

Abstract
Adaptation to low oxygen tension or hypoxia is a critical event in development and tissue homeostasis. Studies by us and others have shown that the fetal growth plate is an avascular tissue with a gradient of oxygenation, and the transcription factor hypoxia-inducible factor-1α (HIF-1α) is essential for its development. In this brief review, we will summarize our current understanding of the role of HIF-1α in fetal growth plate development, and we will discuss yet unanswered questions in the field of hypoxia and endochondral bone formation.
Introduction
Oxygen (O2) is not only an indispensable metabolic substrate in various enzymatic reactions including mitochondrial respiration, but also a regulatory signal that controls stability and activity of the transcription factor hypoxia-inducible factor-1α (HIF-1α), a key mediator of the cellular adaptation to low O2 tension (hypoxia).1,2,3,4,5 The current model predicts that when O2 tension is above 5%, a class of 2-oxoglutarate-dependent and Fe2+-dependent prolyl-4-hydroxylases hydroxylates specific proline residues of the HIF-1α protein. Hydroxylated HIF-1α is then targeted to the proteosome for degradation by the E3 ubiquitin ligase von Hippel-Lindau. When O2 tension drops below 5%, hydroxylation of HIF-1α becomes inefficient for deficiency of substrate; non-hydroxylated HIF-1α protein accumulates in the cytoplasm, translocates into the nucleus and dimerizes with HIF-1β, another member of the HIF family, which differently from HIF-1α is not regulated by levels of O2. In the nucleus, on recruitment of transcriptional co-activators, the HIF-1α binds to hypoxia-responsive elements within the promoters of hypoxia-responsive genes. The products of these genes regulate a variety of biological processes, including angiogenesis, non-oxidative glycolysis and matrix formation.4,5 In addition, HIF-1α controls cell proliferation with modalities that do not involve its transcriptional activity.6 A large body of in vivo and in vitro experimental evidence supports the notion that HIF-1α is important in pathological conditions such as ischemia and cancer, in a variety of physiological processes and in normal development.4,5
Cartilage and bone are connective tissues of diverse embryonic origin.7 The vertebral skeleton is formed by cells that originate from cranial neural crest, somites and lateral plate mesoderm. There are three primary phases of skeletogenesis: migration of cells to the site of future skeletogenesis; formation of mesenchymal condensations; overt differentiation of chondrocytes and osteoblasts within these condensations. Skeletal development depends on two main mechanisms, intramembranous and endochondral.3,8,9,10 The first, in which mesenchymal cells directly differentiate into osteoblasts, is involved in the formation of the flat skull bones. The second, accounting for the development of most other bones, involves a two-stage event, whereby chondrocytes form a matrix template, the fetal growth plate, in which osteoblasts differentiate and initiate the ossification process. The fetal growth plate recapitulates fundamental processes of cell biology with a highly specific temporal and spatial pattern. Chondrocytes within the cartilage anlage synthesize a characteristic extracellular matrix that is enriched in type II collagen. These cells are highly proliferative and pile up to form columns. The most distal cells of the columnar layer exit the cell cycle and differentiate into hypertrophic chondrocytes, which produce type X collagen and mineralize their surrounding matrix. Hypertrophic chondrocytes eventually die or transdifferrentiate into osteoblasts.11 The cartilage anlage is then invaded by blood vessels and replaced by bone, which is enriched in type I collagen. The fetal growth plate is an unique mesenchymal tissue as it is avascular, although it requires the angiogenic switch to be replaced by bone.12,13
Studies by us and others have demonstrated that the fetal growth plate is an avascular tissue with a gradient of oxygenation, and HIF-1α is essential for its development.3,14,15,16,17,18,19,20 It has also been shown that the hypoxia-signaling pathway significantly modulates cartilage regeneration in vitro.21,22,23 More recently, an association between HIF-1α and cartilaginous tumors in humans has been proposed.24 At last, it has been suggested that HIF-2α, another member of the HIF family of transcription factors, is involved in the pathogenesis of osteoarthritis in humans.3,25
In this brief review, we will summarize our current understanding of the role of HIF-1α in fetal growth plate development, and we will discuss yet unresolved questions in the field of hypoxia and endochondral bone formation.
