

Summary
In the aftermath of multiple high-profile cases of chronic traumatic encephalopathy (CTE) in professional American football players, physicians in clinical practice are likely to face an increasing number of retired football players seeking evaluation for chronic neurobehavioral symptoms. Guidelines for the evaluation and treatment of these patients are sparse. Clinical criteria for a diagnosis of CTE are under development. The contribution of CTE vs other neuropathologies to neurobehavioral symptoms in these players remains unclear. Here we describe the experience of our academic memory clinic in evaluating and treating a series of 14 self-referred symptomatic players. Our aim is to raise awareness in the neurology community regarding the different clinical phenotypes, idiosyncratic but potentially treatable symptoms, and the spectrum of underlying neuropathologies in these players.
The retired professional American football player presenting for evaluation of chronic neurobehavioral symptoms poses a challenge to the physician. Many of these players fear development of chronic traumatic encephalopathy (CTE),1 a progressive tauopathy associated with repeated concussive and subconcussive brain traumas.2 The lack of consensus clinical criteria for CTE, however, renders diagnosis during life difficult. Furthermore, professional American football players are at increased risk for many neurodegenerative diseases besides CTE.3 It is therefore not surprising that some symptomatic retired professional football players do not have CTE on autopsy,4 and among those with autopsy-proven CTE, up to 17% are found to have additional comorbid neuropathologies such as Alzheimer disease (AD), frontotemporal lobar degeneration, motor neuron disease, and α-synucleinopathies.2
While we await results of large multicenter prospective studies that will lead to improved clinical diagnostic and management tools for traumatic brain injury (TBI)-related neurodegenerative disease, the physician in practice must act based on the best available evidence. In an effort to guide physicians in clinical practice, we describe the experience of our academic memory clinic in evaluating and treating a series of 14 self-referred retired players. The aims of this case series are to describe the spectrum of neurobehavioral presentations in these retired players, to highlight some of the idiosyncratic but potentially treatable symptoms, and, when possible, to describe the spectrum of underlying neuropathologies.
METHODS
Study design and patients
The University of California, San Francisco (UCSF) is a site for the National Football League's Neurological Care Program and is recommended to former players interested in undergoing neurologic evaluation. We identified all patients who presented to the UCSF Memory and Aging Center between January 2010 and January 2014 reporting at least 1 year of playing professional American football and at least 1 year of neurobehavioral symptoms (n = 14). All patients were evaluated by experienced behavioral neurologists (BLM or GDR) and neuropsychologists (KP or JHK). When appropriate, caregivers were interviewed by an experienced dementia care nurse (JM). See appendix e-1 at Neurology.org/cp for further details.
Standard protocol approvals, registrations, and patient consents
All experimental procedures were approved by the UCSF Human Research Committee. Patients evaluated in the UCSF Memory and Aging Center clinic (n = 8) and the UCSF Alzheimer's Disease Research Center (n = 6) signed a consent allowing chart review and/or research data review for the purposes of anonymized research studies.
Brain imaging
All patients underwent brain MRI (n = 13) or CT (n = 1). Structural neuroimaging was interpreted by a behavioral neurologist (RCG) and a certified neuroradiologist (CPH) who together reviewed patterns of atrophy, ischemic vascular disease, and presence or absence of a cavum septum pellucidum (CSP). Five patients underwent PET imaging at Lawrence Berkeley National Laboratory on a Siemens Biograph 6 PET/CT scanner. All 5 patients underwent PET imaging with 18F-fluorodeoxyglucose (18F-FDG) and one of the following amyloid tracers: 11C-Pittsburgh compound B (11C-PiB, n = 3) or 18F-florbetapir (n = 2). PET scans were visually interpreted by an experienced PET researcher (GDR) who commented on the pattern of metabolic abnormalities on the FDG-PET studies as well as the presence and pattern of amyloid deposition on the amyloid PET studies, using published criteria.5,6 See appendix e-1 for further details.
Identification and reporting of clinical phenotypes
Based on predominant presenting clinical features and/or timing of onset of symptoms, patients were divided into 3 clinical phenotypes. Relevant clinical data are reported at the group level and individually. To protect individual identities, age is reported by decade and football exposure by 5-year increments.
