

Abstract
Background
The Escherichia coli response regulator NarL controls transcription of genes involved in nitrate respiration during anaerobiosis. NarL consists of two domains joined by a linker that wraps around the interdomain interface. Phosphorylation of the NarL N-terminal receiver domain (RD) releases the, otherwise sequestered, C-terminal output domain (OD) that subsequently binds specific DNA promoter sites to repress or activate gene expression. The aim of this study is to investigate the extent to which the NarL OD and RD function independently to regulate transcription, and the affect of the linker on OD function.
Results
NarL OD constructs containing different linker segments were examined for their ability to repress frdA-lacZ or activate narG-lacZ reporter fusion genes. These in vivo expression assays revealed that the NarL OD, in the absence or presence of linker helix α6, constitutively repressed frdA-lacZ expression regardless of nitrate availability. However, the presence of the linker loop α5-α6 reversed this repression and also showed impaired DNA binding in vitro. The OD alone could not activate narG-lacZ expression; this activity required the presence of the NarL RD. A footprint assay demonstrated that the NarL OD only partially bound recognition sites at the narG promoter, and the binding affinity was increased by the presence of the phosphorylated RD. Analytical ultracentrifugation used to examine domain oligomerization showed that the NarL RD forms dimers in solution while the OD is monomeric.
Conclusions
The NarL RD operates as an on-off switch to occlude or release the OD in a nitrate-responsive manner, but has additional roles to directly stimulate transcription at promoters for which the OD lacks independent function. One such role of the RD is to enhance the DNA binding affinity of the OD to target promoter sites. The data also imply that NarL phosphorylation results in RD dimerization and in the separation of the entire linker region from the OD.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-015-0502-9) contains supplementary material, which is available to authorized users.
Background
Bacteria, archaea, and lower eukaryotes rely on two-component signal transduction systems as a major strategy to monitor the extra- and intracellular environment for physical and biological changes [1]. These multi-protein phospho-relay signaling systems allow cellular adaptation to changes in nutrient availability, osmolarity, oxygen, redox potential, light, plus other cell viability and survival determining factors. The two-component paradigm involves phosphoryl transfer from a sensor kinase that detects an environmental change to a cognate response regulator (RR) that executes an adaptive action. RRs of two or more domains usually contain a structurally conserved N-terminal “receiver” domain (RD) that houses the phosphorylation site, and a C-terminal “output” domain (OD) that can have an array of functions. Phosphorylation of the RD elicits a response from the OD, the most prevalent being DNA binding and transcriptional regulation [2, 3].
In Escherichia coli, the availability of nitrate during anaerobiosis triggers the Nar two-component system [4, 5]. Phosphoryl transfer occurs from dual sensor kinases NarX and NarQ to the two RRs NarL and NarP. NarL and NarP are transcription factors, which together regulate a family of genes involved in anaerobic respiration and fermentation [6]. Structures of full-length, unphosphorylated NarL [7, 8] from E. coli depict a canonical N-terminal RD, a DNA-binding C-terminal OD, and a 32-residue linker that joins them (Fig. 1a). In the unphosphorylated conformation, the RD occludes the DNA recognition helix (α9) of the DNA binding OD [7]. The activation mechanism implicit in the structure, whereby phosphorylation serves to disrupt the interdomain interface and to liberate the OD, was also corroborated by biochemical studies [9–11]. Once liberated, the NarL OD binds consensus sequences along promoter regions, with preference for inverted DNA heptamers separated by two base-pairs, called 7-2-7 sites [12]. Structures of the isolated NarL OD bound to DNA [11, 13] revealed that the OD binds 7-2-7 sites as a dimer, whereby dimerization occurs along helix α10. This finding expands the activation mechanism to include the movement of linker-helix α6 so as to render helix α10 available for dimerization.
Fig. 1.
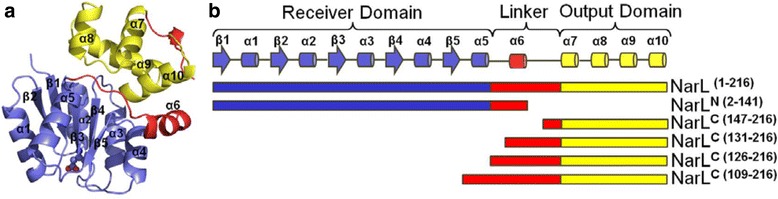
Diagrammatic representation of the NarL constructs used in this study. (a) Ribbon representation of NarL (PDB ID: 1A04), created using Pymol [51]. The RD is colored blue, the linker region in red, and the OD in yellow. The interdomain interface masks the DNA binding elements of the α9 recognition helix, while the linker blocks the α10 dimerization helix. The site of phosphorylation, Asp59, is represented by molecular spheres. The region between helices α6-α7 was too disordered to be resolved. (b) The constructs used in this study are designated by colored bars following the same color scheme as in (a). Residue range is indicated, as is the corresponding secondary structure. Arrows in the secondary structure diagram represent β-strands while cylinders represent α-helices
Most RRs are governed by their oligomeric state before and after phosphorylation, and integral to this process is the oligomeric state of the RD. In RRs that act as transcription factors, phosphorylation usually results in RD dimerization, which serves to either enhance or directly stimulate DNA-binding and, hence, transcriptional activity [14]. Consistently, precluding RD dimerization by mutagenesis compromises these functions [15–18]. Phosphorylation-induced RD dimerization occurs regardless of whether the OD is initially inhibited by the RD in the unphosphorylated state, such as in FixJ and PhoB [16, 19], or is expected to be uninhibited in the unphosphorylated state, such as in OmpR and PhoP [15, 20–23]. The difference is that isolated ODs that are otherwise inhibited in the full-length protein often have some level of intrinsic function. For example, the isolated OD of PhoB activates transcription of phoA and the presence of RD dimers enhances this activity [16, 24]. In contrast, the OmpR OD binds DNA weakly and is unable to activate transcription without its RD [25]. Little is known about the oligomeric state of the NarL RD after phosphorylation, nor of its role in transcriptional regulation. The NarL OD, as shown by Lin and Stewart [26], is able to confer in vivo transcriptional activation at some promoters in the presence of nitrate, such as the napF promoter, and, to a lesser extent, at the yeaR promoter. Thus, there has been evidence that independent function of the isolated NarL OD is variable and promoter dependent, and therefore the role of the RD may be as well. We sought to further investigate this idea by examining the ability of the NarL OD to regulate transcription at two specific promoter regions: the fumarate reductase (frdA) promoter where NarL acts as a repressor in the presence of nitrate, and the nitrate reductase (narG) promoter where NarL acts as an activator in the presence of nitrate.
In this study, we examine the roles of the NarL OD, RD, and linker region in protein function and transcriptional regulation. Protein truncations of the isolated OD and RD, containing various portions of the linker, were created. These constructs were tested for their ability to regulate the in vivo expression of frdA-lacZ or narG-lacZ reporter fusion genes, in the presence or absence of nitrate, as compared to the full-length protein. These assays demonstrated that the NarL OD alone was sufficient to repress frdA-lacZ expression, regardless of nitrate availability. Incremental additions of the linker to the OD reversed frdA-lacZ repression and also impaired DNA binding. At the narG promoter, gene activation required the activities of both the OD and RD. A footprint assay showed that optimal binding of the OD to narG induction sites required the phosphorylated RD, which was also shown to form dimers by analytical ultracentrifugation. The variation of independent function by the NarL OD at the two promoters leads to the conclusion that the NarL RD has promoter specific functions that can extend beyond blocking and releasing the OD.
