

Abstract
DNA damage may compromise genome integrity and lead to cell death. Cells have evolved a variety of processes to respond to DNA damage including damage repair and tolerance mechanisms, as well as damage checkpoints. The DNA damage tolerance (DDT) pathway promotes the bypass of single-stranded DNA lesions encountered by DNA polymerases during DNA replication. This prevents the stalling of DNA replication. Two mechanistically distinct DDT branches have been characterized. One is translesion synthesis (TLS) in which a replicative DNA polymerase is temporarily replaced by a specialized TLS polymerase that has the ability to replicate across DNA lesions. TLS is mechanistically simple and straightforward, but it is intrinsically error-prone. The other is the error-free template switching (TS) mechanism in which the stalled nascent strand switches from the damaged template to the undamaged newly synthesized sister strand for extension past the lesion. Error-free TS is a complex but preferable process for bypassing DNA lesions. However, our current understanding of this pathway is sketchy. An increasing number of factors are being found to participate or regulate this important mechanism, which is the focus of this editorial.
Keywords: DNA damage tolerance, Template switching, DNA damage bypass, DNA replication, Replicative stress, Translesion synthesis, Ubiquitination, Sumoylation
Core tip: DNA damage may compromise genome integrity and lead to cell death. Cells have evolved a variety of processes to respond to DNA damage including damage repair and tolerance mechanisms. The DNA damage tolerance (DDT) pathway promotes the bypass of single-stranded DNA lesions encountered by DNA polymerases during DNA replication. This prevents the stalling of DNA replication. Two mechanistically distinct DDT branches, translesion synthesis and template switching have been characterized. However, our current understanding of DDT is far from complete and that of template switching is especially sketchy. This editorial focuses on recently identified components and regulators of DDT.
DNA DAMAGE TOLERANCE MECHANISMS
The genomic DNA of a living organism is subject to damage by both internal and external chemical and physical agents, leading to various types of lesions. Cells employ DNA damage repair and tolerance/bypass mechanisms as well as damage checkpoints to respond to these lesions[1]. DNA double-stranded breaks (DSBs) can be repaired by homologous recombination (HR) or non-homologous end joining, while single-stranded DNA (ssDNA) lesions are subject to repair by nucleotide excision repair, base excision repair, or DNA mismatch repair[2-6]. DNA damage such as base modification, if left unrepaired in S phase of the cell cycle, stalls the progression of DNA replication, because the replicative polymerases are not able to recognize the modified bases and use them as template for nucleotide incorporation. The DNA damage tolerance (DDT) mechanism (also known as DNA damage bypass or post-replication repair) enables DNA replication to circumvent the lesions, thereby allowing the completion of DNA replication, and leaving the damages to be repaired later[7,8].
Recent studies indicate that DNA damage makes DNA synthesis on both the leading and lagging strands discontinuous as a result of the uncoupling of DNA polymerase and DNA helicase or the reinitiation of DNA synthesis at a distance from the lesion[9-11]. This creates a ssDNA gap behind the replication fork with the DNA lesion at the 3’ end of the gap (Figure 1A). This ssDNA gap can be filled by two distinct DDT mechanisms. One is translesion synthesis (TLS) in which the replicative DNA polymerase is temporarily replaced by a special TLS polymerase pol ζ or η that can replicate across DNA lesions[12] (Figure 1B). As TLS polymerases lack proofreading activity and have flexible active sites that can recognize modified nucleotides, they may incorporate the wrong nucleotides (Figure 1B). As such, TLS synthesis is potentially mutagenic and error-prone. In fact, TLS is a major source of cellular mutagenesis. The other DDT process is template switching (TS) in which the stalled nascent strand switches temporarily to the newly synthesized undamaged sister strand for replication over the lesion (Figure 1C and D). Therefore, TS is an error-free process. Pairing between the two newly synthesized strands in TS is promoted by strand invasion (Figure 1C). The resulting structure is then turned into a sister chromatid junction (SCJ) after gap filling using the damage-free sister strand as a template (Figure 1D). SCJ is later resolved to yield two duplex DNA strands, completing the damage bypass process (Figure 1E). Note that both TLS and TS take place behind the replication fork, and may happen during or after DNA replication. There is evidence suggesting that TS starts early in S phase, whereas TLS doesn’t start until late S phase[13-15].
Figure 1.
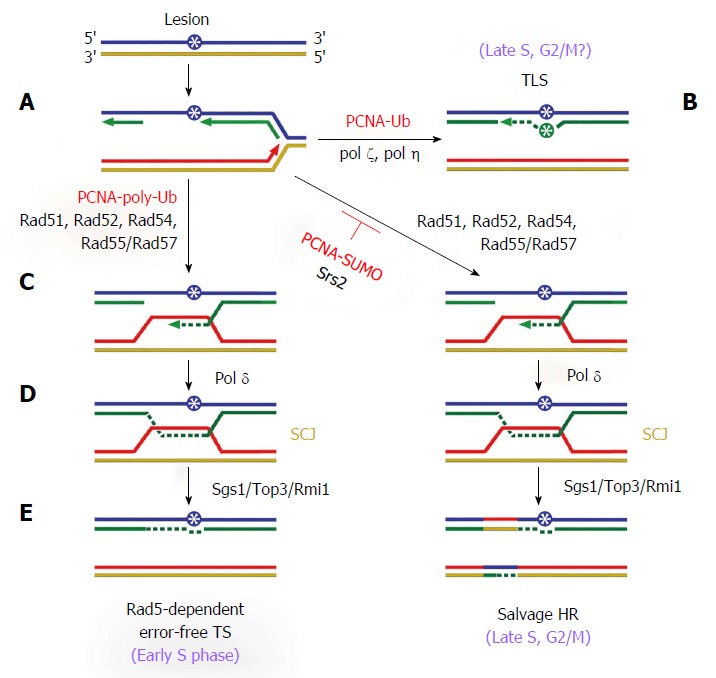
DNA damage tolerance mechanisms. DNA damage (asterisk in blue circle) stalls DNA replication. A: Reinitiation of DNA synthesis results in the formation of a ssDNA gap. Shown is a ssDNA gap on the lagging strand; B: PCNA mono-ubiquitination induces a switch from replicative polymerase to a TLS polymerase Pol ζ or Pol η, resulting in gap filling via TLS, which may incorporate the wrong nucleotide (asterisk in green circle); C-E: PCNA poly-ubiquitination activates the Rad5-dependent error-free TS pathway. Gap filling is achieved by strand invasion mediated by Rad51, Rad52, Rad54, and Rad55/Rad57 and repair synthesis carried out by Pol δ (C), followed by the formation of SCJ (D), and resolution of the SCJ by Sgs1/Top3/Rmi1 (E). DDT can also proceed through the salvage HR pathway that also produces the SCJ intermediate. The salvage pathway is hyper-recombinogenic and prone to crossover as indicated. It is normally inhibited by sumoylated PCNA and Srs2. G2: Gap 2; M: Mitotic; PCNA: Proliferating cell nuclear antigen; S: Synthesis; SUMO: Small ubiquitin-like modifier; TLS: Translesion synthesis; SCJ: Sister chromatid junction; Ub: Ubiquitin.
