temporal changes in the microbiota were not accompanied by a significant change in overall diversity or richness. Thus, the observed compositional changes were not attributable to acquiring a greater diversity of organisms comprising the fecal microbiome but instead to altered abundance of the populations present.
One plausible explanation for the finding that the composition but not diversity changed between times 1 and 2 might was that it reflected a functional change in the microbiota. Indeed, the changes in the fecal microbiota composition between time 1 and time 2 were reflected in the predicted functional pathways of the microbiota. We examined the alterations in the functional pathways of the predicted metagenome in order to determine how fecal microbiota changes may affect the host. Interestingly, many of the predicted functional pathways that were observed to be altered during this time were related to metabolism. There appeared to be a decrease in predicted functional pathways related to metabolism of simple sugars found in milk and to an increase in metabolic pathways related to fermentation and more complex energy metabolism (Fig 8). These metabolic pathway changes suggest that indeed the microbiota is changing to accommodate the changing diet of these foals. It is possible that the changes observed in the functional pathways also influenced immune development and function as age-related immunologic changes have been observed over the same timeframe as the foals in our study [8]. Unfortunately, immune function of foals in this study was not assessed.
Our study had a number of important limitations. First, the sample size was relatively modest. Nevertheless, these data represent the largest group of foals whose microbiome has been evaluated to date. Second, the timing of samples might have been inappropriate. We selected 3 weeks and 5 weeks of age by convenience in order to examine the narrow window after foals’ had acquired a microbiota that was relatively stable but before any confounding effects of illness might have been present. Epidemiological evidence exists that many foals become infected early in life [50, 51], and that foals are more susceptible to infection with R. equi during the first week or 2 of life [52]. Thus, we might have selected sample time-points that were after the age at which susceptibility was most important and, possibly, most apparent; put another way, the critical period for evaluation might have been earlier in life when the composition and diversity of the microbiota were less complex [13]. Third, we examined only the fecal microbiome. Although fecal samples are often used as indicators of the intestinal microbiota, it has been shown that fecal microbiota data do not always correlate with samples taken from regions of the gastrointestinal tract, and that variation in the microbiota occurs among different anatomical sites of the intestinal tract. For example, fecal samples might not have reflected the microbiota inhabiting the intestinal mucosa; the mucosal microbiota might be more important for immune development because it is more intimately associated with the immune cells of the host’s intestinal tract. Finally, our overall hypothesis was that varying composition or reduced diversity of the neonatal foal intestinal microbiome would contribute to susceptibility of developing clinical signs of R. equi pneumonia presumably because of differences in immune function related to differences in the intestinal microbiome. Unfortunately, we did not assess immune function of the study foals. Thus, had we observed a difference in the intestinal microbiota we would have failed to demonstrate a difference in the immunological response. Alternatively, we cannot exclude the possibility that differences in immune function existed between affected and unaffected foals at or between sampling times despite lack of apparent difference in the composition or diversity of the fecal microbiota. Future studies in which immune function and microbiota characterization are performed contemporaneously are recommended to obviate this important limitation.
Our population was limited to a single farm in Texas. We do not know to what extent our results from foals in western Texas may be extrapolated to other populations of foals or even other farms in the same region. Last, we classified the microbiota based on amplification of the V4 hypervariable region of the 16S rRNA gene. While this region has been recommended by some investigators[53], V4 is not considered by all investigators to be ideal for microbiomic analysis of differences by phenotype [54]. Thus, using only the V4 region to classify microbes might have limited our ability to detect differences among foals in the various health groups.
Although we failed to identify differences among our study groups, it remains possible and plausible that changes in the intestinal microbiome influence maturation of the immune system in foals because it has been shown in other species that immune development is strongly influenced by the intestinal microbiota. Thus, our absence of evidence favoring our hypothesis of differences among clinical groups is not the same as evidence of absence of differences among the groups. We did observe a marked and significant difference between a relatively short interval of time that appeared to reflect adaptation to transition from a milk diet to a diet that includes available forage (including hay) and access to concentrate fed to the mare.
Supporting Information
Similarity percentage analysis of the family differences between time 1 and time 2. The first column identifies the family explained by that row, the second column shows average Bray Curtis dissimilarity between time 1 and time 2 for that family, the third column shows the % dissimilarity explained by that family, the fourth column tallies the cumulative Bray Curtis dissimilarity metric for the family thus far represented in the table, and the last three columns show mean abundance at time 1, mean abundance at time 2, and change in mean abundance respectively.