Results
The NarL OD represses frdA expression but is unable to activate narG expression
In the E. coli cell, the presence of nitrate triggers phosphorylation of NarL by NarX. Phosphorylated NarL (NarL-P) subsequently activates genes involved in nitrate respiration and represses the expression of genes encoded for alternative respiratory pathways [4, 27]. At the frdA promoter for the fumarate reductase operon, NarL-P acts as a transcriptional repressor. It binds to several target DNA binding sites that are located near the transcription start site, one of which is a high-affinity 7-2-7 site (Fig. 2a) [28]. To determine whether the NarL OD can confer independent transcriptional regulation at this promoter in vivo, different NarL C-terminal domain (NarLC) constructs were evaluated for their ability to implement nitrate-dependent repression of an frdA-lacZ reporter fusion gene. The NarLC constructs contained various linker lengths (Fig. 1b) , ranging from the shortest, which contained a small portion of the linker (NarLC (147–216)), to the longest, which contained the entire linker region (NarLC (126–216)). An additional construct included helix α5 of the RD (NarLC (109–216)). The NarL N-terminal domain plus linker region (NarLN) was included as a negative control. Wild-type NarL displayed a 15-fold decrease in frdA-lacZ promoter activity in the presence of nitrate, as opposed to the full induction of this gene in the absence of nitrate when NarL is unphosphorylated. In contrast, all NarLC constructs tested at this promoter lacked a nitrate-dependent response as demonstrated by their constitutive repressive behavior (Fig. 2b). Additionally, these derivative proteins displayed two patterns of activity. Those missing the α5-α6 loop (NarLC (147–216) and NarLC (131–216)) bestowed a level of repression comparable to that of wild-type. Those containing the α5-α6 loop (NarLC (126–216) and NarLC (109–216)), resulted in intermediate expression; that is, their ability to repress transcription decreased by 7-fold and 10-fold, respectively, as compared to wild-type NarL. As expected, NarLN was unable to repress expression since it lacked the DNA recognition domain. These results demonstrate that the NarL OD, tolerant of a certain amount of attached linker, is sufficient to repress frdA transcription and does so in a constitutive manner.
Fig. 2.
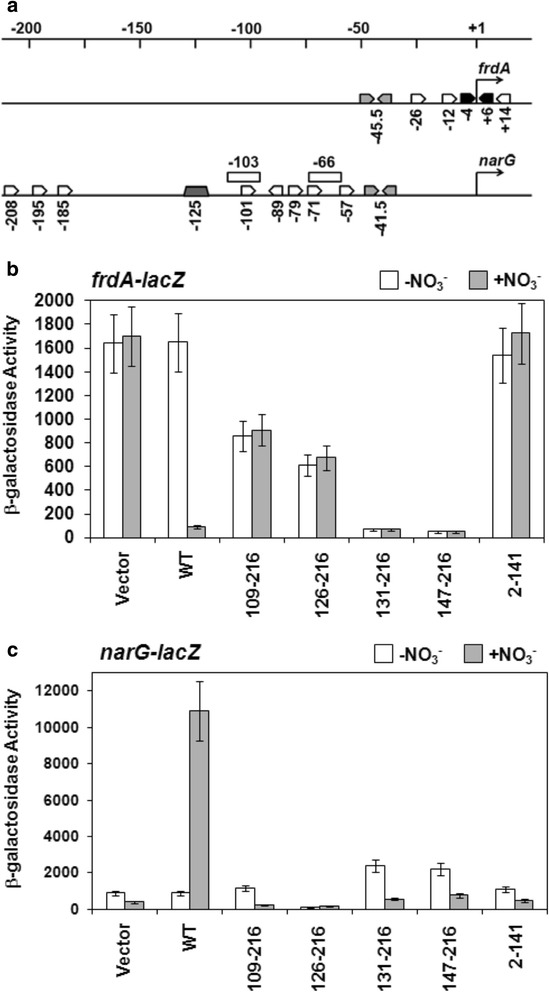
The ability of the NarL protein constructs to repress frdA-lacZ or activate narG-lacZ. (a) Transcription factor binding sites along the frdA and narG promoters. Black inverted arrows represent NarL high-affinity, 7-2-7 binding arrangements; white arrows represent NarL non-7-2-7 sites or NarL single heptameric sites; light gray inverted arrows represent FNR sites, a dark gray trapezoid represents the IHF binding site; white rectangular boxes represent FIS binding sites. The scale denotes nucleotides, and an arrow at the +1 nucleotide represents the transcription start site. Information used to generate this image was taken from the EcoCyc database [52] and references therein. (b) The ability of the NarL proteins to repress frdA-lacZ expression, or (c) to activate narG-lacZ expression. NarL truncated proteins were tested for anaerobic in vivo activity: the indicated lacZ transcriptional reporter fusions were used in E. coli MC4100 strains (ΔnarX/L/Q) harboring pACYC184 plasmid derivatives that contained NarX and either NarL+ or the indicated truncated NarL derivative. Levels of activity are measured in nmol OMPG hydrolyzed per min per mg protein. White bars indicate activity in the absence of nitrate, gray bars indicate activity in the presence of nitrate. Vector refers to parent plasmid pACYC184 run as a control. Error bars (vertical lines) represent one standard deviation, based on the mean value for at least three repeat experiments
At the narG promoter for the nitrate reductase operon, NarL-P acts as a transcriptional activator. Here, the consensus DNA binding sites are located upstream from the transcription start site and lack any 7-2-7 sites (Fig. 2a). When the different NarL proteins were evaluated for their ability to activate transcription of a narG-lacZ reporter fusion gene in vivo, none of the NarLC proteins stimulated transcription relative to wild-type NarL (Fig. 2c). NarLC (147–216) and NarLC (131–216) conferred a slight elevation of activity in the absence of nitrate when compared to the vector control, but showed the same low level expression pattern. In contrast, wild-type NarL gave rise to a 20-fold increase in activity in response to the presence of nitrate. As expected, the NarLN control was unable to activate narG-lacZ gene expression. These results show that the RD is essential for NarL to activate the narG operon. Western blot analysis for these and the above transcription assays showed that all of the NarX and NarL proteins were expressed in vivo, and in relatively uniform concentrations (data not shown).
The NarL RD is required for proper binding to the narG promoter region
One reason for the lack of narG activation by all of the NarLC constructs may be due to poor binding at this promoter region. Previous DNAse I footprint analyses of the narG promoter has revealed two areas of protection by NarL-P: a high affinity site centered in the −89 region and a lower affinity site centered in the −195 region [28, 29]. Occupancy of both regions is necessary for full induction of narG transcription, in addition to other transcription factors, such as FNR and IHF that must also bind to the region [30, 31]. To determine whether the isolated NarL OD is capable of binding these critical regions, a DNAse I protection assay was performed using a 391 base-pair fragment containing the narG promoter region (+155 to −236). Binding patterns of NarLC (147–216), NarL, and NarL-P were compared. NarL-P clearly protected both the −89 and −195 regions (Fig. 3), including regions around −150, while unphosphorylated NarL did not produce any protection pattern. NarLC (147–216) conferred a strong yet different protection pattern at the −89 region, and displayed much weaker protection at the −195 region as compared to NarL-P. This weak interaction primarily resulted from alterations in hypersensitive sites and not by typical zones of protection. Although NarL-P and NarLC (147–216) both bind the −89 site with the same relative affinity, their different binding pattern may be suggestive of improper occupancy by NarLC (147–216). At the −195 region, NarL-P binds more extensively and with a stronger affinity. The compromised binding by the OD at the −195 region signifies at least one reason for the inability of NarLC (147–216) to activate narG-lacZ expression. At this promoter region, the phosphorylated RD is required to impart enhanced binding affinity and full occupancy.
Fig. 3.
