

Abstract
Endogenous glucocorticoids regulate a variety of physiologic processes and are crucial to the systemic stress response. Glucocorticoid receptors are expressed throughout the body, but there is considerable heterogeneity in glucocorticoid sensitivity and induced biological responses across tissues. The immunoregulatory properties of glucocorticoids are exploited in the clinic for the treatment of inflammatory and autoimmune disorders as well as certain hematological malignancies, but adverse side effects hamper prolonged use. Fully understanding the molecular events that shape the physiologic effects of glucocorticoid treatment will provide insight into optimal glucocorticoid treatments, reliable assessment of glucocorticoid sensitivity in patients, and may advance the development of novel GR agonists that exert immunosuppressive effects while avoiding harmful side effects. In this review, we provide an overview of mechanisms that affect glucocorticoid specificity and sensitivity in health and disease, focusing on the distinct isoforms of the glucocorticoid receptor and their unique regulatory and functional properties.
Keywords: glucocorticoid, glucocorticoid receptor, glucocorticoid signaling, hypothalamic-pituitary-adrenal axis, isoforms
A. Introduction
Members of the glucocorticoid family, or “corticosteroids,” have become a clinical mainstay for the treatment of numerous inflammatory and autoimmune disorders, with ~1% of adults in the UK treated with oral glucocorticoids at any given time [1], and an estimated total market size of $10 billion per year [2]. Although the immunoregulatory properties of glucocorticoids are commonly exploited for pharmaceutical use, endogenous glucocorticoids play crucial roles in reestablishing homeostasis in response to physical or psychological stress. Moreover, glucocorticoids play important roles in the development and function of many organ systems including the cardiovascular system, central nervous system, immune system, and gastrointestinal system, and participate in nearly every cellular and molecular network. Microarray studies have revealed that a substantial portion of the genome is targeted by glucocorticoid signaling through its receptor, with some studies indicating that up to 20% of expressed genes are positively or negatively regulated by glucocorticoids [3]. Perhaps, it should not come as a surprise then that the clinical use of glucocorticoids - especially at high doses or for protracted periods of time - is hindered by adverse side effects, including metabolic disorders, osteoporosis, skin atrophy, cushingoid appearance, and growth retardation [2]. Additionally, the effectiveness of glucocorticoid treatment varies among individuals as a result of genetic polymorphisms and/or disease-related activity. Understanding the molecular mechanisms behind glucocorticoid signaling will provide important insight into strategies for: 1.) personalizing glucocorticoid therapies that maximize benefits while minimizing adverse effects, and 2.) developing novel selective glucocorticoid receptor agonists (“SERGRAs”) that exhibit immunosuppressive properties but minimal side effects.
B. Regulation of glucocorticoid synthesis and bioavailability
Glucocorticoids are hormones of the steroid family, which also includes the mineralocorticoids, androgens, estrogens, and progestagens. Steroid hormones are chemically similar and are based on a cyclopentanophenanthrene 4-ring structure. Biologically active steroids are generated from cholesterol through a multienzyme process termed steroidogenesis [4]. The class of steroid produced by distinct endocrine glands is determined by the combination of enzymes and cofactors expressed by the hormone-producing cell. The key enzymes for glucocorticoid synthesis include 17 hydroxylase, 3β-hydroxysteroid dehydrogenase, 21-hydroxylase, and 11β hydroxylase.
Glucocorticoids and the chemically similar mineralocorticoid (aldosterone) are products of the adrenal glands, and are synthesized in the zona fasciculata and the zona glomerulosa, respectively. In humans, the biologically active glucocorticoid is cortisol, whereas corticosterone is the principal mediator of glucocorticoid activity in rodents. Adrenal production of glucocorticoids is regulated by the hypothalamic-pituitary-adrenal (HPA) axis (Fig. 1). The hypothalamus responds to neural, cytokine, and endocrine signals by secreting corticotropin-releasing hormone (CRH) which stimulates adrenocorticotropic hormone (ACTH) release from the anterior pituitary [5]. ACTH induces glucocorticoid synthesis in the adrenal glands and subsequent secretion into the circulation, resulting in systemic effects. Notably, increases in circulating cortisol feed back on the hypothalamus and pituitary, reducing expression of CRH and ACTH, respectively. Thus, the HPA axis operates as a classic negative feedback loop (Fig. 1).
Figure 1.

Regulation of endogenous glucocorticoid synthesis by the hypothalamic-pituitary-adrenal axis in humans. CRH, corticotropin-releasing hormone; ACTH, adrenocorticotropic hormone.
The HPA axis is linked to the circadian clock, thereby coupling glucocorticoid synthesis to diurnal patterns. In humans, blood cortisol concentrations peak in the morning, gradually decrease through the day, and are lowest at night. Oscillations in cortisol secretion are important to bodily processes associated with activity level, as evidenced by signs of hypercortisolism in association with altered daily rhythms (resulting from shift work or trans-time zone travel, for example) [6]. Notably, nocturnal mice and rats, which are commonly used in glucocorticoid research, exhibit an inverse tempo in glucocorticoid profile, with peak plasma levels of corticosterone at night and decreased concentrations during the day. The HPA axis also functions as a component of the systemic stress system, coupling the detection of physical or emotional stress to glucocorticoid release. The HPA axis is also stimulated by pro-inflammatory cytokines, including IL-1, TNFα, IL-6, and the type I interferons (IFNα and IFNβ) [7], which are released in response to pathogens and/or tissue injury. Importantly, cytokine-induced release of cortisol blunts the subsequent inflammatory and immune response, completing a regulatory feedback loop. The importance of HPA regulation is highlighted by the clinical phenotypes associated with overproduction or underproduction of glucocorticoids, which manifest as Cushing’s syndrome and Addison’s disease (adrenal insufficiency), respectively.