(PDF)
The first column provides OTU number, the second column shows taxonomic assignment, the third column provides FDR corrected P-value, and the final 2 columns provide mean proportion for time 1 and time 2 respectively.
(PDF)
The first column list increased KEGG orthologs and the secondond column list decreased KEGG orthologs.
(PDF)
Acknowledgments
The authors acknowledge Whole Systems Genomics Initiative (WSGI) at Texas A&M University for providing computational resources and systems administration support for the WSGI HPC Cluster. The authors thank Dr. Danny Hurych, Kranti Konganti, Joseph Kintzinger, and Brianna Sisson for technical assistance.
Data Availability
Data are available from the NCBI GenBank database submission number SRP060764.
Funding Statement
This work was supported by a grant from Boehringer Ingelheim (Grant number 01072014) http://www.boehringeringelheim.com/. Additional funding was provided by the Link Equine Research Endowment. Fecal sample collection was made possible by a grant from the American Quarter Horse Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Scot E. Dowd is employed by Molecular Research DNA Laboratory (MR DNA). Molecular Research DNA Laboratory (MR DNA) provided support in the form of salary for author SED, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific Cover Letter 2 role of this author is articulated in the ‘author contributions’ section. Glenn P. Blodgett is employed by 6666 Ranch. 6666 Ranch provided support in the form of salary for author GPB, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The specific role of this author is articulated in the ‘author contributions’ section.
References
- 1. Takai S. Epidemiology of Rhodococcus equi infections: a review. Veterinary microbiology. 1997;56(3–4):167–76. Epub 1997/06/16. . [DOI] [PubMed] [Google Scholar]
- 2. Prescott JF. Rhodococcus equi: an animal and human pathogen. Clinical microbiology reviews. 1991;4(1):20–34. Epub 1991/01/01. ; PubMed Central PMCID: PMCPmc358176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Muscatello G, Leadon DP, Klayt M, Ocampo-Sosa A, Lewis DA, Fogarty U, et al. Rhodococcus equi infection in foals: the science of 'rattles'. Equine veterinary journal. 2007;39(5):470–8. Epub 2007/10/04. . [DOI] [PubMed] [Google Scholar]
- 4. Boyd NK, Cohen ND, Lim WS, Martens RJ, Chaffin MK, Ball JM. Temporal changes in cytokine expression of foals during the first month of life. Veterinary immunology and immunopathology. 2003;92(1–2):75–85. Epub 2003/03/12. . [DOI] [PubMed] [Google Scholar]
- 5. Perkins GA, Wagner B. The development of equine immunity: Current knowledge on immunology in the young horse. Equine veterinary journal. 2014. Epub 2014/11/19. 10.1111/evj.12387 . [DOI] [PubMed] [Google Scholar]
- 6. Levy O. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nature reviews Immunology. 2007;7(5):379–90. Epub 2007/04/26. 10.1038/nri2075 . [DOI] [PubMed] [Google Scholar]
- 7. Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nature reviews Immunology. 2004;4(7):553–64. Epub 2004/07/02. 10.1038/nri1394 . [DOI] [PubMed] [Google Scholar]
- 8. Kachroo P, Ivanov I, Seabury AG, Liu M, Chowdhary BP, Cohen ND. Age-related changes following in vitro stimulation with Rhodococcus equi of peripheral blood leukocytes from neonatal foals. PloS one. 2013;8(5):e62879 Epub 2013/05/22. 10.1371/journal.pone.0062879 ; PubMed Central PMCID: PMCPmc3656898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cohen ND, Bourquin JR, Bordin AI, Kuskie KR, Brake CN, Weaver KB, et al. Intramuscular administration of a synthetic CpG-oligodeoxynucleotide modulates functional responses of neutrophils of neonatal foals. PloS one. 2014;9(10):e109865 Epub 2014/10/22. 10.1371/journal.pone.0109865 ; PubMed Central PMCID: PMCPmc4198146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mshvildadze M, Neu J. The infant intestinal microbiome: friend or foe? Early human development. 2010;86 Suppl 1:67–71. Epub 2010/02/02. 10.1016/j.earlhumdev.2010.01.018 ; PubMed Central PMCID: PMCPmc3586601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Murgas Torrazza R, Neu J. The developing intestinal microbiome and its relationship to health and disease in the neonate. Journal of perinatology: official journal of the California Perinatal Association. 2011;31 Suppl 1:S29–34. Epub 2011/04/02. 10.1038/jp.2010.172 . [DOI] [PubMed] [Google Scholar]