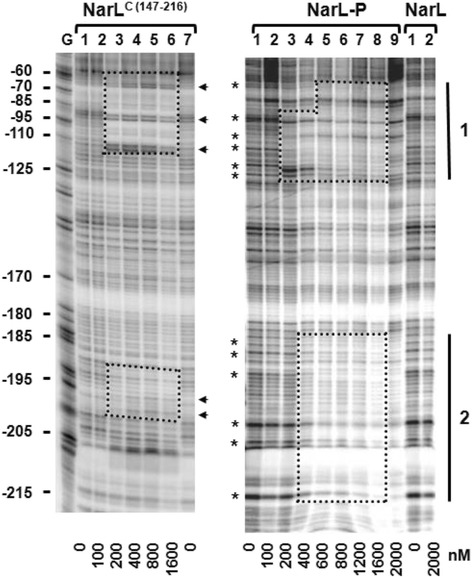
DNase I protection patterns for NarLC (147–216), NarL, and NarL-P. Binding to a 391 base pair fragment containing the narG promoter region is shown. The vertical bars indicate the two major regions of protection seen for NarL-P, which are not as strong or as extensive in NarLC (147–216). Dotted lines highlight areas of protection. Asterisks indicate altered banding patterns or hypersensitive bands for NarL-P. Arrows indicate altered banding patterns for NarLC (147–216). Maxam-Gilbert G-reactions performed on the same DNA fragment were used as size markers. Coordinates relative to the transcription start site are given in base-pairs, and protein concentrations are indicated
Loop α5-α6 inhibits DNA binding of the NarL OD
The impaired frdA-lacZ repression by NarLC constructs containing loop α5-α6 (Fig. 2b) indicates that these proteins weakly bind DNA as compared to NarLC domains lacking this linker segment. To further investigate the effect of loop α5-α6 on DNA binding, we performed an EMSA using the previously constructed oligonucleotide containing the engineered narG −89/−89 sequence [11]. NarLC (147–216), which bestows full frdA-lacZ repression (Fig. 2b), binds to this sequence with equivalent binding affinity as wild-type NarL-P in a DNAse I footprint assay [11], and co-crystallizes with this 7-2-7 site as a dimer [13]. Therefore, this oligonucleotide serves as a useful standard to compare the relative binding affinities of the NarLC constructs used in this study. The EMSA shows that NarLC (131–216), which contains helix α6, bound DNA with equivalent binding affinity as the NarLC (147–216) control (Fig. 4). Consistent with the results of the in vivofrdA-lacZ expression assay, the presence of helix α6 did not impair DNA binding. In contrast, NarLC (126–216) bound DNA less tightly, supporting the hypothesis that loop α5-α6 exerts an inhibition on the NarL OD. The same results were observed by the NarLC truncation proteins when the (His)6-tag was placed at the C-terminal end (data not shown), suggesting that the inhibition by the α5-α6 loop is not due to the (His)6-tag. Furthermore, NarLC (131–216) and NarLC (126–216) are expected to have adopted an overall correct fold (see Discussion).
Fig. 4.
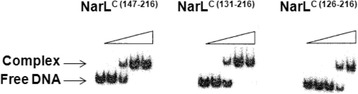
The affect of linker loop α5-α6 on NarLC binding to DNA. An EMSA illustrates the relative binding affinities of the indicated NarLC proteins to a 32P-labeled DNA fragment containing an engineered 7-2-7 narG −89/-89 binding site. Lanes 1–5 show increasing concentrations of the specified NarLC protein (from left to right: 0, 0.25, 0.5, 1.0, and 2.0 μM). The binding affinities of NarLC (147–216) and NarLC (131–216), containing helix α6, are equivalent while that of NarLC (126–216), containing helix α6 and loop α5-α6, is relatively reduced
The NarL RD dimerizes in solution while the NarL OD is monomeric
In several RRs, phosphorylation of the RD enhances DNA binding. This is often accompanied by, or a direct result of, RD dimerization [14, 15, 19, 20]. Therefore, the requirement of the phosphorylated RD to achieve optimal NarL binding at the narG promoter may involve RD dimerization. To determine if the NarL RD is able to dimerize and to gain further insight into the NarL phosphorylation mechanism, analytical ultracentrifugation was applied to NarLN and NarL in native and phosphorylated states, and to the NarLC constructs.
NarLN, containing the RD and linker-helix α6, has a sequence molecular-mass of 16.5 kDa. When examined by analytical ultracentrifugation at a concentration of 60 μM, the NarLN speed dependent molecular-masses were 26.1 kDa (15,000 rpm), 24.9 kDa (18,000 rpm), and 23.4 kDa (22,000 rpm) which were best fit by a monomer and dimer (Table 1 and Fig. 5a). At the corresponding centrifugation speeds, phosphorylated NarLN (NarLN-P) gave apparent molecular masses of 30.8 kDa, 30.2 kDa and 29.0 kDa, and were best fit by a population largely in the dimeric form. When examined at 160 μM, similar results were observed and with reasonably good residuals. Unphosphorylated NarLN had apparent molecular-masses of 25.9 kDa (15,000 rpm) and 23.0 kDa (22,000 rpm), while those for NarLN-P were 30.4 kDa (15,000 rpm) and 27.4 kDa (22,000). The slightly lower-than-expected measured molecular masses for NarLN-P (expected ~32 kDa) were somewhat surprising. NarLN-P autophosphorylates more efficiently with acetyl phosphate than full-length NarL (Additional file 1: Figure S1A), is observably more soluble, and remains phosphorylated for at least 12 hours and gradually dephosphorylates over a period of several days [10]. Although partial loss of phosphorylation in these experiments may have occurred, the observation that unphosphorylated NarLN consistently displayed 50 % dimeric behavior demonstrates that this domain can form dimers when liberated from the OD. Phosphorylation then serves to increase the population of RD dimers.
Table 1.
Summary of sedimentation equilibrium resultsa,b
| Construct | Sequence MMc (kDa)c | Speed (krpm)c | Measured MM at 60 μM (kDa) | Measured MM at 160 μM (kDa) | Predominant Oligomeric State |
|---|---|---|---|---|---|
| NarLC (147–216) | 9.6 | 15 | 9.7 | 12.3 | Monomer |
| 22 | 9.6 | 11.4 | |||
| NarLC (126–216) | 11.8 | 15 | 14.1 | 13.6 | Monomer |
| 22 | 12.7 | 12.4 | |||
| NarL | 23.9 | 11 | 24.0 | 23.9 | Monomer |
| 15 | 22.6 | 23.0 | |||
| NarLN | 16.5 | 15 | 26.1 | 25.9 | Monomer/Dimer |
| 22 | 23.4 | 23.0 | |||
| NarLN-P | 16.6 | 15 | 30.8 | 30.4 | Dimer |
| 22 | 29.0 | 27.4 |
aCertain constructs had additional data collected at other speeds that are not shown in the table but were included in determining the best oligomeric fit (discussed in text)
bAll runs shown were carried out in a buffer solution containing 25 mM Tris, pH 7.5-8.5 and 500 mM NaCl, except for two cases: NarLC (147–214) at 60 μM contained 500 mM (NH4)2SO4, and NarL at 60 μM contained 100 mM NaCl. These changes to salt did not significantly alter the measured molecular masses (see Materials and Methods)
cMM: molecular mass; kDa: kiloDalton; krpm: kilorevolutions per minute
Fig. 5.
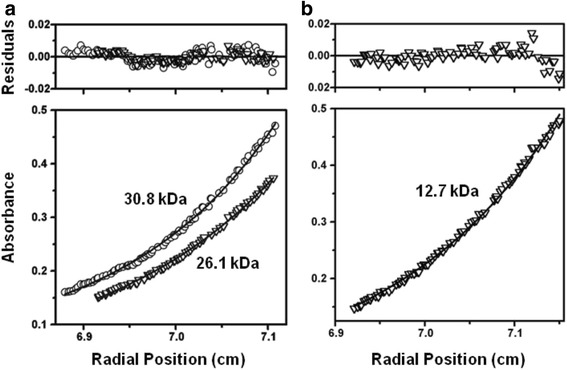
Representative sedimentation equilibrium plots. Plots are single exponential fits with the residuals shown in the upper plot of each panel. Triangles represent unphosphorylated proteins, circles represent phosphorylated proteins. Samples shown were run at a concentration of 60 μM and the measured molecular-masses in kiloDalton (kDa) are indicated. (a) NarLN and NarLN-P after reaching equilibrium at 15,000 rpm and 240 nm. The sequence determined molecular-mass of NarLN is 16.5 kDa, therefore NarLN exists as a monomer and dimer while NarLN-P is mostly a dimer. (b) NarLC (126–216), with a sequence-determined molecular mass of 11.8 kDa, after reaching equilibrium at 22,000 rpm and 280 nm. This construct, as with the shorter NarLC (147–216) construct, is predominantly a monomer
Full-length unphosphorylated NarL was monomeric at 60 μM and 160 μM, with a measured molecular-mass of 24.0 kDa at 11,000 rpm, and with small fitting residuals (Table 1). Its sequence molecular-mass is 23.9 kDa. Consistent results were also observed at the same two concentrations and at 15,000 rpm. Attempts to determine the oligomeric state of NarL-P repeatedly showed that it was dimeric, and perhaps contained a small population of tetramer. However, the results were deemed inconclusive due to the heterogeneity of the sample and the inability to rule out protein aggregation.