Glucocorticoid synthesis plays an important role in the driving systemic cortisol effects, but cortisol activity and availability are also regulated post-secretion and often at the tissue or cellular level [8]. In healthy individuals, ~80–90% of circulating cortisol is bound to corticosteroid-binding globulin (CBG) and ~5–15% is bound to albumin, rendering cortisol in an inactive form; only ~5% of circulating cortisol is bioactive. Thus, the bioavailability of cortisol depends on CBG concentration, with relatively high levels of CBG serving as a buffer against cortisol surges. Glucocorticoid activity is regulated at the cellular level by enzymes of the 11β-hydroxysteroid dehydrogenase (11β-HSD) family. The two members of this family, 11β-HSD1 and 11β-HSD2, regulate the conversion of bioactive cortisol into the inactive precursor cortisone. Under physiologic conditions, 11β-HSD1 favors the conversion of cortisol from cortisone, thereby increasing local glucocorticoid activity beyond that dictated by circulating cortisol levels [9]. 11β-HSD2, in contrast, catalyzes cortisol to cortisone, thereby reducing glucocorticoid activity in cells expressing this enzyme. The expression of 11β-HSD2 is crucial to the proper function of mineralocorticoid-sensitive organs, including the kidneys and pancreas, since cortisol has the capacity to bind and activate the mineralocorticoid receptor; 11β-HSD2 enforces specificity of the mineralocorticoid receptor for aldosterone. Thus, the balance of 11β-HSD1 and 11β-HSD2 expression regulates glucocorticoid activity at the single cell level, and a growing body of evidence indicates that 11β-HSD genes are targets of cytokine signals, with 11β-HSD1 expression enhanced and 11β-HSD2 expression decreased at sites of inflammation [10, 11].
C. The glucocorticoid receptor
Glucocorticoid effects are mediated by the glucocorticoid receptor (GR), the founding member of the nuclear receptor superfamily. GR is a product of a single gene but multiple isoforms are generated through alternative splicing and alternate translation initiation sites. Moreover, the GR protein is a target for posttranslational modifications, including phosphorylation, sumoylation, ubiquitination, and acetylation, which alter GR function [12] (Fig. 2). A growing body of evidence indicates that the sensitivity and specificity of glucocorticoid signaling result, at least in part, from the molecular heterogeneity of GR proteins.
Figure 2.
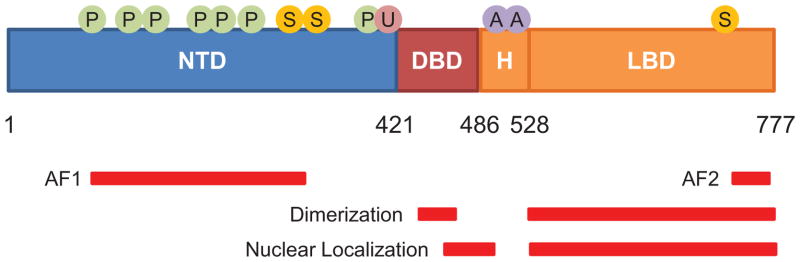
GR structural domains and sites of posttranslational modification. Numbers indicate position of amino acid residues in human GR. The N-terminal transactivation domain (NTD), DNA-binding domain (DBD), hinge region (H), and ligand-binding domain (LBD) for GR are shown. Regions associated with transactivation (AF1 and AF2), receptor dimerization, and nuclear localization are shown by red bars. Also shown are residues subject to translational modification by phosphorylation (P), sumoylation (S), ubiquitination (U), and acetylation (A).
The GR protein comprises three domains: an N-terminal transactivation domain (NTD), a central DNA-binding domain (DBD), and a C-terminal ligand-binding domain (LBD) (Fig. 2)[13]. The NTD contains most sites for posttranslational modification and possesses one of two regions of activation function (AF1); this portion of the protein interacts with coregulators and the transcriptional machinery. The DBD, which is highly conserved across members of the nuclear receptor family, contains two zinc-finger motifs which target the glucocorticoid receptor to DNA. The LBD contains a hydrophobic pocket for glucocorticoid binding, and an AF2 domain for ligand-dependent interactions with coregulators. A flexible hinge region (H) separates the DBD and LBD. GR contains two nuclear localization signals, NL1 and NL2, which are situated at the DBD-H border and within the LBD, respectively.
1. Signaling through the glucocorticoid receptor
In the absence of ligand, GR predominantly localizes to the cytoplasm in a multiprotein complex with chaperones (hsp90, hsp70), members of the FK506 family of immunophilins (FKBP51 and FKBP52), and non-receptor tyrosine kinases including c-Src [14–16]. In this complex, GR is inactive as a transcription factor but exhibits high affinity for glucocorticoid ligands. Ligand binding to GR induces a conformational change that dissociates the multiprotein complex. The effects of ligand-induced dissociation of the GR multiprotein complex are two-fold: 1.) translocation of GR into the nucleus to exert genomic effects, and 2.) liberation of components of the multiprotein complex that integrate into signaling pathways as rapid non-genomic effects (Fig. 3).
Figure 3.

Pathways of glucocorticoid signaling through the GR. GR ligands induce biological changes by binding the GR and inducing genomic and non-genomic effects. Genomic effects of activated GR occur following nuclear translocation and manifest through 3 primary mechanisms: direct binding of GR to DNA via GREs and nGREs to activate or repress transcription (A), tethering to DNA-bound transcription factors to modulate transcription indirectly (B), or composite activity of DNA binding and interaction with adjacent DNA-bound transcription factors to affect transcription (C). Rapid non-genomic effects of GR ligation occur following ligand-induced dissociation of the GR multiprotein complex in the cytoplasm. BTM, basal transcription machinery.
Nuclear translocation of GR results in genomic effects by enhancing or repressing target gene expression. Transactivation is a form of transcriptional regulation in which homodimers of ligand-bound GR bind to sequences of DNA called glucocorticoid-responsive elements (GREs) (Fig. 3). The GRE consensus sequence is GGAACAnnnTGTTCT, an imperfect palindrome containing two half sites with a critical three basepair spacer. One subunit of each GR homodimer binds to each half-site. GREs can be found in the promoters, introns, or exons of target genes, and GR occupation recruits chromatin-remodeling complexes and coregulators that regulate transcription by RNA polymerase II [17–19]. Transcriptional regulation by GR is also mediated by coactivators, including the histone acetyltransferase CBP/p300, the nuclear methylase coactivator-associated arginine methyltransferase, and steroid receptor coactivators. While GR-GRE interactions have been traditionally associated with gene activation, genome-wide analyses have revealed that GR binding to canonical GREs does not necessarily enhance gene transcription and, rather, can lead to suppression of many target genes [20]. Thus, the transcriptional polarity of GR-GRE binding likely depends on regulatory elements outside of the GRE. Recent studies have also revealed that GR can repress transcription by binding to negative glucocorticoid-responsive elements (nGREs) in target gene promoters [21] (Fig. 3).