HIF-1α and the fetal growth plate: facts and challenges
Mammalian embryonic development proceeds in a low O2 environment of ∼2% or lower until the circulatory system develops.1 In adults, gradients of oxygenation (2–9%) are physiological.26 A few adult tissues, such as the kidney medulla and the bone marrow, reach levels of O2 below 1%.26 As mentioned above, HIF-1α is barely detectable when O2 is above 5%, and it progressively accumulates with O2 tension dropping below 5%.1 Stabilization of HIF-1α is one of the hallmarks of hypoxic stress. Hypoxia can be visualized in vivo and in vitro using specific markers such as EF5 or pimonidazole; binding of these hypoxia markers becomes detectable when levels of O2 drop below 2%.1,27,28
Mesenchymal condensations in the limb bud and fetal growth plates have an inner hypoxic region, as shown by staining with EF5 or pimonidazole14,20,29,30 (Figure 1), which is consistent with their anatomic vascularity. Conditional deletion of HIF-1α in either limb bud mesenchyme or fetal chondrocytes leads to marked shortening of the limbs and massive cell death in the inner zone of the developing growth plate.14,16,19,20 The growth plate has been indeed one of the first models to provide clear experimental evidence that in vivo HIF-1α is a survival factor for hypoxic cells. Loss of HIF-1α in limb bud mesenchyme also delays joint specification, differentiation of mesenchymal cells into chondrocytes, and, most likely as a consequence of this early delay, their terminal hypertrophy.14,29 Distinct from HIF-1α, HIF-2α is not as critical for proper growth plate development.31
Figure 1.
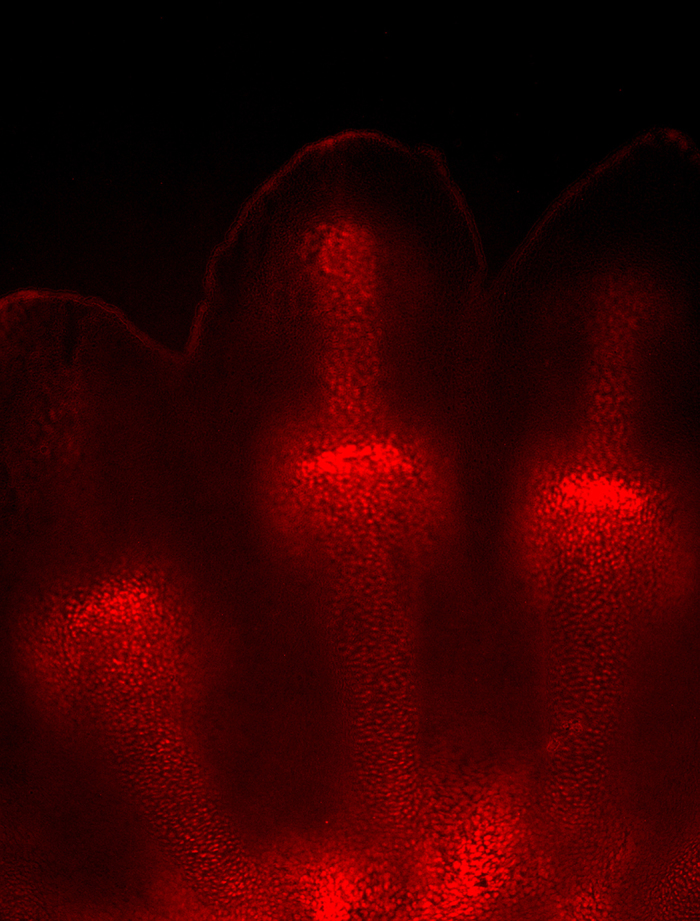
EF5 staining of E13.5 murine forelimb autopod. For additional details, please see text.
Like the fetal growth plate, the nucleus pulposus, a highly hydrated and proteoglycan-enriched tissue forming the inner portion of the intervertebral disc, is an avascular and moderately hypoxic structure.32 As for the growth plate, HIF-1α is crucial for development of the nucleus pulposus as well, as loss of HIF-1α results in its complete disappearance.33
All in all, these findings indicate that HIF-1α has a critical and non-redundant role in the development of avascular and hypoxic tissues. However, a series of important questions remain yet unresolved. We will discuss some of them in the next few paragraphs.