As mentioned, the NarLC (147–216) domain co-crystallizes with a DNA 7-2-7 site as a dimer [11, 13]; but whether this truncation protein can dimerize in the absence of DNA has not been demonstrated. The molecular mass of NarLC (147–216) was determined by sedimentation to be 9.6 kDa at 60 μM and 22,000 rpm, which is identical to its sequence molecular mass. This domain, therefore, remains monomeric in solution (Table 1). No salt conditions tested were found to promote dimerization, and NarLC (147–216) was also predominantly monomeric at 160 μM and 22,000 rpm. To test whether the linker region affects NarLC dimerization, the NarLC (126–216) construct, which contains the entire linker region, was also examined by sedimentation. The molecular mass of NarLC (126–216) was determined at 22,000 rpm and 60 μM to be 12.7 kDa (Table 1 and Fig. 5b) and 12.4 kDa at 160 μM. Compared to its sequence molecular-mass of 11.8 kDa, NarLC (126–216) was also predominately monomeric. Residuals from the exponential fitting were small, indicating little molecular mass heterogeneity. For both NarLC constructs, additional data were taken at speeds of 15,000 rpm and 18,000 rpm, which gave similar results. Taken together, these sedimentation results demonstrate that NarLC, either alone or with the linker attached, is predominantly monomeric in solution.
Discussion
The roles of the NarL RD are promoter specific
Our in vivo and in vitro results indicate that the NarL RD performs different functions to regulate transcription, which depend on the requirements governing specific promoter regions. This idea is summarized in Fig. 6, which depicts the roles of the OD and RD at the preferred sites of the frdA and narG promoters. Transcriptional repression of frdA presumably involves occlusion of RNA polymerase and the transcriptional activator FNR at the transcription start site [32, 33]. According to our results, this repression can be accomplished merely with a liberated OD, which can include linker-helix α6 (Fig. 6a). The NarL RD is not required for these functions. Instead, the role of the RD at this promoter is to prevent uncontrolled gene repression by sequestering the OD in the absence of nitrate. The RD essentially acts as a phosphorylation controlled on-off switch that transitions from the open to closed form of the protein, respectively, in response to nitrate availability. RD dimerization is proposed to occur and may do so to maintain the open form of the protein.
Fig. 6.
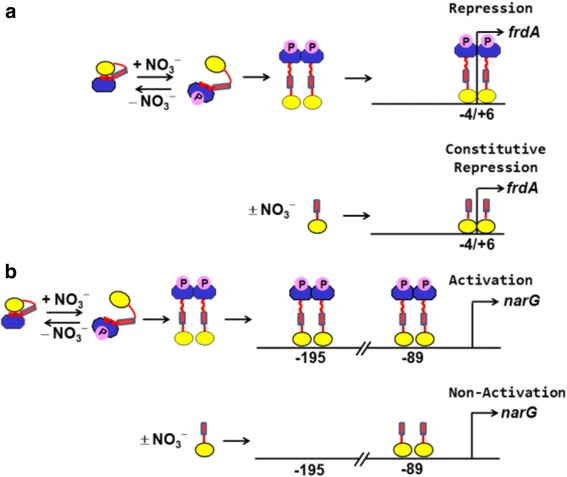
The roles of the OD and RD at the frdA and narG promoter regions. NarL is represented by a cartoon figure: the OD is depicted by a yellow circle; the linker region is in red, with the α5-α6 loop shown as a squiggle and helix α6 as a cylinder; the RD is shown as a blue irregular octagon with phosphorylation represented by a “P”. For simplicity, only preferred binding sites or regions are designated at each promoter, which are not drawn to scale. The transcription start site is indicated by an arrow. (a) The frdA promoter. Top panel: wild-type, phosphorylated, NarL binds to the frdA promoter region, leading to gene repression in a nitrate responsive manner. Bottom panel: NarLC (which may include helix α6) is also able to repress frdA gene expression, but does so in a constitutive manner. The ODs are shown to dimerize at the 7-2-7 (−4/+6) binding region near the transcription start site. (b) The narG promoter. Top panel: wild-type, phosphorylated, NarL binds the essential −89 and −195 regions of the narG promoter to activate gene expression. The RD is presumed to dimerize and may also engage in other protein-protein interactions that are not depicted in the diagram. Bottom panel: NarLC (which may include helix α6) cannot activate the narG promoter due to improper binding at the −195 region. Therefore narG operon expression remains low or non-activated in the absence of a phosphorylated RD
In contrast, the same NarLC constructs that conferred strong constitutive repression of frdA-lacZ (NarLC (147–216 and NarLC (131–216)) were unable to activate narG-lacZ expression and their behavior was not constitutive. Based on our footprint experiment, the lack of narG activation by these constructs is at least attributed to their inability to properly bind the essential −195 region. Thus, release of the sequestered NarL OD alone is insufficient to induce transcription of the narG operon; rather, the presence of the NarL RD is also mandatory for transcriptional activation (Fig. 6b). At this promoter, the NarL RD is required for optimal DNA binding and occupancy, which may be facilitated by RD dimerization. Another possible role for the RD at this promoter, though not exclusive of the first, is that the NarL RD may be required to bend DNA or may be involved in cooperative interactions. Cooperativity at the narG promoter is thought to occur via NarL oligomerization along the DNA in conjunction with DNA bending by IHF [29, 34]. The NarL RD may be required for such functions, and possibly other protein-protein interactions. Similarly, the receiver domain of the RR TodT, a member of the NarL/FixJ family, is required to induce a hairpin bend at its promoter region that is stabilized by IHF and is required for transcription [35].
At the narG promoter, NarL is reminiscent of RRs with unblocked ODs in the pre-phosphorylated state, such as OmpR or UhpA, which can bind DNA (and may initiate basal transcription) but require phosphorylation to enhance binding and bestow full transcriptional activation [36–38]. In other words, sequestering the NarL OD from binding to the narG promoter is not as important as sequestering it from binding to the frdA promoter. Therefore, a response regulator having a sequestered OD does not necessarily indicate that the OD has intrinsic function at all times. Likewise, the OD of DevR (or DosR), a homologue of NarL, is also inhibited in the inactive state, but during hypoxia the DevR OD alone is unable to activate transcription of genes that require cooperativity [39]. Cooperative binding requires the DevR RD. Thus, in situations where the NarL OD has little to no intrinsic function, the NarL RD has an additional regulatory role, the extent of which varies between promoter regions.
Our results are also consistent with the aforementioned NarL studies by Lin and Stewart [26] that showed different extents of OD activity at the napF verses yeaR promoters. NarLC alone was able to fully activate napF and only partially activate yeaR despite both having a 7–2–7 binding site upstream of the transcription start site (−40 to −49 for napF and −38 to −47 for yeaR). This also implies that the functions of the NarL RD depend more on promoter specificity than on the location of the NarL binding sites. Discrepant roles for the RD at different promoter regions have also been observed or implicated in other RRs. Unphosphorylated PhoP, which does not have a blocked OD, can bind DNA derived from the phoP promoter, but requires a phosphorylated RD to bind DNA derived from the msl3 promoter. The RD of FixJ was proposed to be dispensable at the nifA promoter [40], since, when liberated, the OD was able to confer gene activation comparable to wild-type levels. However phosphorylation-induced RD dimerization of FixJ significantly raised the binding affinity of FixJ to fixK and is expected to be vital at this promoter [19].
Helix α6 verses loop α5-α6
We investigated the affects of specific linker segments on NarL OD function and found discrepant activities between OD constructs containing linker helix α6 verses those containing linker loop α5-α6. The presence of helix α6, in NarLC (131–216), bestowed similar behavior to that seen in NarLC (147–216), which lacks this helix and is known to fold correctly from its crystal structure [11]. Since both NarLC (131–216) and NarLC (147–216) showed constitutive repression of frdA-lacZ expression and similar DNA binding in vitro, this indicates that NarLC (131–216) also folded correctly. Supporting evidence that NarLC (131–216) (and NarLC (126–216)) folded correctly stems from the NMR structure of the Erwinia amylovora RcsB OD and linker segment, which shows structural homology to the corresponding region (residues 129–216) in full-length NarL [41]. Therefore, helix α6 in NarLC (131–216) is predicted to form hydrophobic contacts with helix α10, which, in this form, blocks OD dimerization. These modest interactions, however, could render helix α6 flexible and account for its apparent movement that enabled NarLC (131–216) to function properly. Thus, our data support the expected relocation of helix α6 upon NarL phosphorylation that would enable OD dimerization on DNA via helix α10 [11].