This form of regulation occurs through two GR-ligand monomers binding to a nGRE, another palindromic sequence [consensus sequence: CTCC(n)0–2GGAGA] with a shorter and variable spacer of 0–2 basepairs [22]. Although GREs and nGREs are abundant in the genome, global GR recruitment assays reveal that, in any given cell, few GREs are actually occupied by GR [23]. Moreover, the distribution of GR binding to the genome differs substantially across tissues, indicating that chromatin structure dictates which GREs are accessible to GR, contributing to the heterogeneity of cellular responses to glucocorticoids.
Activated GR also has the capacity to associate with or “tether” to another transcription factor bound to its DNA response element, thereby regulating its transcriptional activity. Notably, this form of gene regulation does not require GR to interact with DNA directly. In other cases, however, GR binds to DNA via a GRE while also associating with an adjacent DNA-bound transcription factor, affecting its transcriptional activity in a composite manner (Fig. 3). The most well-studied examples of these forms of regulation are GR-mediated repression of the pro-inflammatory transcription factors NF-κB and AP-1 [24]. Binding of GR to the p65 subunit of NF-κB suppresses induction of pro-inflammatory genes by multiple mechanisms, including interference with coactivators [25] and hindrance of transcriptional machinery [26]. GR also modulates the activity of certain members of the signal transducer and activator of transcription (STAT) family through tethering and composite regulation [27] (Fig. 3).
Additional studies have implicated nongenomic effects of GR activation in rapid cellular responses to glucocorticoids [28]. Nongenomic effects of glucocorticoids do not require protein synthesis, and occur within seconds to minutes of GR activation [29]. In thymocytes, for example, activated GR translocates to mitochondria and regulates apoptosis [30]. Recent studies have also indicated that a membrane-bound form of GR mediates non-genomic effects, modulating signaling through the T cell receptor [31] and regulating neural progenitor cell proliferation [32]. Binding of glucocorticoids to GR not only activates the receptor, but also liberates accessory proteins that participate in secondary signaling cascades. For example, when released from the GR complex, c-Src activates signaling cascades that inhibit phospholipase A2 activity, phosphorylate annexin 1, and impair the release of arachidonic acid [33, 34].
It has become a popular notion that the immunosuppressive effects of glucocorticoid treatment result from repression of pro-inflammatory genes, whereas adverse side effects stem from activation of genes that lead to pathology. Indeed, GR interacts with and suppresses several pro-inflammatory transcription factors, including NF-κB, AP-1, CREB, GATA-1, GATA-3, t-Bet, and Oct-1 [24, 27, 35–37], to exert immunoregulatory effects, whereas glucocorticoids activate genes involved in metabolism, such as phosphoenol pyruvate carboxykinase (a regulator of gluconeogenesis) and tyrosine aminotransferase (important to amino acid catabolism), that likely contribute to side effects, including diabetes mellitus. This strict delineation of repression vs. activation in describing glucocorticoid outcomes, however, is likely an oversimplification of the molecular mechanisms mediating the various effects of glucocorticoid treatment. Additional research is needed to dissect the differential contributions of gene activation and repression to the outcome of glucocorticoid-based therapies.
2. GR splice variants and translational isoforms
GR is encoded by the Nr3c1 gene which spans ~126 kb and contains 9 exons [38, 39]. Exon 1 is not translated, the NTD is encoded by exon 2, the DBD by exons 3 and 4, and the hinge and LBD by exons 5 to 9. GR is expressed throughout the body, but research in recent decades has revealed substantial diversity in the proteins produced from the single GR gene. Heterogeneity in GR proteins results from alternative splicing, of which 5 splice variants have been identified, and the presence of 8 translation initiation sites in the GR mRNA, yielding a combinatorial potential for 40 distinct isoforms of GR protein (Fig. 4). The specificity and sensitivity of various tissues to glucocorticoids likely reflects, to some degree, the profile of GR isoforms expressed by the constituent cells.
Figure 4.

Generation of diverse GR isoforms by alternative splicing and translation initiation sites. Multiple GR isoforms with distinct signaling properties are generated from a single GR primary transcript. Five splice variants (GRα, GRβ, GRγ, GR-A, and GR-P) with 8 potential translational isoforms (A, B, C1, C2, C3, D1, D2, and D3) for each splice variant have been identified.
Full-length GRα is the classic mediator of the glucocorticoid effects described above, but four additional splice variants have been identified: GRβ, GRγ, GR-A, and GR-P. GRα is produced from a primary mRNA transcript in which the terminal end of exon 8 is joined to the beginning of exon 9 (Fig. 4). In contrast, GRβ is translated from a transcript in which exon 8 is spliced to a downstream sequence in exon 9 [40]. Although GRα and GRβ are identical through the N-terminal 727 amino acids, sequence divergence at the C-terminus imbues GRβ with distinct functions and properties. GRβ localizes constitutively to the nucleus and does not bind glucocorticoid agonists, although it has been reported to bind the partial GR antagonist RU-486 [41]. GRβ was originally identified as a dominant negative inhibitor of GRα [40, 42]; mechanisms implicated in GRβ inhibition of GRα include competition for GREs, interference with coregulators, and formation of inactive GRα/GRβ heterodimers [43, 44].
In most tissues and cell lines, GRβ is expressed at lower levels than GRα [45–47] although some cell types, including human neutrophils and certain epithelial cells, contain abundant GRβ [46, 48]. Proinflammatory cytokines and microbial superantigens enhance GRβ expression in mononuclear cells, an effect associated with glucocorticoid resistance [49, 50]. GRβ levels are also elevated in glucocorticoid-resistant cases of asthma [51], rheumatoid arthritis [52], ulcerative colitis [53], and systemic lupus erythematosus [54]. High GRβ expression is also associated with glucocorticoid resistance in acute lymphoblastic leukemia and chronic lymphocytic leukemia [55, 56]. Manipulating GRα/GRβ expression ratios, therefore, may provide a means to modulate glucocorticoid sensitivity. While traditionally thought of as a dominant-negative inhibitor of GRα, GRβ has recently been shown to exert transcriptional activity [41, 57, 58], enhancing and repressing a large cohort of genes, the majority of which are not targets of GRα. These findings suggest that GRα/GRβ expression ratios in tissues and cells are not only indicative of glucocorticoid sensitivity, but may also impact downstream regulation of genes.