The sliding DNA clamp PCNA plays a central role in DDT regulation as its post-translational modifications act as a molecular switch to control the choice of DDT pathways[16]. PCNA is a ring-shaped homotrimer that is loaded to primed DNA template by the clamp loader RFC. It encircles DNA to act as a sliding platform for recruiting many factors involved in DNA replication and DNA damage response. For example, PCNA interacts with replicative DNA polymerases (ε and δ) to serve as their processivity factor. DNA damage such as base modification encountered by the replication fork induces the ubiquitination of PCNA[17] (Figure 2). PCNA is first mono-ubiquitinated at lysine 164 (K164) by the E2-E3 ubiquitinase Rad6/Rad18[17], and can be further poly-ubiquitinated via the formation of K63-linked ubiquitin chains by another E2-E3 ubiquitinase Rad5/Ubc13/Mms2[17] (Figure 2). Mono-ubiquitinated PCNA specifically promotes TLS as it preferentially binds TLS polymerase ζ or η, which is believed to facilitate the replacement of replicative polymerases with TLS polymerases[16,18] (Figure 2). On the other hand, poly-ubiquitinated PCNA activates the Rad5-dependent error-free TS pathway (Figure 2), but the underlying mechanism has yet to be elucidated[16].
Figure 2.
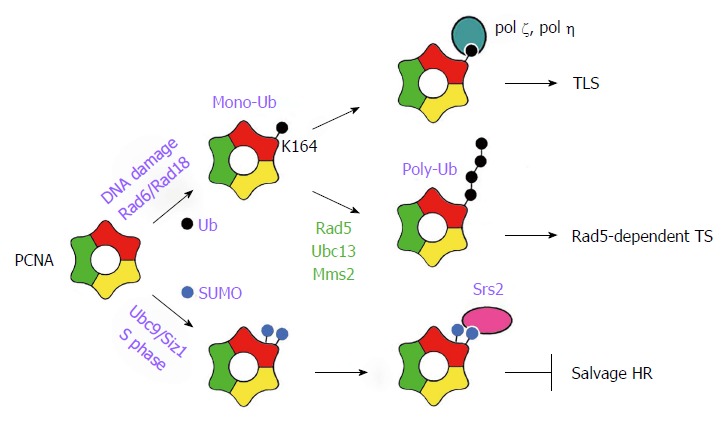
Regulation of DNA damage tolerance by proliferating cell nuclear antigen modification. PCNA is a homotrimer that can be modified at K164 by either ubiquitin or SUMO. It can also be sumoylated at K127. PCNA mono-ubiquitination (mono-Ub) by Rad6/Rad18 facilitates the recruitment of TLS polymerases thereby promoting TLS. Extension of mono-Ub with a K63-linked Ub chain (poly-Ub) by Rad5/Ubc13/Mms2 promotes Rad5-dependent error-free TS pathway. PCNA sumoylation recruits Srs2 that inhibits the salvage HR pathway. HR: Homologous recombination; K: Lysine; PCNA: Proliferating cell nuclear antigen; S: Synthesis; SUMO: Small ubiquitin-like modifier; TLS: Translesion synthesis; TS: Template switching; Ub: Ubiquitin.
COMPONENTS OF THE DDT MACHINERY AND CROSS-TALK BETWEEN DDT AND OTHER DNA DAMAGE RESPONSE MECHANISMS
More and more factors are being implicated in DDT, especially the error-free TS branch of DDT (Table 1). The fact that many of these factors are known to also function in other DNA damage repair and checkpoint mechanisms suggests an extensive crosstalk between DDT and these pathways.
Table 1.