In contrast, the addition of loop α5-α6, such as in NarLC (126–216), compromised frdA-lacZ repression in vivo and weakened DNA binding to a high affinity 7–2–7 site in vitro. This may be explained by the hydrogen bond between M128 in linker loop α5-α6 and G170 in loop α7-α8 of the OD (Fig. 7), which is the only remaining contact for loop α5-α6 once the RD is removed. This hydrogen bond, presumably present in NarLC (126–216), may partially anchor the OD in this region and impart the relative intermediate inhibition observed. By this activation model, release of the OD upon NarL phosphorylation entails severing contacts between the OD and loop α5-α6. This activation mechanism also correlates with our previous EPR studies whereby phosphorylation was proposed to induce a hinge bending movement in the vicinity of the Gly126 [9]. Interestingly, linker involvement at the domain interface and activation mechanism has also been shown in other RRs. In CheB, the linker contributes to the interface and mutations to it can result in methylesterase activity that bypasses phosphorylation [42]. Helix α6 of full-length VraR (which is equivalent to helix α6 in NarL) is thought to stabilize the interdomain interface, and activation by beryllofluoride unwinds this helix as part of releasing the OD [17]. Likewise is observed for the full-length structure of Spr1814 in S. pneumonia where the presumed semi-activated form shows a conformational change of the same helix α6 and the loop which follows [43].
Fig. 7.
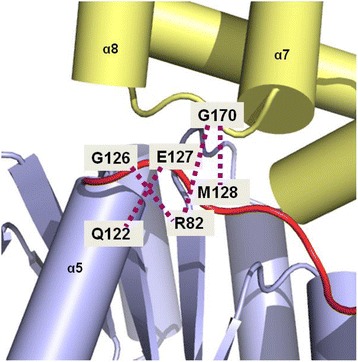
Hydrogen bond contacts in the region of loop α5-α6. Residues 126–128 within the α5-α6 loop (red) form hydrogen bond contacts (purple dotted lines) with both the RD (light blue) and OD (light yellow) of NarL. An interdomain contact between R82 and G170 is also depicted. Removal of the RD abolishes the hydrogen bonds in this region except that of M128 and G170
Conclusions
Based on our results, we propose that phosphorylation of NarL has a dual purpose, to relieve OD inhibition and to enable RD homodimers. Liberation of the OD would entail severing contacts between the RD and OD as wells as between the linker and OD. Once separated, the domains play independent, yet concerted, roles in regulating gene expression. Liberation of the OD alone is sufficient to control some promoters, while control of other promoters requires additional events involving the receiver domain. Thus, the RD can act as a molecular switch or bestow additional complex functions, such as increasing the DNA binding affinity and forming other protein-protein interactions, which render it completely necessary for stimulating transcription.
Methods
Bacterial strains, plasmids, and bacteriophages
The bacterial strains, bacteriophages, and plasmids used in this study are listed in Table 2. All bacterial strains are derived from MC4100 F−araD139 Δ(argF-lac)U169 rpsLl50 relA1 flb-5301 deoCl ptsF25 rbsR [44]. Desired constructs were confirmed with DNA sequence analysis and subsequent subcloning of all pLK63 plasmid derivatives were constructed as previously described [45]. Bacterial strain JM109 and plasmid pQE9 (Qiagen) were used for all protein expression experiments. For protein production and in vitro studies the narL and truncated narL genes were amplified from existing plasmid constructions using PCR technology. TaKaRa Extaq polymerase (Takara Holdings, Inc.), was used for PCR and the amplified products were cloned into the BamHl and Hindlll restriction sites of the pQE9 plasmid. This construction adds an N-terminal histidine epitope to the NarL protein and also results in substitution of Met1 to Gly. NarL is in-frame beginning at Ser2 with the Hindlll restriction site introduced 54 bases downstream from the stop codon of the narL coding sequence. The N-terminal NarL derivative is in-frame beginning at Ser2 with a stop codon introduced at Leu142, immediately followed by a HindIII restriction site. This construct (2–141) is referred to as “NarLN.” The NarL C-terminal expression constructs (referred to as “NarLC”) are in-frame beginning at their indicated residue with the HindIII restriction site introduced 54 bases downstream from the stop codon of the narL coding sequence. NarLC truncation proteins were generated by PCR amplification. The amplified fragments were digested with BamHI and HindIII and then subcloned into the same sites of PQE9 (or digested with NcoI and BglII, and subcloned into the same sites of the PQE60 vector (Qiagen) to add a C-terminal histidine epitope for use in the EMSA experiment only). All restriction endonucleases, polymerases, and other enzymes were purchased from New England Biolabs except where noted.
Table 2.
Strains, plasmids, and phages used in this study
| Strains | Parent | Genotype | Source |
|---|---|---|---|
| CJ236 | F’ cat (pCJ105;M13SCmR) | [53] | |
| dut ung thi relA spoT1 mcrA | |||
| JM109 | endA1 recA1 gyrA96 hsdR17 | [54] | |
| (r k − m k + ) relA1supE44 thi | |||
| [F− traD36 proAB lacI q ZΔM15] Δ(lac-proAB) | |||
| MC4100 | F− Δ(argF-lac) U169 araD139 | [44] | |
| deoC1 relA1 flbB5301 rbsR rpsL50 ptsF25 | |||
| MV1190 | Δ(srl-recA)306:Tn10 (tetR) | BioRad | |
| Δ(lac-proAB) thi supE | |||
| [F: traD36 proAB lacI qZΔM15] | |||
| RC11 | MC4100 | ΔnarXL ΔnarQ::kan recA56 srl::Tn10 tet | [46] |
| Phage | Parent | Genotype | Source |
| M13MK1 | M13mp18 | EcoRI-BamHl 'narX narL + | [46] |
| λLK1 | λRS45 | φ(frdA-lacZ) (hyb) lacY + lacA + | [55] |
| λPC51 | λRS45 | φ(narG-lacZ) (hyb) lacY + IacA + | [56] |
| Plasmid | Parent | Genotype | Source |
| pACYC184 | CmR TcR | [47] | |
| pLK63 | pACYC184 | narX + narL + Cm R | [45] |
| pMJ145 | pLK63 | narX + narL C (147–216) | This study |
| pMJ146 | pLK63 | narX + narL C (131–216) | This study |
| pMJ152 | pLK63 | narX + narL C (126–216) | This study |
| pMJ153 | pLK63 | narX + narL C (109–216) | This study |
| pMJ154 | pLK63 | narX + narL N (2–141) | This study |
| pQE9 | ApR | Qiagen | |
| pMJ05 | pQE9 | narL C (147–216) | This study |
| pMJ99 | pQE9 | narL N (2–141) | This study |
| pMJ104 | pQE9 | narL | This study |
Protein expression and purification
E. coli JM109 cells containing the recombinant over-expression vectors were cultured in 10 mL L Broth with 100 μg/mL ampicillin overnight at 37 °C, and then transferred to 500 mL L Broth with 100 μg/mL ampicillin and grown at 37 °C until the OD600 reached 0.7–1.0. IPTG (Isopropyl-β-D-thiogalactopyranoside) was added to a final concentration of 2 mM. After 3 hours, the cells were harvested by centrifugation at 4000 rpm at 4 °C and then resuspended in buffer (30 mM Tris, pH 8.0, 150 mM NaCl, 2 mM MgCl2), with the addition of DNase (5 mg/mL) and RNase A (10 mg/mL). Cells were broken by passage through a French Press at 12,000 psi. The cell lysate was spun at 16,000 g for 10 min and the supernatant was either incubated with Qiagen Ni-nitrilotriacetic acid resin before loading onto a column, or was loaded onto a 1 mL HiTrap™ chelating Sepharose™ HP column (GE Healthcare). (His)6-tagged NarL proteins were purified according to the manufacturer’s instructions and the proteins were eluted with the above (or related) buffer containing 0.5 M Imidazole. Collected protein fractions were dialyzed and concentrated against a storage buffer (usually 30 mM Tris, pH 7.6, 150 mM KCl, 0.5 mM MgCl2, and 10 % glycerol) using an Amicon Centricon 10, and stored at −80 °C. Protein concentrations were determined using a Bio-Rad protein assay kit with BSA as the protein standard.