The remaining GR splice variants, GRγ, GR-A, and GR-P, were discovered in glucocorticoid-resistant cancer cells, and later found to be expressed in healthy tissues. GRγ, first identified in cancer cells [59, 60] then characterized in blood mononuclear cells [61], results from splicing of the GR primary transcript from an alternative splice donor site in the intron separating exons 3 and 4, resulting in the insertion of a single arginine between the DNA-binding zinc fingers of the DBD [60] (Fig. 4). GRγ functions as transcription factor, but only exhibits ~50% of the activity of GRα for canonical glucocorticoid target genes [59, 60]. GRγ expression in childhood acute lymphoblastic leukemia has been shown to correlate with resistance to dexamethasone treatment [62]. However, a recent study revealed that the arginine insertion of GRγ alters the affinity of the DBD for certain DNA sequences, allowing GRγ to bind degenerate GREs in genes that are not targets of GRα [34]. Thus, GRγ expands the set of genes susceptible to regulation by glucocorticoids.
GR-A and GR-P were discovered in glucocorticoid-resistant multiple myeloma cells as GR isoforms that failed to bind glucocorticoid ligands [63]. GR-A transcripts lack exons 5–7, which encode the amino-terminal half of the LBD, as a result of mRNA splicing from exon 4 to 8 (Fig. 4). Little is known, however, of GR-A’s biological functions. GR-P lacks the carboxy-terminal half of the LBD due to failed splicing at the exon 7/8 boundary (Fig. 4) [63]. GR-P is expressed in normal human tissues and regulates the transcriptional activity of GR-A, and is the predominant splice variant in many glucocorticoid-resistant cancers [64, 65].
In addition to alternative splicing, alternative translation initiation sites further diversify the repertoire of potential GR proteins. Whereas alternative splicing affects the activity of the DBD and/or LBD, alternative translation initiation sites alter the NTD, which is important for interactions with cofactors and transcriptional machinery. Exon 2 of the GR mRNA contains 8 AUG start codons that are conserved across humans, monkeys, rats, and mice (Fig. 4). Thus, a single GRα mRNA transcript can generate eight GRα isoforms with progressively shorter NTDs: GRα-A, GRα-B, GRα-C1, GRα-C2, GRα-C3, GRα-D1, GRα-D2, and GRα-D3 [12, 66]. All 8 translational isoforms have been identified for the GRα splice variant but mRNA transcripts for GRβ, GRγ, GR-A, and GR-P also contain the same array of alternative translation start sites, yielding 8 potential translational isoforms for each GR splice variant.
All GRα translational isoforms bind glucocorticoid ligands and interact with GREs [67], as expected by their intact DBDs and LBDs. However, transfection of individual GRα isoforms into cells has revealed isoform-specific gene sets as targets and distinct capacities to mediate glucocorticoid-induced cell death [16, 67]. In GRE-based reporter assays, the GRα-C3 isoform is the most transcriptionally active of GRα isoforms whereas the GRα-D isoforms are the least active [12]. Moreover, cells expressing GRα-C3 are most sensitive to dexamethasone-induced killing whereas those expressing the GRα-D3 isoform are relatively insensitive to glucocorticoid-induced death [67]. GRα-D does not repress NF-κB activity like other GRα isoforms and fails to inhibit the transcription of certain antiapoptotic genes [68], an effect likely mediated by the truncated NTD of GRα-D isoforms. Moreover, unlike GRα-A, -B, and -C isoforms that are chiefly found in the cytoplasm, unliganded GRα-D isoforms localize to the nucleus [67].
GRα translational isoforms are expressed throughout the body but the relative abundance of each isoform varies substantially across cell types, likely contributing to the tissue and cell-specific effects of glucocorticoids. GRα-C isoforms, for example, are highly expressed in the pancreas, lung, and colon but are expressed at low levels in the liver [12, 67]. GRα-D isoforms, in contrast, are abundant in spleen and bladder, but are relatively scarce in the heart and pancreas. The profile of GR isoforms can also change with maturation or activation within a cell lineage. GRα-D isoforms dominate in immature dendritic cells but GRα-A is most abundant in mature dendritic cells, resulting in differential susceptibility to glucocorticoid-induced cell death [69].
The discovery of multiple GR isoforms has advanced our understanding of the molecular mechanisms behind cell-specific glucocorticoid effects, the diversity of which is amplified by posttranslational modifications and the potential for GR isoforms to operate as monomers, homodimers, and heterodimers. In identifying the GR isoform profile of a tissue or cell type, it may become possible to predict glucocorticoid sensitivity as well as downstream physiological effects.
D. Glucocorticoids in the clinic: history and future
Synthetic glucocorticoids are widely used for the treatment of inflammatory and immune disorders, including rheumatoid arthritis, multiple sclerosis, systemic lupus erythematosus, inflammatory bowel disease, sarcoidosis, and nephrotic syndrome [70]. Glucocorticoids are applied locally to treat dermatitis, asthma, conjunctivitis, and other ophthamolgical disorders. High dose glucocorticoid administration is used for acute inflammatory events such as shock, brain edema, and graft-vs-host disease. The immunosuppressive properties of glucocorticoids are invoked following tissue transplantation to prevent organ rejection, whereas their lympholytic effects are exploited for the treatment of hematological malignancies, including leukemias, lymphomas, and myelomas.
Despite the efficacy of glucocorticoids in the treatment of inflammatory and immune disorders, their utility is limited by harmful side effects of prolonged and/or high dose treatment [2]. These side effects include diabetes, impaired wound healing, skin atrophy, muscle atrophy, susceptibility to infection, activation of latent infections, HPA dysfunction, cataracts, peptic ulcers, hypertension, metabolic syndrome, osteoporosis, and water/electrolyte imbalance. Much effort has been dedicated over the last several decades to enhance glucocorticoid potency while minimizing adverse side effects by modifying the chemical structure of the natural glucocorticoid cortisol (hydrocortisone). As the molecular details of glucocorticoid signaling have been unveiled, novel strategies to optimize glucocorticoid-based therapies have evolved. Manipulation of GRα and GRβ expression also holds promise for sensitizing cells to glucocorticoid effects. Methotrexate, for example, increases the GRα/GRβ ratio in blood mononuclear cells [71], and methotrexate treatment of glucocorticoid-resistant asthma patients sensitizes their circulating T cells to glucocorticoid-induced inhibition [72]. Recent studies in animals have revealed sexual dimorphism in the immunoregulatory effects of glucocorticoids, with dexamethasone inducing stronger anti-inflammatory responses in male rats than in females [73], suggesting that glucocorticoid dosing should take gender in account [74].