RESULTS
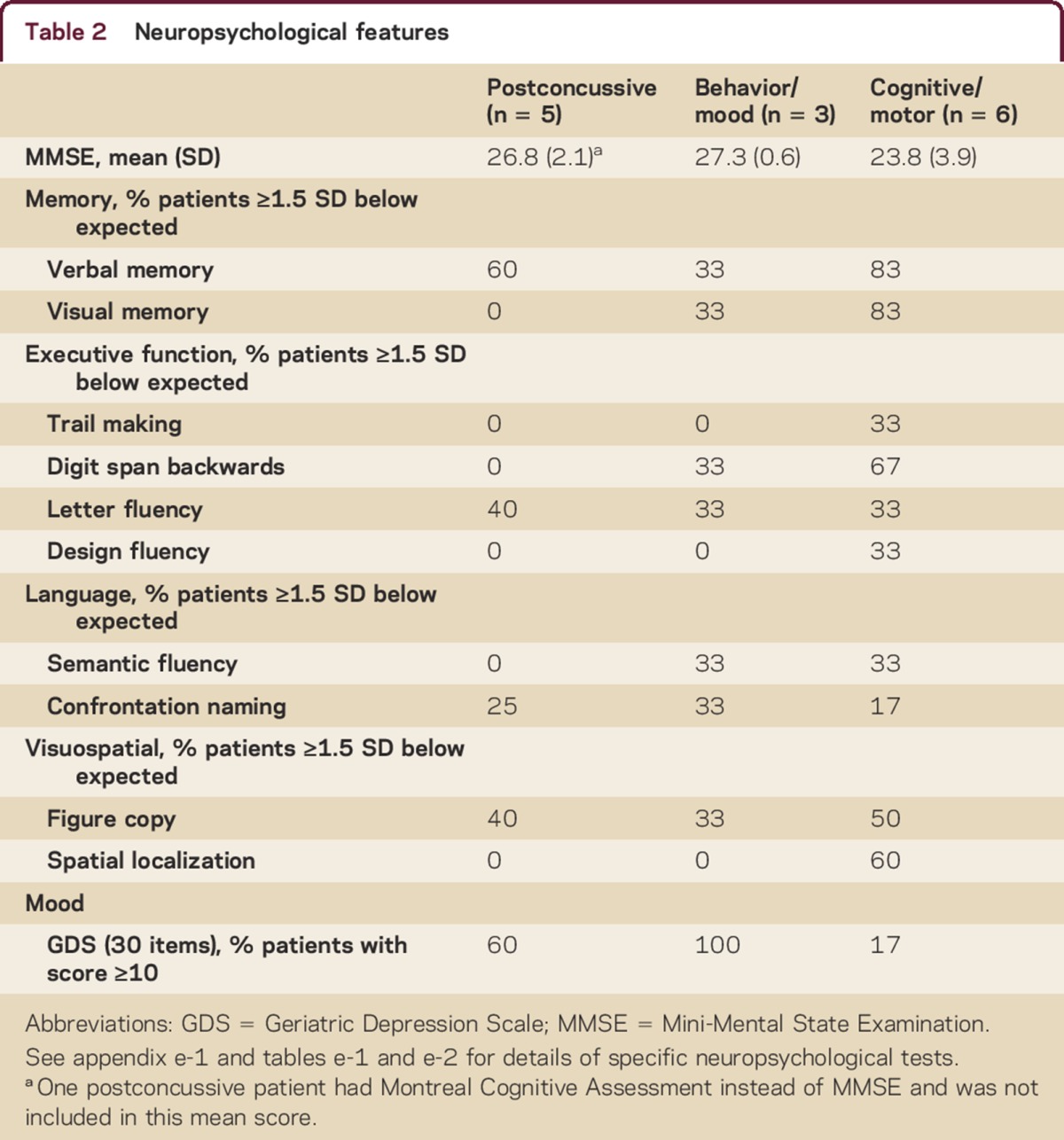
All patients were men. Average age was 53 years (range 20s–70s). We identified 3 phenotypes: young recently retired players with chronic postconcussive symptoms that began during their professional football career, middle-aged players who developed progressive predominantly behavior/mood symptoms an average of 24 years (range approximately 20–30 years) after retirement from professional football, and middle-aged or older players who developed progressive predominantly cognitive/motor symptoms an average of 29 years (range approximately 10–45 years) after retirement from professional football. Group-level clinical features and neuropsychological testing results are provided in tables 1 and 2. See tables e-1 and e-2 for individual-level results and appendix e-1 for a representative vignette of each phenotype. Football exposure and concussion history were based on patient/caregiver report as queried by the evaluating neurologist and recorded in the medical records.
Table 1.