- 12. Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS biology. 2007;5(7):e177 Epub 2007/06/28. 10.1371/journal.pbio.0050177 ; PubMed Central PMCID: PMCPmc1896187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bordin AI, Suchodolski JS, Markel ME, Weaver KB, Steiner JM, Dowd SE, et al. Effects of administration of live or inactivated virulent Rhodococccus equi and age on the fecal microbiome of neonatal foals. PloS one. 2013;8(6):e66640 Epub 2013/06/21. 10.1371/journal.pone.0066640 ; PubMed Central PMCID: PMCPmc3681940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Inman CF, Laycock GM, Mitchard L, Harley R, Warwick J, Burt R, et al. Neonatal colonisation expands a specific intestinal antigen-presenting cell subset prior to CD4 T-cell expansion, without altering T-cell repertoire. PloS one. 2012;7(3):e33707 Epub 2012/03/24. 10.1371/journal.pone.0033707 ; PubMed Central PMCID: PMCPmc3307746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Willing BP, Van Kessel AG. Enterocyte proliferation and apoptosis in the caudal small intestine is influenced by the composition of colonizing commensal bacteria in the neonatal gnotobiotic pig. Journal of animal science. 2007;85(12):3256–66. Epub 2007/09/06. 10.2527/jas.2007-0320 . [DOI] [PubMed] [Google Scholar]
- 16. Myles IA, Fontecilla NM, Janelsins BM, Vithayathil PJ, Segre JA, Datta SK. Parental dietary fat intake alters offspring microbiome and immunity. Journal of immunology (Baltimore, Md: 1950). 2013;191(6):3200–9. Epub 2013/08/13. 10.4049/jimmunol.1301057 ; PubMed Central PMCID: PMCPmc3831371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Faubladier C, Sadet-Bourgeteau S, Philippeau C, Jacotot E, Julliand V. Molecular monitoring of the bacterial community structure in foal feces pre- and post-weaning. Anaerobe. 2014;25:61–6. Epub 2013/12/10. 10.1016/j.anaerobe.2013.11.010 . [DOI] [PubMed] [Google Scholar]
- 18. Chaffin MK, Cohen ND, Blodgett GP, Syndergaard M. Evaluation of ultrasonographic screening methods for early detection of Rhodococcus equi pneumonia in foals. J Equine Vet Sci 2012;32 S20–S1. [Google Scholar]
- 19. Giguère S, Cohen ND, Keith Chaffin M, Slovis NM, Hondalus MK, Hines SA, et al. Diagnosis, Treatment, Control, and Prevention of Infections Caused by Rhodococcus equi in Foals. Journal of veterinary internal medicine / American College of Veterinary Internal Medicine. 2011;25(6):1209–20. 10.1111/j.1939-1676.2011.00835.x [DOI] [PubMed] [Google Scholar]
- 20. Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. The ISME journal. 2012;6(8):1621–4. Epub 2012/03/10. 10.1038/ismej.2012.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods. 2010;7(5):335–6. Epub 2010/04/13. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–200. Epub 2011/06/28. 10.1093/bioinformatics/btr381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–1. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 24. DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Applied and environmental microbiology. 2006;72(7):5069–72. Epub 2006/07/06. 10.1128/aem.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. The ISME journal. 2012;6(3):610–8. Epub 2011/12/03. 10.1038/ismej.2011.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clarke KR. Non-parametric multivariate analyses of changes in community structure. Australian Journal of Ecology. 1993;18(1):117–43. [Google Scholar]
- 27. Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Applied and environmental microbiology. 2005;71(12):8228–35. Epub 2005/12/08. 10.1128/aem.71.12.8228-8235.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bray JR, Curtis JT. An ordination of upland forest communities of southern Wisconsin. Ecological Monographs 1957;27:24. [Google Scholar]
- 29. Hammer O, Harper D, Ryan PD. PAST: Paleon Statictics Software Package for Education and Data Analysis. Palaeontologica Electronica. 2001;4(1):9. [Google Scholar]