Our current working hypothesis is that hypoxic stress modulates differentiation of mesenchymal condensations into chondrocytes and development of the fetal growth plate by stabilizing and activating HIF-1α. The testing of this hypothesis relies on the use of hypoxia markers, such as EF5 or pimonidazole, to evaluate degree of oxygenation of the tissues of interest.27 EF5 and pimonidazole are 2-nitroimidazole-based markers.28 Such compounds are characterized by having a nitro group attached to the imidazole ring structure.28,34 In hypoxic conditions, this nitro group undergoes intracellular reduction to produce an amino group.28,34 One of the intermediate products formed during the reduction is highly reactive and can covalently bind to any cellular protein, forming adducts that can be recognized by specific antibodies.28,34 However, the efficiency of binding of nitroimadozole markers to intracellular proteins is contingent on cellular levels of nitroreductases,28 and this is an obvious limitation of the assay. Direct in vivo measurements of O2 tension in the bone marrow of live mice have been recently achieved using two-photon phosphorescence lifetime microscopy;35 however, the applicability of similar techniques to the study of fetal cartilage needs to be determined.
Even more challenging, hypoxia is not the only regulator of HIF-1α protein accumulation. For example, activation of the mTOR and ERK pathways is known to increase levels of HIF-1α protein by promoting its translation, at least in the context of cancer cells.28
The specific spatial distribution of the massive cell death phenotype observed in the HIF-1α null growth plate undoubtedly indicates that hypoxic chondrocytes require HIF-1α as a survival factor.14,16,19,20 However, it does not unequivocally prove that hypoxia is the only upstream factor controlling HIF-1α protein accumulation and/or activity in the fetal growth plate. The generation and analysis of additional models either in vitro or in vivo will be necessary to address this important issue in a more conclusive manner.
At last, though the experimental evidence clearly points to HIF-1α as a necessary and non-redundant survival and differentiation factor in fetal growth plate development, the detailed molecular mechanisms mediating these functions are still to be fully elucidated. Knowledge of such mechanisms is significant as it may provide novel insights into the biology of both chondrocyte and HIF-1.
Along these lines, studies by us and others demonstrated that HIF-1α improves efficiency of posttranslational modifications of collagens, which are major constituents of the cartilaginous matrix. These modifications include hydroxylation of the proline residues by a family of collagen prolyl 4-hydroxylases (C-P4Hs) distinct from the family of prolyl 4-hydroxylases involved in the destruction of HIFs.36,37 C-P4Hs catalyze the formation of 4-hydroxyprolines within collagen molecules; 4-hydroxyprolines are critical for the stability of the collagen triple helix.36,37 C-P4H-I is the main isoform in most cells, whereas C-P4H-II is highly expressed in chondrocytes. In vitro exposure to hypoxia increases accumulation of both C-P4H-I and C-P4H-II proteins in a HIF-1α-dependent manner.15 Because O2 is an essential substrate for C-P4Hs, a HIF-1α-induced increase in the amount of C-P4Hs could be of special importance in hypoxic tissues that are active in collagen synthesis such as the fetal growth plate as it would guarantee the accumulation of an appropriate amount of matrix despite reduced availability of O2 (Figure 2). A recent study supports the notion that HIF-1α-dependent stimulation of C-P4Hs accumulation and, consequently, formation of an adequate extracellular matrix could be a central event for survival of hypoxic chondrocytes.16 To further test this hypothesis, we recently pursued the in vivo characterization of the global knockouts of C-P4H-I and C-P4H-II. The global knockout of C-P4H-I is embryonic lethal, whereas mutant mice universally lacking C-P4H-II are alive and well with no obvious phenotypic abnormalities.38 Consistent with our hypothesis, whereas heterozygous inactivation of both C-P4H-I and C-P4H-II (p4ha1+/−;p4ha2+/−) or homozygous inactivation of C-P4H-II only (p4ha2−/−) leads to the formation of virtually normal cartilage, growth plates from mice carrying heterozygous inactivation of C-P4H-I combined to homozygous inactivation of C-P4H-II (p4ha1+/−; p4ha2−/−) display an inner cell death phenotype that is reminiscent of what we observed in HIF-1α null growth plates. However, despite an almost 70% decrease of C-P4H enzymatic activity, the inner cell death phenotype observed in the growth plates of p4ha1+/−;p4ha2−/− double mutant mice is extremely mild and transient; moreover, it virtually disappears postnatally, and it does not affect mouse survival. At last, mesenchymal condensations in limb buds isolated from p4ha1+/−;p4ha2−/− double mutant mice do not display any detectable delay of differentiation of mesenchymal cells into chondrocytes. Notably, the chondrodysplasia observed in p4ha1+/−;p4ha2−/− double mutant mice is not secondary to uncompensated ER stress, but it is most likely due to severe matrix abnormalities.38 Overall, these findings suggest that, differently from what has been recently suggested by others16 and from what our own in vitro data imply,15 C-P4Hs have a minor role downstream of HIF-1α in the developing growth plate. Similarly, C-P4Hs do not mediate HIF-1α function as an early differentiation factor in limb bud mesenchyme.