Clinical features
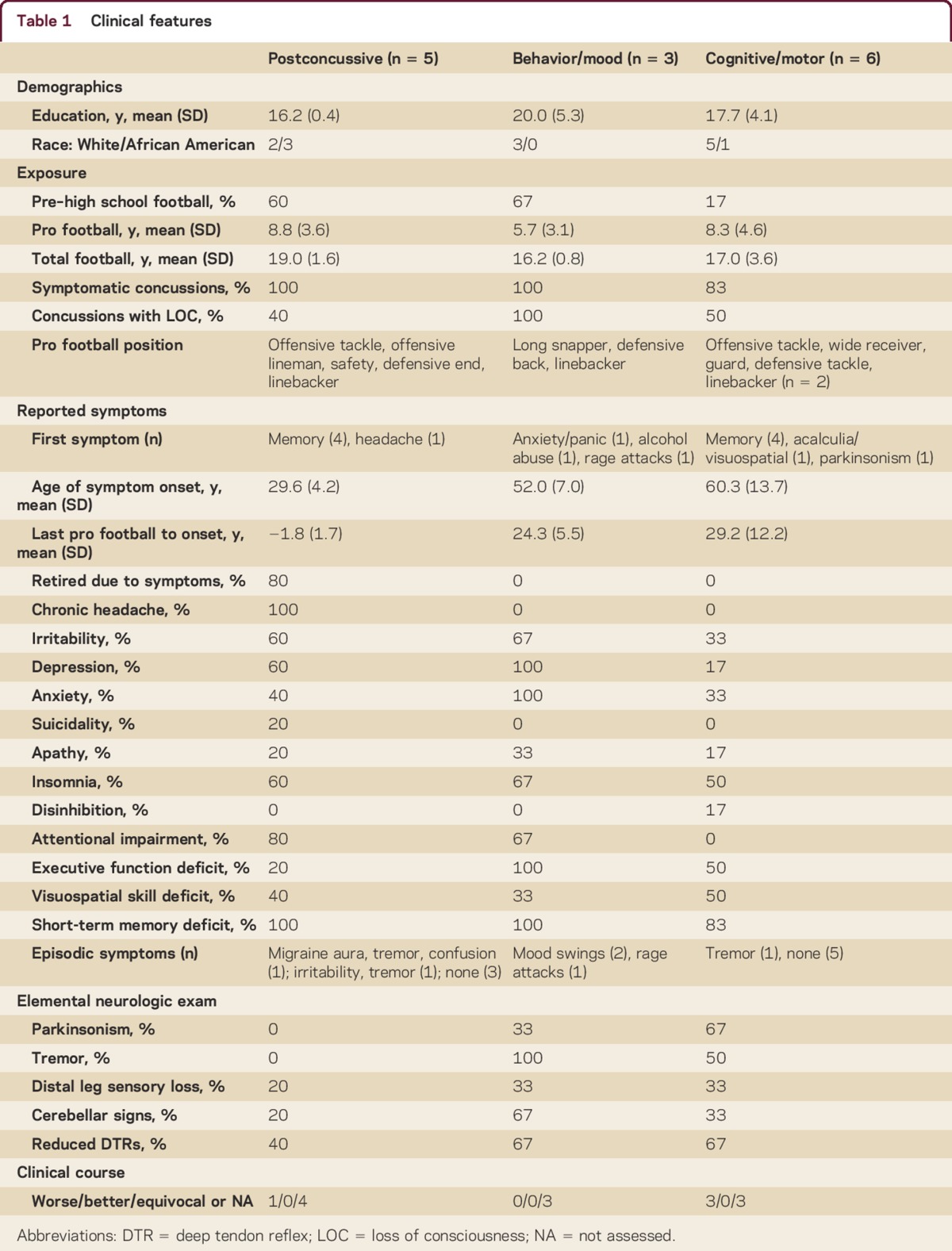
Table 2.
Neuropsychological features
Chronic postconcussive syndrome
Initial presentation
Five patients presented with chronic postconcussive symptoms, including headache, forgetfulness, poor concentration, irritability, depression, apathy, episodic tremor, and insomnia. All patients reported symptom onset during their professional football career. Neuropsychological impairments were observed in all patients, with memory most commonly affected (patients 1, 3, and 5), followed by phonemic fluency (patients 1 and 2), visuospatial dysfunction (patient 1), and confrontation naming (patient 5). Processing speed, set shifting, and design fluency were intact in all patients.