Components and regulators of DNA damage tolerance1
| Factor/complex | Functions/properties |
| Rad6/Rad18 | E2 ubiquitin conjugase/E3 ubiquitin ligase complex; promotes mono-ubiquitination of PCNA at lysine 164 (PCNA-K164) |
| Rad5/Mms2/Ubc13 | E3 ubiquitin ligase/E2 ubiquitin conjugase complex; promotes poly-ubiquitination of PCNA-K164; has ATPase activity |
| Pol30 | Forms a homotrimer (PCNA); its ubiquitination and sumoylation regulates the choice of DDT mechanism |
| Ubc9/Siz1 | E2 SUMO conjugase/E3 SUMO ligase complex; promotes PCNA sumoylation |
| DNA polymerase ζ | TLS polymerase; consists of subunits Rev3, Rev7, Pol31 and Pol32 with Rev3 being the catalytic subunit |
| Rev1 | dCMP transferase; required for TLS by Pol ζ |
| DNA polymerase η (Rad30) | TLS polymerase |
| DNA polymerase δ (Pol3/Pol31/Pol32) | Lagging strand DNA polymerase; Pol3 is the catalytic subunit; promotes repair synthesis in error-free DDT; Pol32 is not essential for DNA replication but is required for repair synthesis |
| Rad51 | Components of HR responsible for homology search and strand invasion; Rad51 molecules bind ssDNA to form a presynaptic filament important for subsequent strand invasion; both Rad55 and Rad57 are paralogs of Rad51; Rad55/Rad57 stabilizes Rad51 nucleoprotein filament and inhibits Srs2 helicase activity |
| Rad52 | |
| Rad54 | |
| Rad55/Rad57 | |
| Srs2 | 3′-5′ helicase and translocase; disrupts Rad51 nucleoprotein filaments; inhibits salvage HR pathway thereby channeling DNA lesions to Rad5 pathway |
| Sgs1/Top3/Rmi1 | Resolves SCJ intermediates generated in error-free DDT |
| RFA (replication factor A) (Rfa1/Rfa2/Rfa3) | ssDNA binding complex; is involved in DNA replication, repair and recombination; RFA-ssDNA nucleofilament helps recruit Rad18 |
| Exo1 | 5’-3’ exonuclease; promotes resection of dsDNA break ends; functions in DNA damage repair, recombination, replication, and telomere integrity; is required for SCJ formation in error-free DDT |
| 9-1-1 complex (Ddc1/Mec3/Rad17) | Ring shaped DNA clamp; is involved in DNA check point signaling; facilitates Pol ζ recruitment; is required for SCJ formation in error-free DDT; interacts with Exo1 |
| MRX complex (Mre11-Rad50-Xrs2) | 3’-5’ exonuclease and ssDNA endonuclease; is potentially involved in both TLS and TS branches of DDT; interacts with Rad18 |
| Shu complex (Shu1/Shu2/ Csm2/Psy3) | Is involved in error-free DDT; interacts with Rad55/Rad57 |
| Ino80 | Chromatin remodeler; associates with replication forks and promotes fork progression; required for Rad18 and Rad51 recruitment to replications forks, PCNA poly-ubiquitination and SCJ formation |
| Elg1 | Unloads PCNA from chromatin during DNA replication; regulates choice of DDT pathway |
| Hmo1 | HMGB homolog; binds ssDNA and DNA with an altered conformation; bends DNA; interacts with Elg1; may help channel DNA lesions to Rad5 pathway; required for SCJ formation in S phase |
| Fun30 | Chromatin remodeler; facilitates long-range resection of dsDNA ends; potentially regulates choice of DDT pathway |
1Listed are DDT factors in S. cerevisiae. ATPase: Adenosine triphosphatase; dCMP: Deoxycytidine monophosphate; DDT: DNA damage tolerance; dsDNA: Double stranded DNA; HMGB: High-mobility group protein B; HR: Homologous recombination; K: Lysine; PCNA: Proliferating cell nuclear antigen; RFA: Replication factor A; S: Synthesis; SCJ: Sister chromatid junction; ssDNA: Single stranded DNA; SUMO: Small ubiquitin-like modifier; TLS: Translesion synthesis; TS: Template switching; Ub: Ubiquitin.
MRX complex has 3’-5’ exonuclease activity and ssDNA endonuclease activity, and is known to act in DSB recognition and processing[19]. Recent epistasis analyses place MRX in both the TLS and TS branches of DDT[20]. Moreover, there is evidence for a physical interaction between MRX and Rad18[20]. These findings suggest that MRX acts early in DDT, likely to process the 3’ end of the ssDNA gap at stalled replication fork and help recruit Rad18[20].
Ino80 chromatin remodeling complex is known to associate with replication forks and aid in fork progression and recovery of stalled forks[21-23]. Ino80 was also found to facilitate Rad18 recruitment to stalled forks, PCNA poly-ubiquitination, Rad51 recruitment and formation of SCJ intermediate of error-free DDT[23]. Therefore, Ino80 plays a role in error-free DDT during DNA replication in S phase. Whether it also plays a role in TLS has not been examined.
Exo1 is a major 5’-3’ nuclease responsible for the resection of dsDNA break ends, and functions in DNA damage repair, recombination, replication, and telomere integrity[24,25]. There is increasing genetic evidence implicating Exo1 and its nuclease activity in error-free TS pathway[20,26-28]. Moreover, Exo1 was found to be required for the formation of SCJ intermediate of error-free DDT[27] .
9-1-1 complex resembles PCNA in assuming a ring structure that can encircle DNA[29]. 9-1-1 has been long established as a DNA damage sensor and a component of checkpoint signaling. It also plays a role in TLS by facilitating the recruitment of TLS polymerase pol ζ[30]. Recent genetic evidence also implicates 9-1-1 in error-free DDT[28]. 9-1-1 is required for SCJ formation[28]. 9-1-1’s function in error-free DDT is separate from its canonical role in G1/S and S phase checkpoint signaling. 9-1-1 is loaded by Rad24 to 5’ end of ssDNA region/gap and is known to associate with many checkpoint and repair proteins[29]. Recently, 9-1-1 was found to also bind Exo1 and stimulate its end resection activity[28,31]. This raises the possibility that 9-1-1 recruits Exo1 to the 5’ end of the ssDNA gap behind stalled replication fork to extend the gap[28].
Error-free DDT is intimately linked to HR as a subset of HR factors has been shown to play roles in it. These include Rad51, Rad52, Rad54, Rad55 and Rad57 that are believed to carry out homology search, strand invasion and pairing of newly synthesized strands from the two sister chromatids[27,32,33]. Rad51 molecules bind the stalled nascent DNA strand to form a presynaptic filament that is later involved in strand invasion[34], and the Rad51 paralogs Rad55 and Rad57 form a complex to facilitate the formation or maintenance of Rad51 filament[35,36].
DNA polymerase δ carries out DNA synthesis for gap filling after the stalled strand switches template during error-free DDT[27]. This results in the formation of a SCJ (Figure 1D) that is resolved by Sgs1/Top3/Rmi1 complex, completing the error-free DDT process[37-39].