Figure 2.
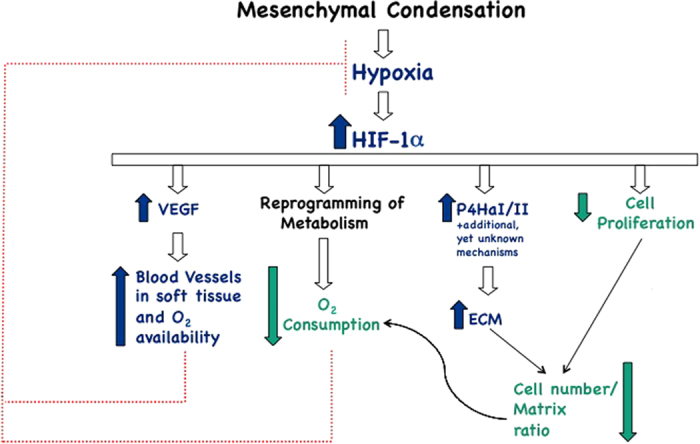
Hypoxia and HIF-1α in growth plate development: a well-orchestrated homeostatic response to keep hypoxia ‘in check'. Blue arrows: positive regulation; green arrows: negative regulation; red dotted lines: negative regulation. For additional details, please see text.
In the course of our investigations we learnt that viable chondrocytes at the periphery of HIF-1α null growth plates are severely more hypoxic than controls.20,30 We then asked the question whether O2 availability to the HIF-1α null growth plate was somehow impaired.
Vascular endothelial growth factor-A (VEGF) is a classical target of HIF-1α and it is expressed not only in hypertrophic chondrocytes, whereas it is a critical regulator of blood vessel invasion and replacement of cartilage by bone,13 but also in the inner hypoxic zone of the fetal growth plate, albeit at low levels.18,39
Loss of VEGF in chondrocytes causes cell death, decreases number of blood vessels in the surrounding soft tissue and increases degree of hypoxia of viable chondrocytes.30,39 VEGF overexpression in chondrocytes fully prevents the inner cell death and the increased hypoxia of VEGF null chondrocytes, most likely by augmenting number of blood vessels in the surrounding soft tissue.30 Hence, VEGF is a survival factor for chondrocytes in all likelihood by increasing O2 availability to these cells (Figure 2). Surprisingly, however, VEGF overexpression in chondrocytes only partially prevents the inner cell death and the increased hypoxia of HIF-1α null growth plates, despite a marked increase of number of blood vessels in the perichondrium and in the surrounding soft tissue.30 Taken together, these findings suggest that VEGF cannot be the main mediator of the HIF-1α role as a survival factor; moreover, it is improbable that decreased O2 availability is the only cause of the severe hypoxia of viable HIF-1α null chondrocytes (Figure 2). Of note, owing to the confounding effect of the massive cell death observed in mutant growth plates deficient in HIF-1α, it is still unclear whether the HIF-1α significantly contributes to modulating VEGF expression in late hypertrophic chondrocytes.
As Newton Harvey stated years ago, the oxygenation status of a cell is the net result of O2 availability and O2 consumption.40 The severe hypoxia of HIF-1α-deficient chondrocytes is not exclusively caused by reduced O2 availability; therefore, it has to be contributed by increased O2 consumption. This possibility is in line with the known biological actions of HIF-1α, as we will discuss in the next paragraph.