Follow-up
Three patients were followed longitudinally for at least 1 year. One (patient 2) demonstrated dramatic improvement in cognitive and behavioral symptoms following treatment of chronic postconcussive migraine headaches with daily flunarizine and periodic inpatient treatment with dihydroergotamine. After 3 years, however, he again began to show cognitive decline despite migraine control (see vignette in appendix e-1). The other 2 patients reported subjective stability of cognitive and behavioral symptoms, with mixed results on follow-up neuropsychological testing.
Delayed-onset progressive behavior/mood syndrome
Initial presentation
Three patients presented with a delayed-onset progressive behavior/mood syndrome characterized by anxiety, depression, irritability, and poor impulse control manifesting as drug/alcohol abuse and mood swings or violent rage attacks. Symptoms began at age 40s–50s, more than 15 years after retirement from professional football. All were noted to have a fine unilateral or bilateral postural tremor on examination. Neuropsychological impairments included memory and confrontation naming (patient 6), working memory and visuospatial function (patient 7), and working memory and semantic fluency (patient 8). All reported significant depression. Processing speed, set shifting, and design fluency were intact in all patients. At baseline all were diagnosed with mild cognitive impairment (MCI).
Follow-up
One patient (patient 7, figure 1) underwent amyloid PET and was diffusely amyloid-positive. This patient presented with depression, mood swings, and alcohol abuse, had a postural tremor and mild limb dysmetria on examination, and had a CSP on structural MRI. Verbal episodic memory and semantic fluency were intact. The one patient (patient 8) with longitudinal follow-up demonstrated near-complete resolution of frequent explosive rage attacks (physical and verbal violence with postevent amnesia) after treatment with lamotrigine (despite an inconclusive video-EEG telemetry admission) but demonstrated progressive cognitive decline ultimately meeting criteria for mild dementia 3 years after presentation (see vignette in appendix e-1).
Figure 1. FDG PET, amyloid PET, and structural neuroimaging in symptomatic retired professional football players suggest a spectrum of underlying neuropathologies.

Fluorodeoxyglucose (FDG) and amyloid PET imaging were obtained on 5/14 patients (cases 7, 9, 11, 12, and 13 corresponding sequentially to rows A-E). Amyloid PET was obtained using either 11C-Pittsburgh Compound-B (PIB) (A, C, and E) or 18F-florbetapir (AV45) (B and D) amyloid tracer. While all 5 had a cavum septum pellucidum (CSP) and abnormal FDG scans, only 2 were diffusely amyloid positive (A and B). One was focally amyloid positive (C, see red arrow), suggesting a traumatic mechanism, and highlighting the complexity of interpreting amyloid positivity after brain trauma. The other 2 were amyloid negative (D and E), suggesting non-AD neuropathologies. Indeed, one has since undergone autopsy consistent with chronic traumatic encephalopathy (CTE) (figure 2). Aside from CSP, additional structural abnormalities were identified including white matter lesions, cortical atrophy, and mammillary body flattening, the latter of which has been described in advanced cases of CTE. Images are displayed in neurologic orientation. DVR = distribution volume ratio; SUVR = standard uptake value ratio.
Delayed-onset progressive cognitive/motor disorder
Initial presentation
Five patients presented with a delayed-onset progressive cognitive disorder characterized primarily by progressive short-term memory impairment and often accompanied by additional impairments in visuospatial, executive, or language function. One patient (patient 14) presented with a delayed-onset slowly progressive levodopa-responsive parkinsonian syndrome with mild cognitive symptoms that began 5 years after the onset of motor symptoms. Irritability, anxiety, and depression were infrequently reported. Symptoms began at age 40s–70s, more than 25 years after retirement from professional football (except in the case of the predominantly motor syndrome, which began less than 10 years after retirement and progressed very slowly). On neuropsychological testing, all 6 patients exhibited memory deficits suggestive of medial temporal lobe dysfunction. Two patients (patients 9 and 10) exhibited a syndrome suggestive of additional parietal dysfunction with prominent set-shifting, working memory, design fluency, and visuospatial deficits. The patient with parkinsonism (patient 14) exhibited spatial localization and visual memory deficits. At baseline 5 were diagnosed with MCI and 1 met criteria for mild dementia.