As mentioned above, the observation that many immunoregulatory properties of glucocorticoids are mediated by gene repression and harmful side effects by gene activation has prompted the search for synthetic GR ligands that induce GR-protein interactions but not GR-DNA binding. Clinical enthusiasm for such GR agonists, referred to as selective glucocorticoid receptor agonists (SERGRAs) or dissociated glucocorticoid receptor agonists (DIGRAs), is based on the observation that a mutated GR with altered DNA-binding capacity (and thus transactivation activity) can still repress inflammation by binding to and inhibiting proinflammatory transcription factors [75, 76]. Using rational drug design, pharmaceutical companies have pursued SEGRAs by modifying known GR ligands in an effort to identify agonists that bind GR and induce conformational changes in the receptor that favor immunosuppressive GR-protein interactions over GR-DNA binding. SEGRAs have shown promise in in vitro and animal models of inflammation, and several are currently in clinical trials [77].
An important challenge in the clinical application of glucocorticoids is the considerable heterogeneity in glucocorticoid responsiveness among individuals, with a substantial portion of the population (up to 30% according to some reports) exhibiting some degree of glucocorticoid resistance [78]. Our expanding knowledge of glucocorticoid signaling has shed light on several potential mechanisms for glucocorticoid resistance, including genetic polymorphisms in the gene encoding GR [79, 80], and effects of proinflammatory signals on GR isoform expression [81]. Tailored glucocorticoid therapies based on empirical assessment of glucocorticoid sensitivity will allow for individual treatment plans that optimize glucocorticoid benefits while minimizing adverse side effects.
Summary
Endogenous and exogenous glucocorticoids are potent regulators of inflammatory and immune processes, but glucocorticoid-based therapies are hampered by adverse side effects and the variability in glucocorticoid responsiveness among individuals. Elucidating the pathways of glucocorticoid signaling has begun to reveal the molecular basis for differential glucocorticoid sensitivity and biological effects, at the tissue level as well as among individuals. The traditional view that circulating glucocorticoid concentrations drive physiologic responses through a single GR protein has been upended by the discovery of diverse GR isoforms. The ongoing characterization of GR isoforms, each exhibiting distinct regulatory and functional profiles, has provided a mechanistic explanation for tissue-specific responses to glucocorticoids. Moreover, altered expression of GR isoforms has been associated with glucocorticoid resistance in the contexts of both inflammation and cancer. Thus, a thorough profiling of GR isoforms in bodily tissues may shed light on glucocorticoid sensitivity on a tissue-by-tissue basis. Moreover, identifying disease-related factors that alter GR isoform expression may allow for fine-tuning of glucocorticoid-based therapies. While some mysteries surrounding glucocorticoid sensitivity and specificity have begun to unravel, more research is needed to advance the development of optimal glucocorticoid treatments.
Practice Points.
At the cellular level, glucocorticoid sensitivity and specificity is influenced by the expression profile of GR isoforms.
Inflammatory and pathological processes modulate cellular GR isoform profiles, altering their sensitivity to glucocorticoids.
Monitoring and/or assessing glucocorticoid sensitivity in individual patients is important for an optimal glucocorticoid treatment plan.
Research agenda.
Determination of genomic and non-genomic effects of individual GR isoforms will facilitate predictions of glucocorticoid sensitivity and effects based on cell-specific GR isoform profile.
Identification and characterization of novel GR agonists that exert anti-inflammatory and immunosuppressive effects with minimal side effects will advance glucocorticoid therapies in the clinic.
Acknowledgments
Support provided by the Intramural Research Program of the National Institutes of Health/National Institute of Environmental Health Sciences.
Abbreviations
- 11β-HSD
11β-hydroxysteroid dehydrogenase
- ACTH
adrenocorticotropic hormone
- AF
activation function
- CRH
corticotropin-releasing hormone
- DBD
DNA-binding domain
- GR
glucocorticoid receptor
- GRE
glucocorticoid-responsive element
- HPA
hypothalamic-pituitary-adrenal
- LBD
ligand-binding domain
- NL
nuclear localization
- NTD
N-terminal transactivation domain
Footnotes
Conflict of Interest Statement
The authors declare that they have no relevant conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.van Staa TP, Leufkens HG, Abenhaim L, Begaud B, Zhang B, Cooper C. Use of oral corticosteroids in the United Kingdom. QJM : monthly journal of the Association of Physicians. 2000;93:105–11. doi: 10.1093/qjmed/93.2.105. [DOI] [PubMed] [Google Scholar]
- 2.Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacology & therapeutics. 2002;96:23–43. doi: 10.1016/s0163-7258(02)00297-8. [DOI] [PubMed] [Google Scholar]
- 3.Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2002;16:61–71. doi: 10.1096/fj.01-0245com. [DOI] [PubMed] [Google Scholar]
- 4.Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocrine reviews. 2011;32:81–151. doi: 10.1210/er.2010-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Webster JI, Tonelli L, Sternberg EM. Neuroendocrine regulation of immunity. Annual review of immunology. 2002;20:125–63. doi: 10.1146/annurev.immunol.20.082401.104914. [DOI] [PubMed] [Google Scholar]
- 6.Kino T, Chrousos GP. Acetylation-mediated epigenetic regulation of glucocorticoid receptor activity: circadian rhythm-associated alterations of glucocorticoid actions in target tissues. Molecular and cellular endocrinology. 2011;336:23–30. doi: 10.1016/j.mce.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.John CD, Buckingham JC. Cytokines: regulation of the hypothalamo-pituitary-adrenocortical axis. Current opinion in pharmacology. 2003;3:78–84. doi: 10.1016/s1471-4892(02)00009-7. [DOI] [PubMed] [Google Scholar]
- 8.Breuner CW, Orchinik M. Plasma binding proteins as mediators of corticosteroid action in vertebrates. The Journal of endocrinology. 2002;175:99–112. doi: 10.1677/joe.0.1750099. [DOI] [PubMed] [Google Scholar]