Keeping hypoxia ‘in check': a working hypothesis
HIF-1α is known to reprogram metabolism and to significantly impair mitochondrial respiration with various modalities. HIF-1α upregulates the expression of glucose transporters and of numerous enzymes of the non-oxidative glycolytic pathway.5 Moreover, HIF-1α shunts pyruvate away from the mitochondria by activating the gene encoding pyruvate dehydrogenase kinase 1 (PDK-1);41,42 PDK-1 phosphorylates pyruvate dehydrogenase, which is the enzyme that converts pyruvate to acetyl coenzyme A for entry into the mitochondrial tricarboxylic acid cycle. In addition, HIF-1α decreases mitochondrial mass by promoting mitochondrial autophagy.43 Furthermore, HIF-1 activates the transcription of the microRNA miR210; miR210 has been shown to block expression of the iron–sulfur cluster assembly proteins that are required for the function of the tricarboxylic acid cycle and of the electron transport chain.44 At last, HIF-1α impairs mitochondrial biogenesis by reducing levels of expression of proliferator-activated receptor-γ coactivator-1α.45 By attenuating mitochondrial respiration, and thus by lowering mitochondrial O2 consumption, HIF-1α may mitigate hypoxia and may also reduce production of reactive oxygen species by the electron transport chain, which in hypoxic conditions paradoxically tends to increase in a HIF-independent manner.46 HIF-1α is a negative regulator of reactive oxygen species accumulation by also inducing the mitochondrial gene NDUFA4L2 (NADH dehydrogenase [ubiquinone]1alphasubcomplex, 4-like 2),47 and by paradoxically optimizing activity of cytochrome c oxidase.48 Taken altogether, HIF-1α deficiency in a hypoxic tissue likely impairs non-oxidative glycolysis, exacerbates hypoxia and augments production of reactive oxygen species. Each of these metabolic changes could significantly affect the ability of hypoxic cells to survive and differentiate.
Not surprisingly, the fetal growth plate is a hypoxic structure that lives on non-oxidative glycolytic metabolism rather than on mitochondrial respiration.20,30 Moreover, us and others demonstrated that expression of messenger RNAs encoding enzymes of the non-oxidative glycolysis and PDK-1 is significantly reduced in HIF-1α-deficient chondrocytes.18,20,30,49
All in all, it is possible that, in addition to support adenosine triphosphate production through non-oxidative glycolysis,49 a key function of HIF-1α in the developing growth plate is to keep hypoxia ‘in check' not only by increasing O2 availability, as discussed above, but also by reducing O2 consumption through reprogramming of metabolism (Figure 2). HIF-1α-dependent reprogramming of metabolism would prevent hypoxic cells from becoming virtually anoxic, a status that would be incompatible with cell survival and differentiation. Thus, HIF-1α could be essential for survival of hypoxic chondrocytes and for timely differentiation of hypoxic mesenchymal cells into chondrocytes at least in part by negatively regulating mitochondrial respiration, and, consequently, mitochondrial O2 consumption. HIF-1α-dependent mitigation of reactive oxygen species production could contribute to this complex homeostatic response.
The hypothesis outlined above implies that, although electron transport chain is essential for viability of well-oxygenated tissues, it is dispensable for tissues that are physiologically hypoxic or ‘functionally anaerobic' and thus live on non-oxidative glycolysis, such as the fetal cartilage.
Moreover, in fetal growth plates deficient in HIF-1α, the proliferation rate of viable chondrocytes is strikingly increased.20 Conversely, loss of von Hippel-Lindau,the E3 ubiquitin ligase that targets HIF-1α to the proteasome for degradation, in growth plate chondrocytes in vivo impairs chondrocyte proliferation, and appears to increase accumulation of matrix in between cells as documented by routine histology.17,18 Along these lines, in vitro exposure of murine primary chondrocytes to hypoxia increases accumulation of type II collagen protein in a HIF-1α-dependent manner and, curiously, without affecting expression of type II collagen messenger RNA.49 All in all, these findings are consistent with the notion that stabilization of HIF-1α impairs proliferation and causes fibrosis.15,50,51,52 HIF-1α could thus be an important regulator of the ratio between extracellular matrix and cell number in the developing growth plates, and this could contribute to keep O2 consumption ‘in check' (Figure 2).
Beyond O2 homeostasis
A ‘pessimist' could argue that hypoxia is an accident, an inevitable mistake that occurs during organogenesis, and Nature tries its best to fix it by turning on an elegantly orchestrated homeostatic response.