Follow-up
Four patients were followed longitudinally and all 4 declined on neuropsychological testing. One (patient 9, figure 1) was diffusely amyloid-positive with a typical AD hypometabolic pattern on FDG-PET, reported subjective improvement in mood and cognition after initiation of donepezil, and remained classified as MCI. Although presenting symptoms were predominantly cognitive, this patient also reported irritability, anxiety, insomnia, and episodic tremor and had a small CSP on structural MRI. The other 3 patients declined functionally, ultimately meeting criteria for dementia, and demonstrated a range of amyloid pathology. Patient 11 showed an unusual region of focal amyloid positivity in the left anterior superior temporal lobe, medial temporal lobe hypometabolism on FDG-PET, and mammillary body flattening and a large CSP on MRI. Patient 12 was amyloid-negative but showed medial temporal and biparietal hypometabolism on FDG-PET and mammillary body flattening and a large CSP on MRI. Patient 13 was amyloid-negative but showed focal hypometabolism in medial temporal regions on FDG-PET and a large CSP on CT. Autopsy revealed tau pathology involving neurons and astrocytes of the hippocampus/medial temporal lobe and brainstem nuclei most consistent with CTE vs primary age-related tauopathy (PART)7 (figure 2 and vignette in appendix e-1). Neuritic plaques typical of AD were not seen.
Figure 2. Brain autopsy of patient 13 revealed tauopathy consistent with chronic traumatic encephalopathy.

Coronal sections of the cerebrum revealed moderate atrophy (hippocampi, entorhinal cortex, posterior frontal cortex) with ex vacuo ventricular enlargement, reduced pigmentation of substantia nigra and locus coeruleus, and absence of the septum pellucidum (A–B). There were tau-positive neurofibrillary tangles (tau is stained brown in C–H) identified in numerous neurons in hippocamal CA3 (C), entorhinal cortex (D), amygdala, subpial temporal cortex (E), and several brainstem nuclei, including the substantia nigra (F), locus coeruleus, dorsal raphe nucleus, superior colliculus, periaqueductal gray, cranial nerve III complex (G), nucleus ambiguus, and dorsal motor nucleus of the vagus. The morphologic features of the tau aggregates ranged from flame-shaped neurofibrillary tangles in cortical and hippocampal neurons to globus neurofibrillary tangles in brainstem neurons. Outside of the above-referenced areas, there were rare clusters of tau protein aggregates in cortical neurons and astrocytes at the depth of the sulcus (H, depicts sulcus in frontal cortex). In addition to the tau pathology, there was prominent TAR DNA-binding protein 43 (TDP-43) proteinopathy (TDP-43 is stained brown in I–J) in the hippocampus CA1 (I), and entorhinal cortex (J), characterized by loss of normal TDP-43 nuclear staining in neurons and scattered TDP-43–positive dystrophic neurites in neuropils. Other contributing neuropathologic features included abundant neurons containing prominent α-synuclein protein aggregates (α-synuclein is stained brown in K–L) in cytoplasm and in dystrophic neurites in substantia nigra (K), ventral tegmental area, locus coeruleus, ventral pontine nuclei, nucleus ambiguus, raphe nucleus, nucleus tractus solitarius, and dorsal motor nucleus of the vagus (L).
Overview of structural neuroimaging findings
There were no contusions, microhemorrhages, fractures, or overt areas of encephalomalacia noted on structural neuroimaging. The most common abnormality was CSP (12/14 patients). Among patients with delayed-onset mood/behavior or cognitive/motor symptoms, 7/9 demonstrated diffuse punctate or patchy T2 hyperintensities in subcortical or periventricular white matter and 7/9 demonstrated cortical atrophy out of proportion to age.
DISCUSSION
In this clinic-based case series, we describe clinical, neuropsychological, and imaging features of a cohort of retired professional American football players presenting with neurobehavioral symptoms. We describe 3 clinical phenotypes but also elucidate subtle phenotypic differences between individuals within each phenotype.
Similar to prior work in autopsy-proven CTE,8 our behavior/mood group was younger and more likely to have anxiety, depression, irritability, and mood swings, whereas the cognitive group was older and more likely to have functional impairment and parkinsonism. Although we chose to categorize players with symptom onset during their career as having “chronic postconcussive syndrome” to highlight the lack of delay between exposure and symptom onset, the progressive course of some of these patients suggests a possible underlying neurodegenerative process and raises the hypothesis that there may be an “early” and “delayed” neurodegenerative phenotype following repeated TBI. While prior studies have reported impairment in executive function,8,9 we found that there was significant variability in this domain. Depression was commonly identified across all groups, consistent with prior reports.10 Mammillary atrophy—frequently reported in advanced cases of CTE in which it is associated with heavy tau burden2—was noted on MRI in 2 of our patients.