- 9.Cooper MS, Stewart PM. 11Beta-hydroxysteroid dehydrogenase type 1 and its role in the hypothalamus-pituitary-adrenal axis, metabolic syndrome, and inflammation. The Journal of clinical endocrinology and metabolism. 2009;94:4645–54. doi: 10.1210/jc.2009-1412. [DOI] [PubMed] [Google Scholar]
- 10.Zbankova S, Bryndova J, Leden P, Kment M, Svec A, Pacha J. 11beta-hydroxysteroid dehydrogenase 1 and 2 expression in colon from patients with ulcerative colitis. Journal of gastroenterology and hepatology. 2007;22:1019–23. doi: 10.1111/j.1440-1746.2006.04529.x. [DOI] [PubMed] [Google Scholar]
- 11.Stegk JP, Ebert B, Martin HJ, Maser E. Expression profiles of human 11beta-hydroxysteroid dehydrogenases type 1 and type 2 in inflammatory bowel diseases. Molecular and cellular endocrinology. 2009;301:104–8. doi: 10.1016/j.mce.2008.10.030. [DOI] [PubMed] [Google Scholar]
- 12*.Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Molecular cell. 2005;18:331–42. doi: 10.1016/j.molcel.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 13.Kumar R, Thompson EB. Gene regulation by the glucocorticoid receptor: structure:function relationship. The Journal of steroid biochemistry and molecular biology. 2005;94:383–94. doi: 10.1016/j.jsbmb.2004.12.046. [DOI] [PubMed] [Google Scholar]
- 14.Grad I, Picard D. The glucocorticoid responses are shaped by molecular chaperones. Molecular and cellular endocrinology. 2007;275:2–12. doi: 10.1016/j.mce.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 15.Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocrine reviews. 1997;18:306–60. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 16.Wu I, Shin SC, Cao Y, Bender IK, Jafari N, Feng G, et al. Selective glucocorticoid receptor translational isoforms reveal glucocorticoid-induced apoptotic transcriptomes. Cell death & disease. 2013;4:e453. doi: 10.1038/cddis.2012.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jenkins BD, Pullen CB, Darimont BD. Novel glucocorticoid receptor coactivator effector mechanisms. Trends in endocrinology and metabolism: TEM. 2001;12:122–6. doi: 10.1016/s1043-2760(00)00357-x. [DOI] [PubMed] [Google Scholar]
- 18.Rosenfeld MG, Glass CK. Coregulator codes of transcriptional regulation by nuclear receptors. The Journal of biological chemistry. 2001;276:36865–8. doi: 10.1074/jbc.R100041200. [DOI] [PubMed] [Google Scholar]
- 19.Lonard DM, O’Malley BW. Expanding functional diversity of the coactivators. Trends in biochemical sciences. 2005;30:126–32. doi: 10.1016/j.tibs.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 20.Uhlenhaut NH, Barish GD, Yu RT, Downes M, Karunasiri M, Liddle C, et al. Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Molecular cell. 2013;49:158–71. doi: 10.1016/j.molcel.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21*.Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. 2011;145:224–41. doi: 10.1016/j.cell.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 22*.Hudson WH, Youn C, Ortlund EA. The structural basis of direct glucocorticoid-mediated transrepression. Nature structural & molecular biology. 2013;20:53–8. doi: 10.1038/nsmb.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23*.John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nature genetics. 2011;43:264–8. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocrine reviews. 2003;24:488–522. doi: 10.1210/er.2002-0006. [DOI] [PubMed] [Google Scholar]
- 25.Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–21. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nissen RM, Yamamoto KR. The glucocorticoid receptor inhibits NFkappaB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes & development. 2000;14:2314–29. doi: 10.1101/gad.827900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liberman AC, Druker J, Perone MJ, Arzt E. Glucocorticoids in the regulation of transcription factors that control cytokine synthesis. Cytokine & growth factor reviews. 2007;18:45–56. doi: 10.1016/j.cytogfr.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 28.Stahn C, Buttgereit F. Genomic and nongenomic effects of glucocorticoids. Nature clinical practice Rheumatology. 2008;4:525–33. doi: 10.1038/ncprheum0898. [DOI] [PubMed] [Google Scholar]
- 29.Haller J, Mikics E, Makara GB. The effects of non-genomic glucocorticoid mechanisms on bodily functions and the central neural system. A critical evaluation of findings. Frontiers in neuroendocrinology. 2008;29:273–91. doi: 10.1016/j.yfrne.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Boldizsar F, Talaber G, Szabo M, Bartis D, Palinkas L, Nemeth P, et al. Emerging pathways of non-genomic glucocorticoid (GC) signalling in T cells. Immunobiology. 2010;215:521–6. doi: 10.1016/j.imbio.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 31.Bartholome B, Spies CM, Gaber T, Schuchmann S, Berki T, Kunkel D, et al. Membrane glucocorticoid receptors (mGCR) are expressed in normal human peripheral blood mononuclear cells and up-regulated after in vitro stimulation and in patients with rheumatoid arthritis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2004;18:70–80. doi: 10.1096/fj.03-0328com. [DOI] [PubMed] [Google Scholar]
- 32.Samarasinghe RA, Di Maio R, Volonte D, Galbiati F, Lewis M, Romero G, et al. Nongenomic glucocorticoid receptor action regulates gap junction intercellular communication and neural progenitor cell proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16657–62. doi: 10.1073/pnas.1102821108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Croxtall JD, Choudhury Q, Flower RJ. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. British journal of pharmacology. 2000;130:289–98. doi: 10.1038/sj.bjp.0703272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34*.Thomas-Chollier M, Watson LC, Cooper SB, Pufall MA, Liu JS, Borzym K, et al. A naturally occurring insertion of a single amino acid rewires transcriptional regulation by glucocorticoid receptor isoforms. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:17826–31. doi: 10.1073/pnas.1316235110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Bosscher K, Vanden Berghe W, Haegeman G. Cross-talk between nuclear receptors and nuclear factor kappaB. Oncogene. 2006;25:6868–86. doi: 10.1038/sj.onc.1209935. [DOI] [PubMed] [Google Scholar]