Alternatively, we would like to propose that the role of hypoxia and HIF-1α in organogenesis, and particularly in endochondral bone development, goes beyond O2 homeostasis. We will briefly discuss some experimental evidences in support of this possibility.
It has been recently reported that HIF-1α and HIF-2α are direct transcriptional regulators of SOX9 expression in murine growth plate chondrocytes and in human articular surface chondrocytes, respectively.14,53,54 SOX9 is the master transcription factor of cartilage, as no cartilage is formed in its absence.55,56,57 Along these lines, it has also been shown that in vitro low O2 tension promotes differentiation of mesenchymal stem cells into chondrocytes.23
More generally, it has been well documented that HIF-1α modulates cell proliferation, accumulation of extracellular matrix, angiogenesis and reprogramming of metabolism, which are all key events in both organogenesis and O2 homeostasis.3,58,59,60
It is thus tempting to speculate that gradients of oxygenation exploit the complex actions of HIF-1α on O2 homeostasis to control tissue development and differentiation. In agreement with this possibility, we have recently reported that loss of von Hippel-Lindau in limb bud mesenchyme, and thus increased HIF transcriptional activity at this site, considerably alters size, shape and overall development of the skeletal elements.17 This finding is consistent with the hypothesis that stabilization of HIF-1α regulates tissue morphogenesis. The testing of this hypothesis, however, requires further studies, as much still needs to be learnt in the fields of hypoxia, HIFs and organogenesis.
Acknowledgments
This work was supported by the NIH RO1 (AR065403-01) grant (to ES). Christophe Merceron received funding from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme (FP7/2007–2013) registered under the Research Executive Agency grant agreement no. 300388.
Footnotes
The authors declare no conflict of interest.
References
- Dunwoodie SL. The role of hypoxia in development of the Mammalian embryo. Dev Cell 2009; 17: 755–773. [DOI] [PubMed] [Google Scholar]
- Giaccia AJ, Simon MC, Johnson R. The biology of hypoxia: the role of oxygen sensing in development, normal function, and disease. Genes Dev 2004; 18: 2183–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes C, Carmeliet G, Schipani E. Hypoxia-driven pathways in bone development, regeneration and disease. Nat Rev Rheumatol 2012; 8: 358–366. [DOI] [PubMed] [Google Scholar]
- Masson N, Ratcliffe PJ. Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer Metab 2014; 2: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 2012; 148: 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiji M, Kageyama Y, Pete E, Horikawa I, Barrett J, Huang L. Hif-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J 2004; 23: 1949–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsenty G. The complexities of skeletal biology. Nature 2003; 423: 316–318. [DOI] [PubMed] [Google Scholar]
- Kronenberg H. Developmental regulation of the growth plate. Nature 2003; 423: 332–336. [DOI] [PubMed] [Google Scholar]
- Provot S, Schipani E. Molecular mechanisms of endochondral bone development. Biochem Biophys Res Commun 2005; 328: 658–665. [DOI] [PubMed] [Google Scholar]
- Zelzer E, Olsen B. The genetic basis for skeletal diseases. Nature 2003; 423: 343–348. [DOI] [PubMed] [Google Scholar]
- Yang L, Tsang KY, Tang HC, Chan D, Cheah KS. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci USA 2014; 111: 12097–12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 1998; 93: 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med 1999; 5: 623–628. [DOI] [PubMed] [Google Scholar]
- Amarilio R, Viukov S, Sharir A, Eshkar-Oren I, Johnson R, Zelzer E. Hif1alpha regulation of Sox9 is necessary to maintain differentiation of hypoxic prechondrogenic cells during early chondrogenesis. Development 2007; 134: 3917–3928. [DOI] [PubMed] [Google Scholar]
- Aro E, Khatri R, Gerard-O'Riley R, Mangiavini L, Myllyharju J, Schipani E. Hypoxia-inducible factor-1 (HIF-1) but not HIF-2 is essential for hypoxic induction of collagen Prolyl 4-hydroxylases in primary newborn mouse epiphyseal growth plate chondrocytes. J Biol Chem 2012; 287: 37134–37144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentovim L, Amarilio R, Zelzer E. HIF1alpha is a central regulator of collagen hydroxylation and secretion under hypoxia during bone development. Development 2012; 139: 4473–4483. [DOI] [PubMed] [Google Scholar]