CSP has been reported in autopsy series of CTE2 and structural neuroimaging studies of patients exposed to repeated TBI11 and was identified in 86% of our patients. It is hypothesized that the CSP seen in contact sport athletes results from a fluid wave generated within the lateral ventricles that disrupts the septum pellucidum at the time of a traumatic hit, and it may be indicative of a hit that is sufficiently forceful to produce neuropathology.
We describe 7 patients with episodic symptoms including confusional episodes, tremor, mood swings, and rage attacks, suggesting that episodic symptoms deserve further study as potentially distinguishing features in patients with repeated TBI. The rage attacks experienced by patient 8 and the “explosivity” noted in autopsy series of CTE2,8 are suggestive of episodic dyscontrol syndrome, for which TBI is the second most common predisposing risk factor.12 Further research is warranted to determine whether symptoms of episodic dyscontrol are a common feature of CTE or other post-TBI neurodegenerative disorders and whether antiepileptic or mood stabilizing agents are effective interventions, as in our patient who responded to lamotrigine.
We describe 5 patients with chronic postconcussive headache symptoms. Even among patients with 1 mild TBI, chronic headache is common, with up to 58% of patients reporting headache 1 year after injury.13 Evidence-based guidelines for the treatment of postconcussive headaches are sparse, given the lack of Class I randomized controlled treatment trials.14,15 Most practice guidelines recommend treating these headaches according to the headache syndrome they phenotypically resemble (usually migraine or tension-type) while making every effort to select an agent that simultaneously addresses other accompanying symptoms.15
As demonstrated by the range of amyloid PET findings in this case series, interpretation of amyloid imaging in TBI-exposed patients is complex. Among patients who die within 1 hour of severe TBI, 30% have amyloid plaques on autopsy.16 A significant minority of autopsy-proven CTE cases have diffuse amyloid plaques that do not meet neuropathologic criteria for AD.2 While amyloid PET is believed to be most sensitive to the dense neuritic plaques of AD, PiB PET may be sensitive to acute post-TBI amyloid deposition even 11 months post-TBI.17 Further research is needed to determine the sensitivity of amyloid PET to the diffuse amyloid plaques described in CTE. We propose that the pattern of early amyloid deposition may prove helpful for differential diagnosis, as in our case of focal amyloid positivity in the anterior temporal region, a region highly susceptible to trauma but not typically a focus of early amyloid deposition in AD.
The tauopathy identified in patient 13 was most pronounced in the hippocampus/medial temporal lobe and brainstem nuclei with only mild involvement of the frontal cortex—nicely correlating with this patient's memory-predominant clinical presentation. Although this pattern is consistent with CTE, it does not follow the proposed progression of CTE, in which hippocampal/medial temporal lobe neurofibrillary pathology occurs in later stages (stage III) along with more widespread cortical neurofibrillary degeneration.2 This atypical pattern may be due to different tissue preparation techniques (e.g., the thin paraffin sections used in this case may underrepresent perivascular tau deposits). Alternatively, the tauopathy in patient 13 may be considered as PART with a Braak stage III distribution.7 These findings highlight the importance of continuing efforts to clarify clinical-pathologic correlates in these players.
This study is limited by its largely cross-sectional nature, referral bias, lack of biomarker data or autopsy confirmation on all patients, and inability to assign clinical diagnoses of CTE, thus highlighting the importance of continuing efforts to establish consensus diagnostic criteria.18 The 3 major clinical phenotypes presented are based on our clinical impression and need to be verified in larger studies. Coverage of CTE in the press may prompt patients with any degree of football exposure to seek neurologic evaluation, thus more research is warranted regarding the long-term risk and clinical spectrum of patients with elementary school, high school, or college football exposure.
CONCLUSIONS AND RECOMMENDATIONS
There is some evidence that TBI may lead to abnormal neuronal aggregation of multiple proteins, such as amyloid-β, tau, and α-synuclein, that are implicated in an array of neurodegenerative diseases.19 We hypothesize that the specific mechanics of a single or repeated TBI will determine the site of maximal neuronal injury, while additional risk and protective factors will determine the subsequent type and progression of neurodegenerative changes, ultimately producing symptoms when the cognitive, behavioral, or motor reserve has been exhausted.