SALVAGE HOMOLOGOUS RECOMBINATION PATHWAY OF DDT
Besides the Rad5-dependent error-free DDT pathway, there is an alternative HR-mediated DDT mechanism referred to as the salvage HR pathway that does not depend on PCNA ubiquitination[27,28,40-42]. This pathway, if allowed to act during DNA replication, would generate hyper-recombinogenic intermediates that may lead to chromosome rearrangement and genome instability. The existence of the salvage HR pathway was suggested by the finding that RAD52 is a high copy suppressor of replicative stress-sensitivity of a mutant in which PCNA cannot be ubiquitinated[40]. Consistently, deletion of the “antirecombinase” Srs2 suppresses the sensitivity of cells lacking Rad6 to DNA damage[43]. This suppression is dependent on the HR repair pathway since it is blocked when RAD51, RAD52, RAD54, RAD55 or RAD57 is absent[43]. Moreover, this suppression was found to be specific for Rad5-dependent DDT[40,44-46]. These findings are consistent with the notion that the salvage HR pathway functions independently of the Rad5-dependent error-free TS pathway to bypass DNA lesions, and is normally hindered by Srs2.
Srs2 is a DNA helicase that has the ability to disrupt Rad51-ssDNA nucleofilament, and is thus called an “antirecombinase”[47,48]. The role of Srs2 in inhibiting recombination during replication is closely linked to the sumoylation of PCNA at K164 or K127 by Ubc9/Siz1[40,45]. Unlike PCNA ubiquitination that is induced by DNA damage, PCNA sumoylation is independent of DNA damage[17]. Sumoylation of PCNA is cell cycle dependent as it only occurs in S phase. Srs2 was found to preferentially interact with sumoylated PCNA, leading to the notion that sumoylated PCNA recruits Srs2 to replicating DNA to prevent unwanted recombination during normal DNA replication, and blocks salvage HR pathway in the presence of replicative stress[40,45].
The Rad5-dependent pathway and salvage HR pathway both employ template switching to bypass DNA lesions encountered by the replication fork. As such, it is not surprising that these two pathways share many components such as HR factors Rad51, Rad52, Rad54, Rad55 and Rad57. Both pathways generate SCJ intermediates that are resolved by Sgs1/Top3/Rmi1[27,49]. It was found recently that the 9-1-1 complex is required for SCJ formation in both the Rad5-dependent and salvage HR pathways[28], suggesting that the putative 9-1-1 mediated processing of the ssDNA gap is needed for each pathway.
Despite their mechanistic similarity, the Rad5 and salvage HR pathways clearly differ in that the former is dependent on Rad5/Ubc13/Mms2 mediated PCNA poly-ubiquitination and the latter is not. In addition, they are distinguishable regarding their timing of action in the cell cycle. There is evidence indicating that the Rad5 pathway operates in early S phase, whereas salvage pathway acts in late S or G2/M phase[28,41,49]. This is presumably because the salvage HR pathway is normally kept in check in S phase by sumoylated PCNA and Srs2, and only becomes derepressed when the level of PCNA sumoylation starts to decline in late S phase[28,49,50]. Moreover, the salvage pathway is hyper-recombinogenic and thus has a high propensity to yield chromosomal rearrangement, but the Rad5 pathway is not. This might, at least in part, stem from the fact that salvage pathway acts in the absence of Srs2 while the Rad5 pathway operates in the presence of Srs2. Since Srs2 is known to suppress crossover in HR and promote noncrossover HR[51-53], it is likely that the salvage pathway is prone to crossover and the Rad5 pathway is free of crossover. Lastly, Srs2 has recently been found to also limit D-loop extension, or repair synthesis, during HR and template switching[54], which may also contribute to the prevention of DNA rearrangement in Rad5 dependent error-free DDT.
REGULATION OF THE CHOICE OF DDT PATHWAY
The choice of DDT pathway greatly impacts genome integrity as mutagenesis by TLS and hyper-recombination by salvage HR pathway can lead to the accumulation of harmful mutations and chromosomal rearrangements[55,56]. There is increasing evidence for the existence of multiple factors/mechanisms that ensure the preferential employment of the Rad5-dependent error-free pathway for DNA damage bypass. First, TLS, Rad5 pathway and salvage HR pathway seem to operate within distinct time windows in relation to DNA replication, with Rad5 pathway acting in early S phase, and TLS and salvage HR pathway acting in late S or G2/M phase[13,14,28,41]. Thus Rad5 pathway is the earliest DDT mechanism available to deal with DNA lesions encountered by replication forks (Table 1).
Secondly, Srs2 is believed to channel DNA lesions to Rad5 pathway by inhibiting salvage HR pathway. This is based on the finding that Srs2 deletion (or blocking PCNA sumoylation) suppresses rad5Δ hypersensitivity to DNA damage in a Rad51-dependent fashion[40,44,45]. Since the salvage HR pathway is normally kept in check in S phase by PCNA sumoylation and Srs2, lack of Srs2 function would allow the salvage HR pathway to work ectopically in S phase. Alternatively, or in addition, lack of Ssr2 function increases the “scheduled” function of salvage HR pathway in late S and G2/M phase. Regardless of its possible timing, the derepression of the salvage HR pathway allows rad5Δ cells to effectively bypass DNA lesions and stay viable, albeit at a cost of genome stability.
As Srs2 has a general antirecombinase activity of disrupting Rad51-ssDNA nucleofilament, it should in theory also inhibit the Rad5 pathway as it inhibits the salvage HR pathway. However, Rad5 pathway is apparently not subject to inhibition by Srs2. What then prevents Srs2 from blocking Rad5 pathway? Recent discoveries on potential functions of the Shu complex and Rad55/Rad57 in error-free DDT provided a possible answer to this question. The Shu complex consists of Shu1, Shu2, Psy3 and Csm2, and genetic evidence has implicated it in error-free DNA damage repair and DDT[57-59]. The Csm2 subunit of Shu complex associates with the auxiliary HR factor Rad55/Rad57. Importantly, Rad55/Rad57 promotes the formation of Rad51 nucleofilament and may counter the anti-recombination function of Srs2[35,36,60-62]. It is possible that Shu complex contributes to error-free DDT by recruiting Rad55/Rad57[63]. Shu complex itself may be recruited to stalled DNA replication fork directly or indirectly by PCNA poly-ubiquitination. In short, Shu complex may help prevent Srs2 from inhibiting Rad5-dependent pathway. The notion that Srs2 specifically inhibits the salvage HR pathway is in full accordance with srs2Δ mediated suppression of the hypersensitivity to replicative stress of cells lacking Rad5 pathway[40,44,45]. An alternative explanation for why Rad5 pathway is not subject to inhibition by Srs2 is that ubiquitination of K164 of PCNA by Rad5/Ubc13/Mms2 competes with PCNA-K164 sumoylation that is necessary for Srs2 recruitment[18].