- Mangiavini L, Merceron C, Araldi E, Khatri R, Gerard-O'Riley R, Wilson TL et al. Loss of VHL in mesenchymal progenitors of the limb bud alters multiple steps of endochondral bone development. Dev Biol 2014; 393: 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfander D, Kobayashi T, Knight MC, Zelzer E, Chan DA, Olsen BR et al. Deletion of Vhlh in chondrocytes reduces cell proliferation and increases matrix deposition during growth plate development. Development 2004; 131: 2497–2508. [DOI] [PubMed] [Google Scholar]
- Provot S, Schipani E. Fetal growth plate: a developmental model of cellular adaptation to hypoxia. Ann N Y Acad Sci 2007; 1117: 26–39. [DOI] [PubMed] [Google Scholar]
- Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev 2001; 15: 2865–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adesida AB, Grady LM, Khan WS, Millward-Sadler SJ, Salter DM, Hardingham TE. Human meniscus cells express hypoxia inducible factor-1alpha and increased SOX9 in response to low oxygen tension in cell aggregate culture. Arthritis Res Ther 2007; 9: R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanichai M, Ferguson D, Prendergast PJ, Campbell VA. Hypoxia promotes chondrogenesis in rat mesenchymal stem cells: a role for AKT and hypoxia-inducible factor (HIF)-1alpha. J Cell Physiol 2008; 216: 708–715. [DOI] [PubMed] [Google Scholar]
- Merceron C, Vinatier C, Portron S, Masson M, Amiaud J, Guigand L et al. Differential effects of hypoxia on osteochondrogenic potential of human adipose-derived stem cells. Am J Physiol Cell Physiol 2010; 298: C355–C364. [DOI] [PubMed] [Google Scholar]
- Vissers LE, Fano V, Martinelli D, Campos-Xavier B, Barbuti D, Cho TJ et al. Whole-exome sequencing detects somatic mutations of IDH1 in metaphyseal chondromatosis with D-2-hydroxyglutaric aciduria MC-HGA). Am J Med Genet A 2011; 155A: 2609–2616. [DOI] [PubMed] [Google Scholar]
- Saito T, Fukai A, Mabuchi A, Ikeda T, Yano F, Ohba S et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat Med 2010; 16: 678–686. [DOI] [PubMed] [Google Scholar]
- Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev 2008; 9: 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch CJ. Measurement of absolute oxygen levels in cells and tissues using oxygen sensors and 2-nitroimidazole EF5. Methods Enzymol 2002; 352: 3–31. [DOI] [PubMed] [Google Scholar]
- Kizaka-Kondoh S, Konse-Nagasawa H. Significance of nitroimidazole compounds and hypoxia-inducible factor-1 for imaging tumor hypoxia. Cancer Sci 2009; 100: 1366–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provot S, Zinyk D, Gunes Y, Kathri R, Le Q, Kronenberg HM et al. Hif-1alpha ;regulates differentiation of limb bud mesenchyme and joint development. J Cell Biol 2007; 177: 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes C, Araldi E, Haigh K, Khatri R, Van Looveren R, Giaccia AJ et al. VEGF-independent cell-autonomous functions of HIF-1alpha regulating oxygen consumption in fetal cartilage are critical for chondrocyte survival. J Bone Miner Res 2012; 27: 596–609. [DOI] [PubMed] [Google Scholar]
- Araldi E, Khatri R, Giaccia AJ, Simon MC, Schipani E. Lack of HIF-2alpha in limb bud mesenchyme causes a modest and transient delay of endochondral bone development. Nat Med 2011; 17: 25–26 author reply 27–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DC, Adams CS, Albert TJ, Shapiro IM, Evans SM, Koch CJ. In situ oxygen utilization in the rat intervertebral disc. J Anat 2007; 210: 294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merceron C, Mangiavini L, Robling A, Wilson TL, Giaccia AJ, Shapiro IM et al. Loss of HIF-1alpha in the notochord results in cell death and complete disappearance of the nucleus pulposus. PloS One 2014; 9: e110768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsman MR, Mortensen LS, Petersen JB, Busk M, Overgaard J. Imaging hypoxia to improve radiotherapy outcome. Nat Rev Clin Oncol 2012; 9: 674–687. [DOI] [PubMed] [Google Scholar]