- 30. Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature biotechnology. 2013;31(9):814–21. Epub 2013/08/27. 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42(Database issue):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cohen ND, Smith KE, Ficht TA, Takai S, Libal MC, West BR, et al. Epidemiologic study of results of pulsed-field gel electrophoresis of isolates of Rhodococcus equi obtained from horses and horse farms. American journal of veterinary research. 2003;64(2):153–61. Epub 2003/02/27. . [DOI] [PubMed] [Google Scholar]
- 33. Morton AC, Begg AP, Anderson GA, Takai S, Lammler C, Browning GF. Epidemiology of Rhodococcus equi strains on Thoroughbred horse farms. Applied and environmental microbiology. 2001;67(5):2167–75. Epub 2001/04/25. 10.1128/aem.67.5.2167-2175.2001 ; PubMed Central PMCID: PMCPmc92851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cohen ND, Carter CN, Scott HM, Chaffin MK, Smith JL, Grimm MB, et al. Association of soil concentrations of Rhodococcus equi and incidence of pneumonia attributable to Rhodococcus equi in foals on farms in central Kentucky. American journal of veterinary research. 2008;69(3):385–95. Epub 2008/03/04. 10.2460/ajvr.69.3.385 . [DOI] [PubMed] [Google Scholar]
- 35. Grimm MB, Cohen ND, Slovis NM, Mundy GD, Harrington JR, Libal MC, et al. Evaluation of fecal samples from mares as a source of Rhodococcus equi for their foals by use of quantitative bacteriologic culture and colony immunoblot analyses. American journal of veterinary research. 2007;68(1):63–71. Epub 2007/01/04. 10.2460/ajvr.68.1.63 . [DOI] [PubMed] [Google Scholar]
- 36. Buntain S, Carter CN, Kuskie KR, Smith JL, R S, Chaffin MK, et al. Frequency of Rhodococcus equi in feces of mares in central Kentucky. J Equine Vet Sci 2010; 30:4. [Google Scholar]
- 37. Muscatello G, Anderson GA, Gilkerson JR, Browning GF. Associations between the ecology of virulent Rhodococcus equi and the epidemiology of R. equi pneumonia on Australian thoroughbred farms. Applied and environmental microbiology. 2006;72(9):6152–60. Epub 2006/09/08. 10.1128/aem.00495-06 ; PubMed Central PMCID: PMCPmc1563629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Muscatello G, Gerbaud S, Kennedy C, Gilkerson JR, Buckley T, Klay M, et al. Comparison of concentrations of Rhodococcus equi and virulent R. equi in air of stables and paddocks on horse breeding farms in a temperate climate. Equine veterinary journal. 2006;38(3):263–5. Epub 2006/05/19. . [DOI] [PubMed] [Google Scholar]
- 39. Cohen ND, Kuskie KR, Smith JL, Slovis NM, Brown SE 2nd, Stepusin RS, et al. Association of airborne concentration of virulent Rhodococcus equi with location (stall versus paddock) and month (January through June) on 30 horse breeding farms in central Kentucky. American journal of veterinary research. 2012;73(10):1603–9. Epub 2012/09/28. 10.2460/ajvr.73.10.1603 . [DOI] [PubMed] [Google Scholar]
- 40. Kuskie K, Smith JL, Sinha S, Catrer CN, Chaffin MK, Slovis NM, et al. Associations between the exposure to airborne virulent Rhodococcus equi and the incidence of R. equi pneumonia among individual foals. J Equine Vet Sci 2011. 2011;31:6. [Google Scholar]
- 41. Cohen ND, Chaffin MK, Kuskie KR, Syndergaard MK, Blodgett GP, Takai S. Association of perinatal exposure to airborne Rhodococcus equi with risk of pneumonia caused by R equi in foals. American journal of veterinary research. 2013;74(1):102–9. Epub 2012/12/29. 10.2460/ajvr.74.1.102 . [DOI] [PubMed] [Google Scholar]
- 42. Venner M, Rodiger A, Laemmer M, Giguere S. Failure of antimicrobial therapy to accelerate spontaneous healing of subclinical pulmonary abscesses on a farm with endemic infections caused by Rhodococcus equi. Veterinary journal (London, England: 1997). 2012;192(3):293–8. Epub 2011/09/20. 10.1016/j.tvjl.2011.07.004 . [DOI] [PubMed] [Google Scholar]
- 43. Venner M, Astheimer K, Lammer M, Giguere S. Efficacy of mass antimicrobial treatment of foals with subclinical pulmonary abscesses associated with Rhodococcus equi. Journal of veterinary internal medicine / American College of Veterinary Internal Medicine. 2013;27(1):171–6. Epub 2013/01/03. 10.1111/jvim.12030 . [DOI] [PubMed] [Google Scholar]
- 44. Venner M, Credner N, Lammer M, Giguere S. Comparison of tulathromycin, azithromycin and azithromycin-rifampin for the treatment of mild pneumonia associated with Rhodococcus equi. The Veterinary record. 2013;173(16):397 Epub 2013/10/10. 10.1136/vr.101867 . [DOI] [PubMed] [Google Scholar]