Interestingly, similar to Srs2 deletion, deletion of Elg1, Hmo1 or Fun30 also suppresses hypersensitivity to replicative stress of cells lacking Rad5 pathway, suggesting that these three functionally diverse proteins also regulate the choice of DDT pathway[49,64,65]. Elg1 is homologous to Rfc1, the largest subunit of PCNA loader RFC[16]. Elg1 associates with the other subunits of RFC (Rfc2 thorough Rfc5) to form a RFC like complex (Elg1-RLC) that is known to play a role in genome stability[66]. Elg1 deletion causes an abnormal accumulation of sumoylated and unmodified PCNA on replicating chromatin[64,67]. Elg1 physically interacts with PCNA with a preference for sumoylated PCNA[64]. Elg1-RLC can unload unmodified and sumoylated PCNA from chromatin during DNA replication[64,67]. Given that sumoylated PCNA recruits Srs2, it is not surprising that Srs2 also accumulates on chromatin in the absence of Elg1[64]. Although these findings suggest a role of Elg1 in regulating the error-free DDT pathway by controlling the level of chromatin-associated PCNA, they do not readily provide an explanation of elg1Δ mediated suppression of rad5Δ hypersensitivity to replicative stress.
Hmo1 is a chromatin architecture protein homologous to HMGB (High mobility group box) protein[68]. It preferentially binds ssDNA or DNA with altered conformations, and has the ability to bend DNA[69,70]. Hmo1 was found to physically interact with Elg1[49]. However, genetic evidence suggests Hmo1 to act in parallel with Elg1 (and Srs2) in regulating DDT pathway choice[49]. Hmo1 is suggested to facilitate the channeling of DNA lesions to the Rad5-dependent pathway and prevent the salvage HR pathway and TLS. There is evidence suggesting that the regulatory function of Hmo1 in DDT is dependent on its DNA bending activity[49]. In addition to its potential role in regulating DDT pathway choice, Hmo1 was suggested to function specifically in the Rad5-dependent pathway. This was based on the finding that Hmo1 is required for SCJ formation in S phase, which coincides with the time window of Rad5 pathway, but is not required for SCJ formation in late S or G2/M phase of the cell cycle. It was thought that Hmo1 facilitates SCJ formation by binding and stabilizing sister chromatid bridges and hemicatenanes formed at replication forks with ssDNA gaps[49]. Note that unlike Hmo1, Elg1 and Srs2 are not required for SCJ formation, and are therefore unlikely to be participants of the error-free DDT process[49].
Fun30 is a chromatin remodeler that has been shown to facilitate long-range resection of dsDNA ends that proceeds HR[71-73]. This is believed to underlie its function in promoting DSB repair[71-74]. We obtained evidence that implicates Fun30 in the regulation of DDT[65]. Fun30 deletion suppresses rad5Δ hypersensitivity to replicative stress, which is unlikely due to a reduction in DNA end resection[65]. This suppression is dependent on Rad51 but not TLS or DNA damage repair pathways including nucleotide excision repair, base excision repair and non-homologous end joining[65]. It is possible that Fun30 negatively regulates the salvage HR pathway as Srs2 does.
WORKING MODEL FOR DDT MACHINERY
We incorporate recent findings on DDT described above in a working model as follows. When a DNA lesion stalls the replication fork in S phase, a ssDNA gap forms behind the fork (Figure 1A). RFA then binds ssDNA to form RFA-ssDNA nucleofilament that recruits Rad18, thereby starting the DDT process[11,75]. RFA-ssDNA filament also activates the replication checkpoint[76]. Ino80 chromatin remodeler that normally associates with replication forks to promote fork progression remodels chromatin at stalled replication fork, resulting in a chromatin environment that facilitates Rad18 recruitment. MRX complex may process the ssDNA gap by resecting its 3’ end, and also by helping to recruit Rad18 via physical interaction.
Rad18 recruited to the ssDNA gap then binds Rad6 to form a E2-E3 ubiquitinase that mono-ubiquitinates K164 on PCNA that is associated with the 3’ end of the gap. At this point, mono-ubiquitinated PCNA can lead to TLS or error free TS branches of DDT. On the one hand, mono-ubiquitinated PCNA can promote TLS by facilitating the replacement of replicative polymerases bound to PCNA with TLS polymerases (Figure 1B). 9-1-1 also plays a role in TLS by facilitating the recruitment of TLS polymerase pol ζ. On the other hand, Rad6/Rad18 complex may recruit Rad5/Ubc13/Mms2 E2-E3 ubiquitinase via physical interaction between Rad18 and Rad5[77]. Rad5/Ubc13/Mms2 further ubiquitinates mono-ubiquitinated PCNA. Poly-ubiquitinated PCNA then signals for error-free TS. 9-1-1 loaded to the 5’ end of the ssDNA gap facilitates error-free TS by recruiting Exo1 for gap expansion, which is presumably important for subsequent events in error-free DDT.
Rad51 binds to the stalled nascent DNA strand to form a Rad51-ssDNA presynaptic filament. The extent and stability of this filament is determined by a balance between the opposing actions of Srs2 and Rad55/Rad57. Shu complex may help recruit Rad55/Rad57 and Rad51. With the help of Rad52 and Rad54, the Rad51 presynaptic filament then searches for homology and invades the DNA duplex of the sister chromatid, and pairs with the nascent complementary sister strand (Figure 1C). Hmo1 may facilitate strand invasion by binding and stabilizing sister chromatid bridges and hemicatenanes.
After annealing with its sister strand, the nascent strand from the damaged DNA duplex is extended by replicative DNA polymerase pol δ over the DNA lesion (Figure 1C). This D-loop extension or repair synthesis is limited and transient, as it is hindered by Srs2. After the completion of repair synthesis, the 3’ end of the newly synthesized sequence switches back to the ssDNA gap on the damaged DNA. This effectively fills the ssDNA gap and results in the formation of an SCJ structure (Figure 1D). SCJ is then resolved by Sgs1/Top3/Rmi1 complex to form two duplex DNA strands (Figure 1E), concluding the error-free TS process.