β-galactosidase assays
Strain RC11 was lysogenized with either λLK1 (frdA-lacZ) or λPC51 (narG-lacZ) reporter fusions and is used in all in vivo β-galactosidase experiments as previously described [46]. Plasmids containing the NarL constructs, pLK63, are derived from the parent plasmid, pACYC184 (Table 2), which has been shown to exist in low cellular concentration (10–15 copies per cell) [47]. Values reported represent the averages of at least three experiments, and a standard deviation of less than 15 %.
DNAse I footprinting assay
DNase I footprinting assays were performed essentially as described [48]. A 391 base-pair EcoRI-BamHI fragment derived from pIS35 was used as DNA template for the footprinting assays [31]. This fragment contains the narG promoter region extending from nucleotides −236 to +155 relative to the start of transcription. The fragment was isolated and end-labeled by fill-in reactions using Klenow DNA polymerase fragment and [32P]-dATP (3000 Ci/mmole). Purified NarL or NarLC (147–216) (0–2000 nM) and labeled DNA template (1.4 nM) were incubated in 20 μL complex formation buffer (10 mM Tris, pH 7.6, 50 mM KCl, 7.5 mM MgCl2, 3 mM CaCl2, 100 μg/mL BSA) for 30 min at 22 °C. For assays using phosphorylated NarL, the protein was first phosphorylated for 20 min (50 mM HEPES, pH 8.0, 50 mM KCl, 5 mM MgCl2, 2 mM dithiothreitol, 25 mM acetyl phosphate) and 25 mM acetyl phosphate was also added to the individual reaction mixtures. DNase I (500 pg, GE Healthcare) was then added to each reaction mixture for 30 s. The digestion reaction was terminated with the addition of 15 μL stop solution (34 mM EDTA, 6.5 M ammonium acetate). Samples were precipitated, washed with 70 % ethanol, and resusupended in 5 μL sample loading buffer (90 % formamide, 1xTBE, 0.05 % bromophenol blue, 0.05 % xylene cyanol) before separating on 8 M urea, 6 % polyacrylamide electrophoresis gels. The G sequencing reaction ladders were created using an alternate protocol to the Maxim-Gilbert sequencing reaction [48]. Gel exposure, development, and quantitation were performed using Molecular Dynamics PhosphorImager 445 SI and accompanying software.
Electrophoretic-mobility shift assay (EMSA)
Products from the PCR reaction, using the pIS69 [11] plasmid as a template against OSB5 [11] and OSB6 [11] primers, were digested with EcoRI. The resulting 183 base-pair oligonucleotide contained the engineered, 7−2−7 palindromic narG −89/−89 site, 5’-TACCCCTAAAGGGGTA-3’ (heptamer sites underlined). The oligonucleotide was extracted from gels and labeled with 32P using the Klenow fragment of DNA polymerase (New England Biolabs) and purified using a Qiagen PCR clean-up kit. The EMSA was performed, as previously described [13], by incubating the 32P-labeled DNA probes with NarL protein constructs for 10 min at room temperature. The reaction mixtures were immediately run on a 6 % non-denaturing polyacrylamide gel electrophoresis (Invitrogen) and visualized by autoradiography.
Sample preparation for sedimentation equilibrium experiments
Phosphorylated NarL (NarL-P) or phosphorylated NarLN (NarLN-P) was prepared by incubating 246 μM protein with phosphorylation buffer (40 mM KCl, 50 mM Tris, pH 7.5, 40 mM MgCl2, 2.5 % glycerol) and acetyl phosphate (at a 1200:1 molar ratio of acetyl phosphate to protein) for 10 min at room temperature. Since NarLN and NarLN–P are indistinguishable by gel electrophoresis, our method of phosphorylating NarLN was validated using radioactive acetyl-phosphate, which showed that NarLN phosphorylates more efficiently than NarL (Additional file 1: Figure S1A) and is expected to be maintained for up to several days [10]. The phosphorylation level of NarL-P used for sedimentation equilibrium was routinely evaluated on a 20 % native polyacrylamide gel electrophoresis (2 μg loaded per well) using a Phast Gel System (GE Healthcare), and revealed NarL-P yields above 70 % (Additional file 1: Figure S1B) as measured by densitometry (AlphaImager densitometer, Alpha Innotech Corp.). Small-scale electrophoretic studies of NarL-P demonstrated its relative stability for up to one week at 4 °C (data not shown). All samples used for sedimentation equilibrium were prepared fresh for each run, placed in microdialysis buttons (Hampton) using a 1,000 molecular-mass-cutoff membrane (Spectrum), and dialyzed against several changes of buffer (25 mM Tris, pH 7.5 and 500 mM NaCl). To avoid precipitation of NarL-P, dialysis began at 750 mM NaCl. Samples were then diluted to 60 μM or 160 μM. NarLC (147–216) at 60 μM, however, was dialyzed in the buffer solution containing 150 mM NaCl or 500 mM (NH4)2SO4. Both samples were tested and showed that different salt concentrations produced no significant difference in their sedimented molecular masses. Similarly, dialyzing NarL–P in the buffer solution containing 100 mM NaCl or 500 mM NaCl did not significantly change the results.
Sedimentation equilibrium
Sedimentation equilibrium runs were performed at 4 °C in a Beckman Optima XL-A analytical ultracentrifuge using absorption optics. Samples were examined in 3 mm double sector, 12 mm double sector and 12 mm six sector cells at an appropriate wavelength (240, 280, or 295 nm) to ensure the absorbance was sufficient to give a good signal-to-noise ratio and the maximum absorbance was within the linear range of the instrument (less than 1.35 OD). Sedimentation equilibrium profiles were measured at 11,000, 15,000 and 18,000 rpm for NarL and NarL–P and 15000, 18,000 and 22,000 rpm for NarLC (147–216), NarLC (126–216), NarLN and NarLN-P. For low wavelength (240 nm) scans, a baseline was determined by pelleting the protein at 50,000 rpm. The data were initially fit with nonlinear, least-squares exponential for a single ideal species using the Beckman Origin-based software (Version 3.01) to give a weight-average molecular mass of all species in solution. When concentration and speed dependence of the molecular masses indicated association behavior, multiple runs (at least 4, including two concentrations and two different speeds) were analyzed using the “multifit” option of the Beckman global analysis software. The monomeric sequence molecular mass and various models (monomer-dimer, monomer-tetramer, and so on) were tested to see which would give the best fit to the data. Partial specific volumes of 0.740 for NarL, 0.733 for NarLN, 0.734 for NarLC (147–216) and 0.739 for NarLC (126–216) calculated from the amino acid composition and corrected to 4 °C were used [49, 50]. The calculated effect of the phosphate group on the partial specific volume was negligible, and thus ignored.
Acknowledgements
We thank Professor Carla Koehler, Jeffrey H. Zhang, Wojciech Bartkowski, Ann E. Maris, Mary L. Kopka, and Professor Richard E. Dickerson for support and input. Ultracentrifugation studies were performed in the UCLA-DOE Biochemistry Instrumentation facility. This work was supported by grants from the National Institutes of Health, part of which were from Public Health service grants AI21678 and GM31299.