- 36.Liberman AC, Refojo D, Druker J, Toscano M, Rein T, Holsboer F, et al. The activated glucocorticoid receptor inhibits the transcription factor T-bet by direct protein-protein interaction. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2007;21:1177–88. doi: 10.1096/fj.06-7452com. [DOI] [PubMed] [Google Scholar]
- 37.Rogatsky I, Ivashkiv LB. Glucocorticoid modulation of cytokine signaling. Tissue antigens. 2006;68:1–12. doi: 10.1111/j.1399-0039.2006.00599.x. [DOI] [PubMed] [Google Scholar]
- 38.Encio IJ, Detera-Wadleigh SD. The genomic structure of the human glucocorticoid receptor. The Journal of biological chemistry. 1991;266:7182–8. [PubMed] [Google Scholar]
- 39.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome research. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bamberger CM, Bamberger AM, de Castro M, Chrousos GP. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. The Journal of clinical investigation. 1995;95:2435–41. doi: 10.1172/JCI117943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewis-Tuffin LJ, Jewell CM, Bienstock RJ, Collins JB, Cidlowski JA. Human glucocorticoid receptor beta binds RU-486 and is transcriptionally active. Molecular and cellular biology. 2007;27:2266–82. doi: 10.1128/MCB.01439-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oakley RH, Sar M, Cidlowski JA. The human glucocorticoid receptor beta isoform. Expression, biochemical properties, and putative function. The Journal of biological chemistry. 1996;271:9550–9. doi: 10.1074/jbc.271.16.9550. [DOI] [PubMed] [Google Scholar]
- 43.Oakley RH, Jewell CM, Yudt MR, Bofetiado DM, Cidlowski JA. The dominant negative activity of the human glucocorticoid receptor beta isoform. Specificity and mechanisms of action. The Journal of biological chemistry. 1999;274:27857–66. doi: 10.1074/jbc.274.39.27857. [DOI] [PubMed] [Google Scholar]
- 44.Charmandari E, Chrousos GP, Ichijo T, Bhattacharyya N, Vottero A, Souvatzoglou E, et al. The human glucocorticoid receptor (hGR) beta isoform suppresses the transcriptional activity of hGRalpha by interfering with formation of active coactivator complexes. Molecular endocrinology. 2005;19:52–64. doi: 10.1210/me.2004-0112. [DOI] [PubMed] [Google Scholar]
- 45.de Castro M, Elliot S, Kino T, Bamberger C, Karl M, Webster E, et al. The non-ligand binding beta-isoform of the human glucocorticoid receptor (hGR beta): tissue levels, mechanism of action, and potential physiologic role. Molecular medicine. 1996;2:597–607. [PMC free article] [PubMed] [Google Scholar]
- 46.Oakley RH, Webster JC, Sar M, Parker CR, Jr, Cidlowski JA. Expression and subcellular distribution of the beta-isoform of the human glucocorticoid receptor. Endocrinology. 1997;138:5028–38. doi: 10.1210/endo.138.11.5501. [DOI] [PubMed] [Google Scholar]
- 47.Pujols L, Mullol J, Roca-Ferrer J, Torrego A, Xaubet A, Cidlowski JA, et al. Expression of glucocorticoid receptor alpha- and beta-isoforms in human cells and tissues. American journal of physiology Cell physiology. 2002;283:C1324–31. doi: 10.1152/ajpcell.00363.2001. [DOI] [PubMed] [Google Scholar]
- 48*.Strickland I, Kisich K, Hauk PJ, Vottero A, Chrousos GP, Klemm DJ, et al. High constitutive glucocorticoid receptor beta in human neutrophils enables them to reduce their spontaneous rate of cell death in response to corticosteroids. The Journal of experimental medicine. 2001;193:585–93. doi: 10.1084/jem.193.5.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hauk PJ, Hamid QA, Chrousos GP, Leung DY. Induction of corticosteroid insensitivity in human PBMCs by microbial superantigens. The Journal of allergy and clinical immunology. 2000;105:782–7. doi: 10.1067/mai.2000.105807. [DOI] [PubMed] [Google Scholar]
- 50*.Webster JC, Oakley RH, Jewell CM, Cidlowski JA. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6865–70. doi: 10.1073/pnas.121455098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamid QA, Wenzel SE, Hauk PJ, Tsicopoulos A, Wallaert B, Lafitte JJ, et al. Increased glucocorticoid receptor beta in airway cells of glucocorticoid-insensitive asthma. American journal of respiratory and critical care medicine. 1999;159:1600–4. doi: 10.1164/ajrccm.159.5.9804131. [DOI] [PubMed] [Google Scholar]
- 52.Chikanza IC. Mechanisms of corticosteroid resistance in rheumatoid arthritis: a putative role for the corticosteroid receptor beta isoform. Annals of the New York Academy of Sciences. 2002;966:39–48. doi: 10.1111/j.1749-6632.2002.tb04200.x. [DOI] [PubMed] [Google Scholar]
- 53.Honda M, Orii F, Ayabe T, Imai S, Ashida T, Obara T, et al. Expression of glucocorticoid receptor beta in lymphocytes of patients with glucocorticoid-resistant ulcerative colitis. Gastroenterology. 2000;118:859–66. doi: 10.1016/s0016-5085(00)70172-7. [DOI] [PubMed] [Google Scholar]
- 54.Piotrowski P, Burzynski M, Lianeri M, Mostowska M, Wudarski M, Chwalinska-Sadowska H, et al. Glucocorticoid receptor beta splice variant expression in patients with high and low activity of systemic lupus erythematosus. Folia histochemica et cytobiologica / Polish Academy of Sciences, Polish Histochemical and Cytochemical Society. 2007;45:339–42. [PubMed] [Google Scholar]
- 55.Koga Y, Matsuzaki A, Suminoe A, Hattori H, Kanemitsu S, Hara T. Differential mRNA expression of glucocorticoid receptor alpha and beta is associated with glucocorticoid sensitivity of acute lymphoblastic leukemia in children. Pediatric blood & cancer. 2005;45:121–7. doi: 10.1002/pbc.20308. [DOI] [PubMed] [Google Scholar]
- 56.Shahidi H, Vottero A, Stratakis CA, Taymans SE, Karl M, Longui CA, et al. Imbalanced expression of the glucocorticoid receptor isoforms in cultured lymphocytes from a patient with systemic glucocorticoid resistance and chronic lymphocytic leukemia. Biochemical and biophysical research communications. 1999;254:559–65. doi: 10.1006/bbrc.1998.9980. [DOI] [PubMed] [Google Scholar]