CONCLUSION
More than four decades of studies of DNA damage tolerance/bypass have yielded significant advances in our knowledge of this important process including the characterization of the TLS polymerases and the discovery of PCNA ubiquitination as a molecular switch for TLS and TS pathways. However, the current understanding of DDT especially error-free DDT is far from complete, and many questions remain unanswered: (1) What determines the choice between TLS and Rad5-dependent TS? Specifically, what triggers poly-ubiquitination of mono-ubiquitinated PCNA? What promotes the interaction between Rad5 and Rad18? (2) How PCNA poly-ubiquitination activates downstream TS events? Does it directly recruit a component(s) involved in error-free DDT? (3) Are the three subunits of PCNA subject to independent or coordinated ubiquitination and sumoylation? (4) Does stalling DNA replication induce changes in local chromatin structure that are necessary for error-free DDT? (5) How do Elg1, Hmo1 and Fun30 regulate the choice of DDT pathway? (6) Are recently identified DDT components MRX complex, 9-1-1, Shu complex and Hmo1 directly or indirectly involved in DDT? (7) Does ssDNA gap at stalled replication fork have to be expanded by MRX complex and/or Exo1 for DDT to occur? (8) What determines the putative time windows in the cell cycle in which TLS, Rad5 pathway and salvage HR pathway operate? (9) What prevents the Rad5 pathway from being hyper-recombinogenic? and (10) Does 9-1-1 complex associate with the 5’ end of the ssDNA gap at stalled replication fork and help recruit DDT components such as Exo1? Addressing these questions would help yield a comprehensive understanding of the molecular mechanism and regulation of DDT.
Footnotes
Supported by United States National Science Foundation, No. MCB-1158008.
Conflict-of-interest statement: Xin Bi declares that no conflict of interest exists.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: March 3, 2015
First decision: May 13, 2015
Article in press: August 3, 2015
P- Reviewer: Wang QE S- Editor: Tian YL L- Editor: A E- Editor: Wang CH
References
- 1.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krejci L, Altmannova V, Spirek M, Zhao X. Homologous recombination and its regulation. Nucleic Acids Res. 2012;40:5795–5818. doi: 10.1093/nar/gks270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiruvella KK, Liang Z, Wilson TE. Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol. 2013;5:a012757. doi: 10.1101/cshperspect.a012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krokan HE, Bjørås M. Base excision repair. Cold Spring Harb Perspect Biol. 2013;5:a012583. doi: 10.1101/cshperspect.a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nouspikel T. DNA repair in mammalian cells: Nucleotide excision repair: variations on versatility. Cell Mol Life Sci. 2009;66:994–1009. doi: 10.1007/s00018-009-8737-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 7.Ghosal G, Chen J. DNA damage tolerance: a double-edged sword guarding the genome. Transl Cancer Res. 2013;2:107–129. doi: 10.3978/j.issn.2218-676X.2013.04.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sale JE. Competition, collaboration and coordination--determining how cells bypass DNA damage. J Cell Sci. 2012;125:1633–1643. doi: 10.1242/jcs.094748. [DOI] [PubMed] [Google Scholar]
- 9.Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- 10.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208–219. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 12.Goodman MF, Woodgate R. Translesion DNA polymerases. Cold Spring Harb Perspect Biol. 2013;5:a010363. doi: 10.1101/cshperspect.a010363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waters LS, Walker GC. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase. Proc Natl Acad Sci USA. 2006;103:8971–8976. doi: 10.1073/pnas.0510167103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lang GI, Murray AW. Mutation rates across budding yeast chromosome VI are correlated with replication timing. Genome Biol Evol. 2011;3:799–811. doi: 10.1093/gbe/evr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daigaku Y, Davies AA, Ulrich HD. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 2010;465:951–955. doi: 10.1038/nature09097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 17.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 18.Stelter P, Ulrich HD. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003;425:188–191. doi: 10.1038/nature01965. [DOI] [PubMed] [Google Scholar]
- 19.D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol. 2002;3:317–327. doi: 10.1038/nrm805. [DOI] [PubMed] [Google Scholar]
- 20.Ball LG, Hanna MD, Lambrecht AD, Mitchell BA, Ziola B, Cobb JA, Xiao W. The Mre11-Rad50-Xrs2 complex is required for yeast DNA postreplication repair. PLoS One. 2014;9:e109292. doi: 10.1371/journal.pone.0109292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimada K, Oma Y, Schleker T, Kugou K, Ohta K, Harata M, Gasser SM. Ino80 chromatin remodeling complex promotes recovery of stalled replication forks. Curr Biol. 2008;18:566–575. doi: 10.1016/j.cub.2008.03.049. [DOI] [PubMed] [Google Scholar]