- Spencer JA, Ferraro F, Roussakis E, Klein A, Wu J, Runnels JM et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014; 508: 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myllyharju J, Schipani E. Extracellular matrix genes as hypoxia-inducible targets. Cell Tissue Res 2010; 339: 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipani E. Posttranslational modifications of collagens as targets of hypoxia and Hif-1alpha in endochondral bone development. Ann N Y Acad Sci 2010; 1192: 317–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aro E, Salo AM, Khatri R, Finnilä M, Miinalainen I, Sormunen R et al. Severe extracellular matrix abnormalities and chondrodysplasia in mice lacking collagen prolyl 4-hydroxylase isoenzyme II in combination with a reduced amount of isoenzyme I. J Biol Chem 2015; 290: 16964–16978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelzer E, Mamluk R, Ferrara N, Johnson R, Schipani E, Olsen B. VEGFA is necessary for chondrocyte survival during bone development. Development 2004; 131: 2161–2171. [DOI] [PubMed] [Google Scholar]
- Harvey E. The oxygen consumption of luminous bacteria. J Gen Physiol 1928; 11: 469–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006; 3: 177–185. [DOI] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006; 3: 187–197. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008; 283: 10892–10903. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab 2009; 10: 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Ma Z, Zhao C, Wang Y, Wu G, Xiao J et al. HIF-1alpha and HIF-2alpha are critically involved in hypoxia-induced lipid accumulation in hepatocytes through reducing PGC-1alpha-mediated fatty acid beta-oxidation. Toxicol Lett 2014; 226: 117–123. [DOI] [PubMed] [Google Scholar]
- Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 2000; 275: 25130–25138. [DOI] [PubMed] [Google Scholar]
- Tello D, Balsa E, Acosta-Iborra B, Fuertes-Yebra E, Elorza A, Ordonez A et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting Complex I activity. Cell Metab 2011; 14: 768–779. [DOI] [PubMed] [Google Scholar]
- Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007; 129: 111–122. [DOI] [PubMed] [Google Scholar]
- Pfander D, Cramer T, Schipani E, Johnson R. HIF-1alpha controls extracellular matrix synthesis by epiphyseal chondrocytes. J Cell Sci 2003; 116: 1819–1826. [DOI] [PubMed] [Google Scholar]
- Higgins DF, Biju MP, Akai Y, Wutz A, Johnson RS, Haase VH. Hypoxic induction of Ctgf is directly mediated by Hif-1. Am J Physiol Renal Physiol 2004; 287: F1223–F1232. [DOI] [PubMed] [Google Scholar]
- Chen YY, Li YL, Li CJ, Chen LP, Jiang YP, Gao H et al. Studying the growth of cervical carcinoma nests and angiogenesis by immunostaining, quantitation and three-dimensional structural analysis. Anal Quant Cytol Histol 2000; 22: 80–84. [PubMed] [Google Scholar]
- Erler J, Bennewith K, Nicolau M, Dornhofer N, Kong C, Le Q et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006; 440: 1222–1226. [DOI] [PubMed] [Google Scholar]
- Lafont JE, Talma S, Hopfgarten C, Murphy CL. Hypoxia promotes the differentiated human articular chondrocyte phenotype through SOX9-dependent and -independent pathways. J Biol Chem 2008; 283: 4778–4786. [DOI] [PubMed] [Google Scholar]
- Robins JC, Akeno N, Mukherjee A, Dalal RR, Aronow BJ, Koopman P et al. Hypoxia induces chondrocyte-specific gene expression in mesenchymal cells in association with transcriptional activation of Sox9. Bone 2005; 37: 313–322. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Lefebvre V. Unraveling the transcriptional regulatory machinery in chondrogenesis. J Bone Miner Metab 2011; 29: 390–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre V, Huang W, Harley VR, Goodfellow PN, de Crombrugghe B. SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol Cell Biol 1997; 17: 2336–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet 1999; 22: 85–89. [DOI] [PubMed] [Google Scholar]
- Shyh-Chang N, Daley GQ, Cantley LC. Stem cell metabolism in tissue development and aging. Development 2013; 140: 2535–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell 2012; 11: 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslaa T, Teitell MA. Pluripotent stem cell energy metabolism: an update. EMBO J 2015; 34: 138–153. [DOI] [PMC free article] [PubMed] [Google Scholar]