Abbreviations
- EMSA
Electrophoretic mobility shift assay
- EPR
Electroparamagnetic resonance
- FIS
Factor for inversion stimulation
- FNR
Regulator of fumarate and nitrate reduction
- IHF
Integration host factor
- NMR
Nuclear magnetic resonance
- OD
Output domain
- RD
Receiver domain
- RR
Response regulator
- NarLN
NarL receiver domain with portion of the linker
- NarLC
NarL output domain with portion(s) of the linker
- NarLN-P
Phosphorylated NarL receiver domain with portion of the linker
- NarL-P
Phosphorylated full length NarL protein
Additional file
NarL and NarLN phosphorylation reactions. (A) The phosphorylation time course for full length NarL and NarLN. Each protein (25 μM) was incubated with radiolabeled acetyl phosphate (25 mM) at room temperature. Samples of NarL-P (solid line) or NarLN-P (dotted line) were taken at the indicated time points. Units are expressed as PhosphoImager (PI). (B) NarL phosphorylation reactions. NarL (246 μM) was phosphorylated with different ratios of acetyl phosphate (AP) at room temperature and run on the depicted 20 % native Phast gel; each lane contains ~2 μg. Lane 1- NarL only; Lane 2- NarL-P at 400:1 AP: NarL; Lane 3- NarL-P at 600:1 AP: NarL; Lane 4- NarL-P at 800:1 AP:NarL; Lane 5- NarL-P at 1200:1 AP:NarL; Lane 6- NarL-P at 400:1 AP:NarL plus 175 mM additional KCl. Lanes 5 and 6 are 76.7 and 80 % phosphorylated, respectively, as determined from an AlphaImager densitometer. Lane 5 represents the reaction used the sedimentation equilibrium experiments; lane 6 represents an improved version that was also used in the experiments. (DOCX 58 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests that might be perceived to influence the results and discussion reported in this article.
Authors’ contributions
MJ performed the transcription and footprinting assays, ZM performed the EMSA, GK and MP performed the sedimentation studies. GK wrote the manuscript with input from RPG and contributions from the other authors. RPG conceived of the study and participated in its design and coordination. All authors read and approved the final manuscript.
References
- 1.Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annu Rev Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 2.Gao R, Stock AM. Biological insights from structures of two-component proteins. Annu Rev Microbiol. 2009;63:133–154. doi: 10.1146/annurev.micro.091208.073214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galperin MY. Diversity of structure and function of response regulator output domains. Curr Opin Microbiol. 2010;13:150–159. doi: 10.1016/j.mib.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gunsalus RP. Control of electron flow in Escherichia coli: coordinated transcription of respiratory pathway genes. J Bacteriol. 1992;174:7069–7074. doi: 10.1128/jb.174.22.7069-7074.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stewart V. Dual interacting two-component regulatory systems mediate nitrate- and nitrite-regulated gene expression in Escherichia coli. Res Microbiol. 1994;145:450–454. doi: 10.1016/0923-2508(94)90093-0. [DOI] [PubMed] [Google Scholar]
- 6.Constantinidou C, Hobman JL, Griffiths L, Patel MD, Penn CW, Cole JA, Overton TW. A reassessment of the FNR regulon and transcriptomic analysis of the effects of nitrate, nitrite, NarXL, and NarQP as Escherichia coli K12 adapts from aerobic to anaerobic growth. J Biol Chem. 2006;281:4802–4815. doi: 10.1074/jbc.M512312200. [DOI] [PubMed] [Google Scholar]
- 7.Baikalov I, Schröder I, Kaczor-Grzeskowiak M, Grzeskowiak K, Gunsalus RP, Dickerson RE. Structure of the Escherichia coli Response Regulator NarL. Biochemistry. 1996;35:11053–11061. doi: 10.1021/bi960919o. [DOI] [PubMed] [Google Scholar]
- 8.Baikalov I, Schröder I, Kaczor-Grzeskowiak M, Cascio D, Gunsalus RP, Dickerson RE. NarL dimerization? Suggestive evidence from a new crystal form. Biochemistry. 1998;37:3665–3676. doi: 10.1021/bi972365a. [DOI] [PubMed] [Google Scholar]
- 9.Zhang JH, Xiao G, Gunsalus RP, Hubbell WL. Phosphorylation triggers domain separation in the DNA binding response regulator NarL. Biochemistry. 2003;42:2552–2559. doi: 10.1021/bi0272205. [DOI] [PubMed] [Google Scholar]
- 10.Eldridge AM, Kang H-S, Johnson E, Gunsalus R, Dahlquist FW. Effect of phosphorylation on the interdomain interaction of the response regulator, NarL. Biochemistry. 2002;41:15173–15180. doi: 10.1021/bi026254+. [DOI] [PubMed] [Google Scholar]
- 11.Maris AE, Sawaya MR, Kaczor-Grzeskowiak M, Jarvis MR, Bearson SMD, Kopka ML, Schröder I, Gunsalus RP, Dickerson RE. Dimerization allows DNA target site recognition by the NarL response regulator. Nat Struct Biol. 2002;9:771–778. doi: 10.1038/nsb845. [DOI] [PubMed] [Google Scholar]
- 12.Darwin AJ, Tyson KL, Busby SJ, Stewart V. Differential regulation by the homologous response regulators NarL and NarP of Escherichia coli K-12 depends on DNA binding site arrangement. Mol Microbiol. 1997;25:583–595. doi: 10.1046/j.1365-2958.1997.4971855.x. [DOI] [PubMed] [Google Scholar]
- 13.Maris AE, Kaczor-Grzeskowiak M, Ma Z, Kopka ML, Gunsalus RP, Dickerson RE. Primary and secondary modes of DNA recognition by the NarL two-component response regulator. Biochemistry. 2005;44:14538–14552. doi: 10.1021/bi050734u. [DOI] [PubMed] [Google Scholar]
- 14.Gao R, Stock AM. Molecular strategies for phosphorylation-mediated regulation of response regulator activity. Curr Opin Microbiol. 2010;13:160–167. doi: 10.1016/j.mib.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakashima K, Kanamaru K, Aiba H, Mizuno T. Signal transduction and osmoregulation in Escherichia coli. J Bacteriol. 1991;266:10775–10780. [PubMed] [Google Scholar]
- 16.Mack TR, Gao R, Stock AM. Probing the roles of the two different dimers mediated by the receiver domain of the response regulator PhoB. J Mol Biol. 2009;389:349–364. doi: 10.1016/j.jmb.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leonard PG, Golemi-Kotra D, Stock AM. Phosphorylation-dependent conformational changes and domain rearrangments in Staphylococcus aureus VraR activation. Proc Natl Acad Sci U S A. 2013;110:8525–8530. doi: 10.1073/pnas.1302819110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Birck C, Samama J, Hulett FM. Residue R113 is essential for PhoP dimerization and function: a residue buried in the asymmetric PhoP dimer interface determined in the PhoPN three-dimensional crystal structure. J Bacteriol. 2003;185:262–273. doi: 10.1128/JB.185.1.262-273.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Da Re S, Schumacher J, Rousseau P, Fourment J, Ebel C, Kahn D. Phosphorylation-induced dimerization of the FixJ receiver domain. Mol Microbiol. 1999;34:504–511. doi: 10.1046/j.1365-2958.1999.01614.x. [DOI] [PubMed] [Google Scholar]
- 20.Barbieri C, Wu T, Stock A. Comprehensive analysis of OmpR phosphorylation, dimerization, and DNA binding supports a canonical model for activation. J Mol Biol. 2013;425:1612–1626. doi: 10.1016/j.jmb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu W, Hulett FM. Bacillus subtilis PhoP binds to the phoB tandem promoter exclusively within the phosphate starvation-inducible promoter. J Bacteriol. 1997;179:6302–6310. doi: 10.1128/jb.179.20.6302-6310.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rhee JE, Sheng W, Morgan LK, Nolet R, Liao X, Kenney LJ. Amino acids important for DNA recognition by the response regulator OmpR. J Biol Chem. 2008;283:8664–8677. doi: 10.1074/jbc.M705550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshida T, Qin L, Egger LA, Inouye M. Transcription regulation of ompF and ompC by a single transcription factor, OmpR. J Biol Chem. 2006;281:17114–17123. doi: 10.1074/jbc.M602112200. [DOI] [PubMed] [Google Scholar]