- 57.Kino T, Manoli I, Kelkar S, Wang Y, Su YA, Chrousos GP. Glucocorticoid receptor (GR) beta has intrinsic, GRalpha-independent transcriptional activity. Biochemical and biophysical research communications. 2009;381:671–5. doi: 10.1016/j.bbrc.2009.02.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kelly A, Bowen H, Jee YK, Mahfiche N, Soh C, Lee T, et al. The glucocorticoid receptor beta isoform can mediate transcriptional repression by recruiting histone deacetylases. The Journal of allergy and clinical immunology. 2008;121:203–8. e1. doi: 10.1016/j.jaci.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 59.Kasai Y. Two naturally-occurring isoforms and their expression of a glucocorticoid receptor gene from an androgen-dependent mouse tumor. FEBS letters. 1990;274:99–102. doi: 10.1016/0014-5793(90)81339-p. [DOI] [PubMed] [Google Scholar]
- 60.Ray DW, Davis JR, White A, Clark AJ. Glucocorticoid receptor structure and function in glucocorticoid-resistant small cell lung carcinoma cells. Cancer research. 1996;56:3276–80. [PubMed] [Google Scholar]
- 61.Rivers C, Levy A, Hancock J, Lightman S, Norman M. Insertion of an amino acid in the DNA-binding domain of the glucocorticoid receptor as a result of alternative splicing. The Journal of clinical endocrinology and metabolism. 1999;84:4283–6. doi: 10.1210/jcem.84.11.6235. [DOI] [PubMed] [Google Scholar]
- 62.Beger C, Gerdes K, Lauten M, Tissing WJ, Fernandez-Munoz I, Schrappe M, et al. Expression and structural analysis of glucocorticoid receptor isoform gamma in human leukaemia cells using an isoform-specific real-time polymerase chain reaction approach. British journal of haematology. 2003;122:245–52. doi: 10.1046/j.1365-2141.2003.04426.x. [DOI] [PubMed] [Google Scholar]
- 63.Moalli PA, Pillay S, Krett NL, Rosen ST. Alternatively spliced glucocorticoid receptor messenger RNAs in glucocorticoid-resistant human multiple myeloma cells. Cancer research. 1993;53:3877–9. [PubMed] [Google Scholar]
- 64.de Lange P, Segeren CM, Koper JW, Wiemer E, Sonneveld P, Brinkmann AO, et al. Expression in hematological malignancies of a glucocorticoid receptor splice variant that augments glucocorticoid receptor-mediated effects in transfected cells. Cancer research. 2001;61:3937–41. [PubMed] [Google Scholar]
- 65.Krett NL, Pillay S, Moalli PA, Greipp PR, Rosen ST. A variant glucocorticoid receptor messenger RNA is expressed in multiple myeloma patients. Cancer research. 1995;55:2727–9. [PubMed] [Google Scholar]
- 66.Lu NZ, Cidlowski JA. Glucocorticoid receptor isoforms generate transcription specificity. Trends in cell biology. 2006;16:301–7. doi: 10.1016/j.tcb.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 67*.Lu NZ, Collins JB, Grissom SF, Cidlowski JA. Selective regulation of bone cell apoptosis by translational isoforms of the glucocorticoid receptor. Molecular and cellular biology. 2007;27:7143–60. doi: 10.1128/MCB.00253-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gross KL, Oakley RH, Scoltock AB, Jewell CM, Cidlowski JA. Glucocorticoid receptor alpha isoform-selective regulation of antiapoptotic genes in osteosarcoma cells: a new mechanism for glucocorticoid resistance. Molecular endocrinology. 2011;25:1087–99. doi: 10.1210/me.2010-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69*.Cao Y, Bender IK, Konstantinidis AK, Shin SC, Jewell CM, Cidlowski JA, et al. Glucocorticoid receptor translational isoforms underlie maturational stage-specific glucocorticoid sensitivities of dendritic cells in mice and humans. Blood. 2013;121:1553–62. doi: 10.1182/blood-2012-05-432336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. The New England journal of medicine. 2005;353:1711–23. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 71.Goecke IA, Alvarez C, Henriquez J, Salas K, Molina ML, Ferreira A, et al. Methotrexate regulates the expression of glucocorticoid receptor alpha and beta isoforms in normal human peripheral mononuclear cells and human lymphocyte cell lines in vitro. Molecular immunology. 2007;44:2115–23. doi: 10.1016/j.molimm.2006.07.303. [DOI] [PubMed] [Google Scholar]
- 72.Corrigan CJ, Shiner R, Shakur BH, Ind PW. Methotrexate therapy in asthma increases T cell susceptibility to corticosteroid inhibition. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2003;33:1090–6. doi: 10.1046/j.1365-2222.2003.t01-1-01723.x. [DOI] [PubMed] [Google Scholar]
- 73.Duma D, Collins JB, Chou JW, Cidlowski JA. Sexually dimorphic actions of glucocorticoids provide a link to inflammatory diseases with gender differences in prevalence. Science signaling. 2010;3:ra74. doi: 10.1126/scisignal.2001077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Quinn M, Ramamoorthy S, Cidlowski JA. Sexually dimorphic actions of glucocorticoids: beyond chromosomes and sex hormones. Annals of the New York Academy of Sciences. 2014;1317:1–6. doi: 10.1111/nyas.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75*.Reichardt HM, Tuckermann JP, Gottlicher M, Vujic M, Weih F, Angel P, et al. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. The EMBO journal. 2001;20:7168–73. doi: 10.1093/emboj/20.24.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heck S, Kullmann M, Gast A, Ponta H, Rahmsdorf HJ, Herrlich P, et al. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. The EMBO journal. 1994;13:4087–95. doi: 10.1002/j.1460-2075.1994.tb06726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gessi S, Merighi S, Borea PA. Glucocorticoid’s pharmacology: past, present and future. Current pharmaceutical design. 2010;16:3540–53. doi: 10.2174/138161210793797915. [DOI] [PubMed] [Google Scholar]
- 78.Barnes PJ, Adcock IM. Glucocorticoid resistance in inflammatory diseases. Lancet. 2009;373:1905–17. doi: 10.1016/S0140-6736(09)60326-3. [DOI] [PubMed] [Google Scholar]
- 79.Manenschijn L, van den Akker EL, Lamberts SW, van Rossum EF. Clinical features associated with glucocorticoid receptor polymorphisms. An overview. Annals of the New York Academy of Sciences. 2009;1179:179–98. doi: 10.1111/j.1749-6632.2009.05013.x. [DOI] [PubMed] [Google Scholar]
- 80.van Oosten MJ, Dolhain RJ, Koper JW, van Rossum EF, Emonts M, Han KH, et al. Polymorphisms in the glucocorticoid receptor gene that modulate glucocorticoid sensitivity are associated with rheumatoid arthritis. Arthritis research & therapy. 2010;12:R159. doi: 10.1186/ar3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kino T, Su YA, Chrousos GP. Human glucocorticoid receptor isoform beta: recent understanding of its potential implications in physiology and pathophysiology. Cellular and molecular life sciences : CMLS. 2009;66:3435–48. doi: 10.1007/s00018-009-0098-z. [DOI] [PMC free article] [PubMed] [Google Scholar]