- 45. Hildebrand F, Venner M, Giguere S. Efficacy of gamithromycin for the treatment of foals with mild to moderate bronchopneumonia. Journal of veterinary internal medicine / American College of Veterinary Internal Medicine. 2015;29(1):333–8. Epub 2015/01/27. 10.1111/jvim.12504 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pickard JM, Maurice CF, Kinnebrew MA, Abt MC, Schenten D, Golovkina TV, et al. Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature. 2014;514(7524):638–41. Epub 2014/10/03. 10.1038/nature13823 ; PubMed Central PMCID: PMCPmc4214913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dougal K, de la Fuente G, Harris PA, Girdwood SE, Pinloche E, Newbold CJ. Identification of a core bacterial community within the large intestine of the horse. PloS one. 2013;8(10):e77660 Epub 2013/11/10. 10.1371/journal.pone.0077660 ; PubMed Central PMCID: PMCPmc3812009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Crowell-Davis SL, Houpt KA, Carnevale J. Feeding and drinking behavior of mares and foals with free access to pasture and water. Journal of animal science. 1985;60(4):883–9. Epub 1985/04/01. . [DOI] [PubMed] [Google Scholar]
- 49. Argenzio RA, Southworth M, Stevens CE. Sites of organic acid production and absorption in the equine gastrointestinal tract. The American journal of physiology. 1974;226(5):1043–50. Epub 1974/05/01. . [DOI] [PubMed] [Google Scholar]
- 50. Horowitz ML, Cohen ND, Takai S, Becu T, Chaffin MK, Chu KK, et al. Application of Sartwell's model (lognormal distribution of incubation periods) to age at onset and age at death of foals with Rhodococcus equi pneumonia as evidence of perinatal infection. Journal of veterinary internal medicine / American College of Veterinary Internal Medicine. 2001;15(3):171–5. Epub 2001/05/31. . [DOI] [PubMed] [Google Scholar]
- 51. Chaffin MK, Cohen ND, Martens RJ. Chemoprophylactic effects of azithromycin against Rhodococcus equi-induced pneumonia among foals at equine breeding farms with endemic infections. Journal of the American Veterinary Medical Association. 2008;232(7):1035–47. Epub 2008/04/03. 10.2460/javma.232.7.1035 . [DOI] [PubMed] [Google Scholar]
- 52. Sanz M, Loynachan A, Sun L, Oliveira A, Breheny P, Horohov DW. The effect of bacterial dose and foal age at challenge on Rhodococcus equi infection. Veterinary microbiology. 2013;167(3–4):623–31. Epub 2013/10/22. 10.1016/j.vetmic.2013.09.018 . [DOI] [PubMed] [Google Scholar]
- 53. Mizrahi-Man O, Davenport ER, Gilad Y. Taxonomic classification of bacterial 16S rRNA genes using short sequencing reads: evaluation of effective study designs. PloS one. 2013;8(1):e53608 Epub 2013/01/12. 10.1371/journal.pone.0053608 ; PubMed Central PMCID: PMCPmc3538547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guo F, Ju F, Cai L, Zhang T. Taxonomic precision of different hypervariable regions of 16S rRNA gene and annotation methods for functional bacterial groups in biological wastewater treatment. PloS one. 2013;8(10):e76185 Epub 2013/10/23. 10.1371/journal.pone.0076185 ; PubMed Central PMCID: PMCPmc3797802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Similarity percentage analysis of the family differences between time 1 and time 2. The first column identifies the family explained by that row, the second column shows average Bray Curtis dissimilarity between time 1 and time 2 for that family, the third column shows the % dissimilarity explained by that family, the fourth column tallies the cumulative Bray Curtis dissimilarity metric for the family thus far represented in the table, and the last three columns show mean abundance at time 1, mean abundance at time 2, and change in mean abundance respectively.
(PDF)
The first column provides OTU number, the second column shows taxonomic assignment, the third column provides FDR corrected P-value, and the final 2 columns provide mean proportion for time 1 and time 2 respectively.
(PDF)
The first column list increased KEGG orthologs and the secondond column list decreased KEGG orthologs.
(PDF)
Data Availability Statement
Data are available from the NCBI GenBank database submission number SRP060764.