The physician evaluating a symptomatic retired professional football player should consider any combination of CTE, AD, frontotemporal lobar degeneration, motor neuron disease, or α-synucleinopathy in the differential diagnosis. If AD pathology is suspected, the patient may benefit from treatment with an acetylcholinesterase inhibitor. If parkinsonism is identified, a trial of dopaminergic therapy may be beneficial. And finally, regardless of the underlying neuropathology, symptoms such as chronic headache or episodic dyscontrol should be evaluated and treated aggressively.
Supplementary Material
ACKNOWLEDGMENT
The authors thank our patients and their families for contributing to research on traumatic brain injury and neurodegenerative disease. The authors also thank Dr. William Jagust for PET scanning and acknowledge administrative and technical support from Trishna Subas, Shirley Reeder, Kristen Norton, Dr. James O'Neill, and Dr. Suzanne Baker.
Footnotes
Supplemental data at Neurology.org/cp
AUTHOR CONTRIBUTIONS
R.C. Gardner: design of the study, analysis of the data, drafting and revising the manuscript for intellectual content. K. Possin: design of the study, analysis of the data, drafting and revising the manuscript for intellectual content. C.P. Hess: analysis of the data, revising the manuscript for intellectual content. E.J. Huang: analysis of the data, figure preparation, revising the manuscript for intellectual content. L.T. Grinberg: analysis of the data, revising the manuscript for intellectual content. A.L. Nolan: analysis of the data, revising the manuscript for intellectual content. B.I. Cohn-Sheehy: analysis of the data, revising the manuscript for intellectual content. P.M. Ghosh: revising the manuscript for intellectual content. S. Lanata: revising the manuscript for intellectual content. J. Merrilees: revising the manuscript for intellectual content. J.H. Kramer: revising the manuscript for intellectual content. M.S. Berger: revising the manuscript for intellectual content. B.L. Miller: revising the manuscript for intellectual content. K. Yaffe: revising the manuscript for intellectual content. G.D. Rabinovici: design of the study, analysis of the data, revising the manuscript for intellectual content.
STUDY FUNDING
The following directly funded this work: Alzheimer's Association (GDR), Avid Radiopharmaceuticals (GDR), John Douglas French Alzheimer's Foundation (GDR), Hellman Family Foundation (GDR), UCSF ADRC (P50 AG023501) (GDR, BLM), and State Center grants (BLM). The following indirectly funded this work: Department of Veterans Affairs Advanced Fellowship in Mental Illness Research/Treatment (RCG), UCSF Pepper Center Research Career Development Core (RCG), Tau Consortium (GDR), NIA K23-AG037566 (KP), NIA R01-AG045611 (GDR), NIA K23-AG037566 (KP).
DISCLOSURES
R.C. Gardner has received funding for travel from the American Academy of Neurology and the Alzheimer's Association and has received research support from the Department of Veterans Affairs, NIH/NIA, and UCSF Pepper Center Research Career Development Core. K. Possin has received speaker honoraria from SAFRA (a nonprofit educational program for nurses), serves as a consultant to Acupera, and receives research support from NIH/NIA, Michael J. Fox Foundation, Hellman Family Foundation, and Alzheimer's Association. C.P. Hess serves on the editorial boards of American Journal of Neuroradiology and PLoS ONE; serves as a consultant for Imaging Endpoints; receives research support from General Electric and NIH (NICHD, National Institute of Neurological Disorders and Stroke); and has served as an expert witness in medico-legal proceedings. E.J. Huang serves as an Associate Editor for The Journal of Neuroscience and on the editorial board of Brain Pathology. L.T. Grinberg has received funding for travel from Controversies in Neurology (comtecmed) and speaker honoraria from Israeli Neurological Association; serves as an Associate Editor for Frontiers in Dementia and Cell and Tissue Banking; serves as a consultant for AVID Radiopharmaceuticals; and receives research support from NIH/NIA, John Douglas French Alzheimer's Foundation, Alzheimer's Association, and Rainwater Foundation. A.L. Nolan, B.I. Cohn-Sheehy, and P.M. Ghosh report no disclosures. S. Lanata receives research support from the NIH. J. Merrilees reports no disclosures. J.H. Kramer served as consulting editor for the Journal of the International Neuropsychological Society, receives publishing royalties for California Verbal Learning Test (Pearson, Inc., 2001), and receives research support from the NIH. M.S. Berger serves on a scientific advisory board and receives stock/stock options in Ivivi Health Sciences and serves as a consultant for Plamaco Kinetics. B.L. Miller serves on scientific advisory boards for Tau Consortium, John Douglas French Foundation, The Larry Hillblom Foundation, National Institute for Health Research in Dementia (UK), FasterCures (a center of the Milken Institute), and Tangled Bank Studios; serves as Editor of Neurocase and on the editorial boards of Cambridge University Press and Guilford Publications, Inc.; receives publishing royalties for Behavioral Neurology of Dementia (Cambridge University Press, 2009), Handbook of Neurology (Elsevier, 2009), and The Human Frontal Lobes (Guilford, 2008); and receives research support from NIH/NIA and CMS. K. Yaffe serves on scientific advisory boards for Takeda, Inc., NIH, Beeson, and Alzheimer's Association; serves as an Associate Editor for International Review of Psychiatry and Journal of Gerontology: Medical Sciences and on the editorial board of Journal of Prevention of Alzheimer's Disease; receives research support from NIH (NIA, NIDDK, NIMH, NHLBI), Department of Defense, Department of Veterans Affairs, Bright Focus Foundation, and Alzheimer’s Association; and has participated in medico-legal cases. G.D. Rabinovici has received funding for travel and/or speaker honoraria from Alzheimer's Association, GE Healthcare, Rockpointe, Tau Consortium, and Medscape and receives research support from Avid Radiopharmaceuticals/Eli Lilly, NIH/NIA, Alzheimer's Association, John Douglas French Alzheimer's Foundation, Hellman Family Foundation, and Tau Consortium. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.