- 24.Ellison DW, McCleary WR. The unphosphorylated receiver domain of PhoB silences the activity of its output domain. J Bacteriol. 2000;182:6592–6597. doi: 10.1128/JB.182.23.6592-6597.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walthers D, Tran VK, Kenney LJ. Interdomain linkers of homologous response regulators determine their mechanism of action. J Bacteriol. 2003;185:317–324. doi: 10.1128/JB.185.1.317-324.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin AV, Stewart V. Functional roles for the GerE-family carboxyl-terminal domains of nitrate response regulators NarL and NarP of Escherichia coli K-12. Microbiology. 2010;156(Pt 10):2933–2943. doi: 10.1099/mic.0.040469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Egan SM, Stewart V. Mutational analysis of nitrate regulatory gene narL in Escherichia coli K-12. J Bacteriol. 1991;173:4424–4432. doi: 10.1128/jb.173.14.4424-4432.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Kustu S, Stewart V. In vitro interaction of nitrate-responsive regulatory protein NarL with DNA target sequences in the fdnG, narG, narK and frdA operon control regions of Escherichia coli K-12. J Mol Biol. 1994;241:150–165. doi: 10.1006/jmbi.1994.1485. [DOI] [PubMed] [Google Scholar]
- 29.Walker M, DeMoss J. NarL-phosphate must bind to multiple upstream sites to activate transcription the narG promoter of Escherichia coli. Mol Microbiol. 1994;14:633–641. doi: 10.1111/j.1365-2958.1994.tb01302.x. [DOI] [PubMed] [Google Scholar]
- 30.Rabin RS, Collins LA, Stewart V. In vivo requirement of integration host factor for nar (nitrate reductase) operon expression in Escherichia coli K-12. Proc Natl Acad Sci U S A. 1992;89:8701–8705. doi: 10.1073/pnas.89.18.8701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schröder I, Darie S, Gunsalus RP. Activation of the Escherichia coli nitrate reductase (narGHJI) operon by NarL and Fnr requires Integration Host Factor. J Biol Chem. 1993;268:771–774. [PubMed] [Google Scholar]
- 32.Bearson SMD, Albrecht JA, Gunsalus RP. Oxygen and nitrate-dependent regulation of dmsABC operon expression in Escherichia coli: sites for Fnr and NarL protein interactions. BMC Microbiol. 2002;2:13. doi: 10.1186/1471-2180-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Partridge JD, Browning DF, Xu M, Newnham LJ, Scott C, Roberts RE, Poole RK, Green J. Characterization of the Escherichia coli K-12 ydhYVWXUT operon: regulation by FNR, NarL and NarP. Microbiology. 2008;154:608–618. doi: 10.1099/mic.0.2007/012146-0. [DOI] [PubMed] [Google Scholar]
- 34.Darwin AJ, Li J, Stewart V. Analysis of nitrate regulatory protein NarL-binding sites in the fdnG and narG operon control regions of Escherichia coli K-12. Mol Microbiol. 1996;20:621–632. doi: 10.1046/j.1365-2958.1996.5491074.x. [DOI] [PubMed] [Google Scholar]
- 35.Lacal J, Guazzaroni M-E, Gutierrez-del-Arroyo P, Busch A, Velez M, Krell T, Ramos JL. Two levels of cooperativeness in the binding of TodT to the tod operon promoter. J Mol Biol. 2008;384:1037–1047. doi: 10.1016/j.jmb.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 36.Webber CA, Kadner RJ. Involvement of the amino-terminal phosphorylation module of UhpA in activation of uhpT transcription in Escherichia coli. Mol Microbiol. 1997;24:1039–1048. doi: 10.1046/j.1365-2958.1997.4021765.x. [DOI] [PubMed] [Google Scholar]
- 37.Aiba H, Mizuno T. Phosphorylation of a bacterial activator protein, OmpR, by a protein kinase, EnvZ, stimulates the transcription of the ompF and ompC genes in Escherichia coli. FEBS Lett. 1990;261:19–22. doi: 10.1016/0014-5793(90)80626-T. [DOI] [PubMed] [Google Scholar]
- 38.Head CG, Tardy A, Kenney LJ. Relative binding affinities of OmpR and OmpR-phosphate at the ompF and ompC regulatory sites. J Mol Biol. 1998;281:857–870. doi: 10.1006/jmbi.1998.1985. [DOI] [PubMed] [Google Scholar]
- 39.Gautam US, Chauhan S, Tyagi JS. Determinants outside the DevR C-terminal domain are essential for cooperativity and robust activation of dormancy genes in Mycobacterium tuberculosis. PLoS One. 2011;6 doi: 10.1371/journal.pone.0016500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Da Re S, Bertagnoli S, Fourment J, Reyrat JM, Kahn D. Intramolecular signal transduction within the FixJ transcriptional activator: in vitro evidence for the inhibitory effect of the phosphorylatable regulatory domain. Nucleic Acids Res. 1994;22:1555–61. doi: 10.1093/nar/22.9.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pristovsek P, Sengupta K, Löhr F, Schäfer B, von Trebra MW, Rüterjans H, Bernhard F. Structural analysis of the DNA-binding domain of the Erwinia amylovora RcsB protein and its interaction with the RcsAB box. J Biol Chem. 2003;278:17752–17759. doi: 10.1074/jbc.M301328200. [DOI] [PubMed] [Google Scholar]
- 42.Anand GS, Goudreau PN, Stock AM. Activation of methylesterase CheB: evidence of a dual role for the regulatory domain. Biochemistry. 1998;37:14038–14047. doi: 10.1021/bi980865d. [DOI] [PubMed] [Google Scholar]
- 43.Park A, Moon J, Oh J, Lee K, Chi Y. Crystal structure of the response regulator spr1814 from Streptococcus pneumoniae reveals unique interdomain contacts among NarL family proteins. Biochem Biophys Res Commmun. 2013;434:65–69. doi: 10.1016/j.bbrc.2013.03.065. [DOI] [PubMed] [Google Scholar]
- 44.Silhavy T, Berman M, Enquist L. Experiments with gene fusions. New York: Cold Spring Harbor; 1984. [Google Scholar]
- 45.Kalman LV, Gunsalus RP. Identification of a second gene involved in global regulation of fumarate reductase and other nitrate-controlled genes for anaerobic respiration in Escherichia coli. J Bacteriol. 1989;171:3810–3816. doi: 10.1128/jb.171.7.3810-3816.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cavicchioli R, Schröder I, Constanti M, Gunsalus RP. The NarX and NarQ sensor-transmitter proteins of Escherichia coli each require two conserved histidines for nitrate-dependent signal transduction to NarL. J Bacteriol. 1995;177:2416–2424. doi: 10.1128/jb.177.9.2416-2424.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang A, Cohen S. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol. 1978;134:1141–1156. doi: 10.1128/jb.134.3.1141-1156.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perbal B. A Practical Guide to Molecular Cloning. New York: John Wiley & Sons; 1988. [Google Scholar]
- 49.Cohn E, Edsall J. Density and apparent specific volume of proteins. Proteins, Amin Acids Pept as Ions Dipolar Ions (Edited by Cohn eJ Edsall JT) Reinhold Publ Corp New York 1943:370–381.
- 50.Laue T, Shah B, Ridgeway T, Pelletier S. Computer-Aided Interpretation of Analytical Sedimentation Data for Proteins. In: Rowe AJ, Hort JC, editors. Anal Ultracentrifugation Biochem Polym Sci SE Harding. Cambridge: Gt Britain, R Soc Chem; 1992. pp. 90–125. [Google Scholar]
- 51.Schrödinger L. The PyMOL molecular graphics system. Version 13.
- 52.Keseler IM, Collado-Vides J, Santos-Zavaleta A, Peralta-Gil M, Gama-Castro S, Muñiz-Rascado L, Bonavides-Martinez C, Paley S, Krummenacker M, Altman T, Kaipa P, Spaulding A, Pacheco J, Latendresse M, Fulcher C, Sarker M, Shearer AG, Mackie A, Paulsen I, Gunsalus RP, Karp PD. EcoCyc: a comprehensive database of Escherichia coli biology. Nucleic Acids Res. 2011;39(Database issue):D583–D590. doi: 10.1093/nar/gkq1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raleigh E, Lech K, Brent R. Current Protocols in Molecular Biology (F.M. Ausubel et al. ed.). 1989, vol. 1.4: Wiley Interscience, New York.
- 54.Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;1:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 55.Kalman LV, Gunsalus RP. The frdR gene of Escherichia coli globally regulates several operons involved in anaerobic growth in response to nitrate. J Bacteriol. 1988;170:623–629. doi: 10.1128/jb.170.2.623-629.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cotter P, Darie S, Gunsalus R. The effect of iron limitation on expression of the aerobic and anaerobic electron transport pathway genes in Escherichia coli. FEMS Microbiol Lett. 1992;100:227–232. doi: 10.1111/j.1574-6968.1992.tb14045.x. [DOI] [PubMed] [Google Scholar]