- 22.Vincent JA, Kwong TJ, Tsukiyama T. ATP-dependent chromatin remodeling shapes the DNA replication landscape. Nat Struct Mol Biol. 2008;15:477–484. doi: 10.1038/nsmb.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falbo KB, Alabert C, Katou Y, Wu S, Han J, Wehr T, Xiao J, He X, Zhang Z, Shi Y, et al. Involvement of a chromatin remodeling complex in damage tolerance during DNA replication. Nat Struct Mol Biol. 2009;16:1167–1172. doi: 10.1038/nsmb.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tran PT, Erdeniz N, Symington LS, Liskay RM. EXO1-A multi-tasking eukaryotic nuclease. DNA Repair (Amst) 2004;3:1549–1559. doi: 10.1016/j.dnarep.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 25.Mimitou EP, Symington LS. DNA end resection--unraveling the tail. DNA Repair (Amst) 2011;10:344–348. doi: 10.1016/j.dnarep.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tran PT, Fey JP, Erdeniz N, Gellon L, Boiteux S, Liskay RM. A mutation in EXO1 defines separable roles in DNA mismatch repair and post-replication repair. DNA Repair (Amst) 2007;6:1572–1583. doi: 10.1016/j.dnarep.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vanoli F, Fumasoni M, Szakal B, Maloisel L, Branzei D. Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genet. 2010;6:e1001205. doi: 10.1371/journal.pgen.1001205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karras GI, Fumasoni M, Sienski G, Vanoli F, Branzei D, Jentsch S. Noncanonical role of the 9-1-1 clamp in the error-free DNA damage tolerance pathway. Mol Cell. 2013;49:536–546. doi: 10.1016/j.molcel.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 29.Eichinger CS, Jentsch S. 9-1-1: PCNA’s specialized cousin. Trends Biochem Sci. 2011;36:563–568. doi: 10.1016/j.tibs.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Sabbioneda S, Minesinger BK, Giannattasio M, Plevani P, Muzi-Falconi M, Jinks-Robertson S. The 9-1-1 checkpoint clamp physically interacts with polzeta and is partially required for spontaneous polzeta-dependent mutagenesis in Saccharomyces cerevisiae. J Biol Chem. 2005;280:38657–38665. doi: 10.1074/jbc.M507638200. [DOI] [PubMed] [Google Scholar]
- 31.Ngo GH, Balakrishnan L, Dubarry M, Campbell JL, Lydall D. The 9-1-1 checkpoint clamp stimulates DNA resection by Dna2-Sgs1 and Exo1. Nucleic Acids Res. 2014;42:10516–10528. doi: 10.1093/nar/gku746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prakash S, Sung P, Prakash L. DNA repair genes and proteins of Saccharomyces cerevisiae. Annu Rev Genet. 1993;27:33–70. doi: 10.1146/annurev.ge.27.120193.000341. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H, Lawrence CW. The error-free component of the RAD6/RAD18 DNA damage tolerance pathway of budding yeast employs sister-strand recombination. Proc Natl Acad Sci USA. 2005;102:15954–15959. doi: 10.1073/pnas.0504586102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holthausen JT, Wyman C, Kanaar R. Regulation of DNA strand exchange in homologous recombination. DNA Repair (Amst) 2010;9:1264–1272. doi: 10.1016/j.dnarep.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 35.Liu J, Renault L, Veaute X, Fabre F, Stahlberg H, Heyer WD. Rad51 paralogues Rad55-Rad57 balance the antirecombinase Srs2 in Rad51 filament formation. Nature. 2011;479:245–248. doi: 10.1038/nature10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Godin S, Wier A, Kabbinavar F, Bratton-Palmer DS, Ghodke H, Van Houten B, VanDemark AP, Bernstein KA. The Shu complex interacts with Rad51 through the Rad51 paralogues Rad55-Rad57 to mediate error-free recombination. Nucleic Acids Res. 2013;41:4525–4534. doi: 10.1093/nar/gkt138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta-Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 2005;19:339–350. doi: 10.1101/gad.322605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernstein KA, Shor E, Sunjevaric I, Fumasoni M, Burgess RC, Foiani M, Branzei D, Rothstein R. Sgs1 function in the repair of DNA replication intermediates is separable from its role in homologous recombinational repair. EMBO J. 2009;28:915–925. doi: 10.1038/emboj.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cejka P, Plank JL, Dombrowski CC, Kowalczykowski SC. Decatenation of DNA by the S. cerevisiae Sgs1-Top3-Rmi1 and RPA complex: a mechanism for disentangling chromosomes. Mol Cell. 2012;47:886–896. doi: 10.1016/j.molcel.2012.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pfander B, Moldovan GL, Sacher M, Hoege C, Jentsch S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature. 2005;436:428–433. doi: 10.1038/nature03665. [DOI] [PubMed] [Google Scholar]
- 41.Branzei D, Vanoli F, Foiani M. SUMOylation regulates Rad18-mediated template switch. Nature. 2008;456:915–920. doi: 10.1038/nature07587. [DOI] [PubMed] [Google Scholar]
- 42.Minca EC, Kowalski D. Multiple Rad5 activities mediate sister chromatid recombination to bypass DNA damage at stalled replication forks. Mol Cell. 2010;38:649–661. doi: 10.1016/j.molcel.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schiestl RH, Prakash S, Prakash L. The SRS2 suppressor of rad6 mutations of Saccharomyces cerevisiae acts by channeling DNA lesions into the RAD52 DNA repair pathway. Genetics. 1990;124:817–831. doi: 10.1093/genetics/124.4.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Broomfield S, Xiao W. Suppression of genetic defects within the RAD6 pathway by srs2 is specific for error-free post-replication repair but not for damage-induced mutagenesis. Nucleic Acids Res. 2002;30:732–739. doi: 10.1093/nar/30.3.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Papouli E, Chen S, Davies AA, Huttner D, Krejci L, Sung P, Ulrich HD. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell. 2005;19:123–133. doi: 10.1016/j.molcel.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 46.Marini V, Krejci L. Srs2: the “Odd-Job Man” in DNA repair. DNA Repair (Amst) 2010;9:268–275. doi: 10.1016/j.dnarep.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Le Cam E, Fabre F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature. 2003;423:309–312. doi: 10.1038/nature01585. [DOI] [PubMed] [Google Scholar]
- 48.Antony E, Tomko EJ, Xiao Q, Krejci L, Lohman TM, Ellenberger T. Srs2 disassembles Rad51 filaments by a protein-protein interaction triggering ATP turnover and dissociation of Rad51 from DNA. Mol Cell. 2009;35:105–115. doi: 10.1016/j.molcel.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gonzalez-Huici V, Szakal B, Urulangodi M, Psakhye I, Castellucci F, Menolfi D, Rajakumara E, Fumasoni M, Bermejo R, Jentsch S, et al. DNA bending facilitates the error-free DNA damage tolerance pathway and upholds genome integrity. EMBO J. 2014;33:327–340. doi: 10.1002/embj.201387425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karras GI, Jentsch S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell. 2010;141:255–267. doi: 10.1016/j.cell.2010.02.028. [DOI] [PubMed] [Google Scholar]