REFERENCES
- 1.Fainaru-Wada M, Fainaru S. League of Denial: The NFL, Concussions, and the Battle for Truth. New York, NY: Crown Publishing Group; 2013. [Google Scholar]
- 2.McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013;136:43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lehman EJ, Hein MJ, Baron SL, Gersic CM. Neurodegenerative causes of death among retired National Football League players. Neurology 2012;79:1970–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hazrati LN, Tartaglia MC, Diamandis P, et al. Absence of chronic traumatic encephalopathy in retired football players with multiple concussions and neurological symptomatology. Front Hum Neurosci 2013;7:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA 2011;305:275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rabinovici GD, Rosen HJ, Alkalay A, et al. Amyloid vs FDG-PET in the differential diagnosis of AD and FTLD. Neurology 2011;77:2034–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014;128:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stern RA, Daneshvar DH, Baugh CM, et al. Clinical presentation of chronic traumatic encephalopathy. Neurology 2013;81:1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amen DG, Newberg A, Thatcher R, et al. Impact of playing American professional football on long-term brain function. J Neuropsychiatry Clin Neurosci 2011;23:98–106. [DOI] [PubMed] [Google Scholar]
- 10.Guskiewicz KM, Marshall SW, Bailes J, et al. Recurrent concussion and risk of depression in retired professional football players. Med Sci Sports Exerc 2007;39:903–909. [DOI] [PubMed] [Google Scholar]
- 11.Orrison WW, Hanson EH, Alamo T, et al. Traumatic brain injury: a review and high-field MRI findings in 100 unarmed combatants using a literature-based checklist approach. J Neurotrauma 2009;26:689–701. [DOI] [PubMed] [Google Scholar]
- 12.Elliott FA. Neurological findings in adult minimal brain dysfunction and the dyscontrol syndrome. J Nerv Ment Dis 1982;170:680–687. [DOI] [PubMed] [Google Scholar]
- 13.Lucas S, Hoffman JM, Bell KR, Dikmen S. A prospective study of prevalence and characterization of headache following mild traumatic brain injury. Cephalalgia 2014;34:93–102. [DOI] [PubMed] [Google Scholar]
- 14.Zasler ND. Sports concussion headache. Brain Inj 2015;29:207–220. [DOI] [PubMed] [Google Scholar]
- 15.Watanabe TK, Bell KR, Walker WC, Schomer K. Systematic review of interventions for post-traumatic headache. PM R 2012;4:129–140. [DOI] [PubMed] [Google Scholar]
- 16.Roberts GW, Gentleman SM, Lynch A, Graham DI. beta A4 amyloid protein deposition in brain after head trauma. Lancet 1991;338:1422–1423. [DOI] [PubMed] [Google Scholar]
- 17.Hong YT, Veenith T, Dewar D, et al. Amyloid imaging with carbon 11-labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol 2014;71:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jordan BD. The clinical spectrum of sport-related traumatic brain injury. Nat Rev Neurol 2013;9:222–230. [DOI] [PubMed] [Google Scholar]
- 19.Uryu K, Chen XH, Martinez D, et al. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol 2007;208:185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.