- 51.Ira G, Malkova A, Liberi G, Foiani M, Haber JE. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell. 2003;115:401–411. doi: 10.1016/s0092-8674(03)00886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robert T, Dervins D, Fabre F, Gangloff S. Mrc1 and Srs2 are major actors in the regulation of spontaneous crossover. EMBO J. 2006;25:2837–2846. doi: 10.1038/sj.emboj.7601158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miura T, Shibata T, Kusano K. Putative antirecombinase Srs2 DNA helicase promotes noncrossover homologous recombination avoiding loss of heterozygosity. Proc Natl Acad Sci USA. 2013;110:16067–16072. doi: 10.1073/pnas.1303111110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burkovics P, Sebesta M, Sisakova A, Plault N, Szukacsov V, Robert T, Pinter L, Marini V, Kolesar P, Haracska L, et al. Srs2 mediates PCNA-SUMO-dependent inhibition of DNA repair synthesis. EMBO J. 2013;32:742–755. doi: 10.1038/emboj.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shor E, Weinstein J, Rothstein R. A genetic screen for top3 suppressors in Saccharomyces cerevisiae identifies SHU1, SHU2, PSY3 and CSM2: four genes involved in error-free DNA repair. Genetics. 2005;169:1275–1289. doi: 10.1534/genetics.104.036764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ball LG, Zhang K, Cobb JA, Boone C, Xiao W. The yeast Shu complex couples error-free post-replication repair to homologous recombination. Mol Microbiol. 2009;73:89–102. doi: 10.1111/j.1365-2958.2009.06748.x. [DOI] [PubMed] [Google Scholar]
- 59.Mankouri HW, Ngo HP, Hickson ID. Shu proteins promote the formation of homologous recombination intermediates that are processed by Sgs1-Rmi1-Top3. Mol Biol Cell. 2007;18:4062–4073. doi: 10.1091/mbc.E07-05-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sung P. Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes Dev. 1997;11:1111–1121. doi: 10.1101/gad.11.9.1111. [DOI] [PubMed] [Google Scholar]
- 61.Bernstein KA, Reid RJ, Sunjevaric I, Demuth K, Burgess RC, Rothstein R. The Shu complex, which contains Rad51 paralogues, promotes DNA repair through inhibition of the Srs2 anti-recombinase. Mol Biol Cell. 2011;22:1599–1607. doi: 10.1091/mbc.E10-08-0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sasanuma H, Tawaramoto MS, Lao JP, Hosaka H, Sanda E, Suzuki M, Yamashita E, Hunter N, Shinohara M, Nakagawa A, et al. A new protein complex promoting the assembly of Rad51 filaments. Nat Commun. 2013;4:1676. doi: 10.1038/ncomms2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu X, Ball L, Chen W, Tian X, Lambrecht A, Hanna M, Xiao W. The yeast Shu complex utilizes homologous recombination machinery for error-free lesion bypass via physical interaction with a Rad51 paralogue. PLoS One. 2013;8:e81371. doi: 10.1371/journal.pone.0081371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Parnas O, Zipin-Roitman A, Pfander B, Liefshitz B, Mazor Y, Ben-Aroya S, Jentsch S, Kupiec M. Elg1, an alternative subunit of the RFC clamp loader, preferentially interacts with SUMOylated PCNA. EMBO J. 2010;29:2611–2622. doi: 10.1038/emboj.2010.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bi X, Yu Q, Siler J, Li C, Khan A. Functions of Fun30 chromatin remodeler in regulating cellular resistance to genotoxic stress. PLoS One. 2015;10:e0121341. doi: 10.1371/journal.pone.0121341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aroya SB, Kupiec M. The Elg1 replication factor C-like complex: a novel guardian of genome stability. DNA Repair (Amst) 2005;4:409–417. doi: 10.1016/j.dnarep.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 67.Kubota T, Nishimura K, Kanemaki MT, Donaldson AD. The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol Cell. 2013;50:273–280. doi: 10.1016/j.molcel.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 68.Lu J, Kobayashi R, Brill SJ. Characterization of a high mobility group 1/2 homolog in yeast. J Biol Chem. 1996;271:33678–33685. doi: 10.1074/jbc.271.52.33678. [DOI] [PubMed] [Google Scholar]
- 69.Kamau E, Bauerle KT, Grove A. The Saccharomyces cerevisiae high mobility group box protein HMO1 contains two functional DNA binding domains. J Biol Chem. 2004;279:55234–55240. doi: 10.1074/jbc.M409459200. [DOI] [PubMed] [Google Scholar]
- 70.Bauerle KT, Kamau E, Grove A. Interactions between N- and C-terminal domains of the Saccharomyces cerevisiae high-mobility group protein HMO1 are required for DNA bending. Biochemistry. 2006;45:3635–3645. doi: 10.1021/bi0522798. [DOI] [PubMed] [Google Scholar]
- 71.Costelloe T, Louge R, Tomimatsu N, Mukherjee B, Martini E, Khadaroo B, Dubois K, Wiegant WW, Thierry A, Burma S, et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature. 2012;489:581–584. doi: 10.1038/nature11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen X, Cui D, Papusha A, Zhang X, Chu CD, Tang J, Chen K, Pan X, Ira G. The Fun30 nucleosome remodeller promotes resection of DNA double-strand break ends. Nature. 2012;489:576–580. doi: 10.1038/nature11355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eapen VV, Sugawara N, Tsabar M, Wu WH, Haber JE. The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol Cell Biol. 2012;32:4727–4740. doi: 10.1128/MCB.00566-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neves-Costa A, Will WR, Vetter AT, Miller JR, Varga-Weisz P. The SNF2-family member Fun30 promotes gene silencing in heterochromatic loci. PLoS One. 2009;4:e8111. doi: 10.1371/journal.pone.0008111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Davies AA, Huttner D, Daigaku Y, Chen S, Ulrich HD. Activation of ubiquitin-dependent DNA damage bypass is mediated by replication protein a. Mol Cell. 2008;29:625–636. doi: 10.1016/j.molcel.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 77.Ulrich HD, Jentsch S. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. EMBO J. 2000;19:3388–3397. doi: 10.1093/emboj/19.13.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]