

Abstract
Muscle weakness is an important phenotype of many diseases that is linked to impaired locomotion and increased mortality. The force that a muscle can generate is determined predominantly by muscle size, fiber type and the excitation–contraction coupling process. Here we describe methods for the histological assessment of whole muscle to determine fiber cross-sectional area and fiber type, determination of changes in myocyte size using C2C12 cells, in vivo functional tests and measurement of contractility in dissected whole muscles. The extensor digitorum longus and soleus muscles are ideally suited for whole-muscle contractility, and dissection of these muscles is described.
Introduction
Assessment of physiological muscle function helps identify potential mechanisms involved in muscle disease and the effect of treatments. The use of experimental mouse models is a central component for understanding the causes of muscle diseases and the development and testing of therapeutics. In addition, myoblasts isolated from mouse muscle have been used to identify intracellular signaling mechanisms in muscle that would be difficult to perform in living animals. In this report, we describe the methods useful for muscle studies ranging from myoblasts through histological assessment of muscle tissue to whole-muscle contractility. In situ and in vivo methods of muscle strength measurement are also possible.1,2 These methods maintain an intact blood and nerve supply. In vivo methods can be performed repeatedly in a longitudinal study, whereas whole-muscle contractility and in situ methods are end of experiment studies.
Muscle excision and histological analysis
Muscle histological techniques provide a powerful means for analysis of morphological parameters such as fiber cross-sectional area (CSA) and muscle fiber type. These are among the first considerations made when assessing skeletal muscle mass and muscle phenotype. The use of in vitro culture systems (for example, C2C12 myoblasts) has also been used to identify and characterize factors responsible for maintaining muscle size or driving atrophic programs in muscle. Beyond basic characterization and classification of muscle and in vitro cultures, in vivo measurements of muscle strength are invaluable to assess muscle function, as well as therapeutic approaches for disease. Finally, muscle contractility is used to measure the force generation of isolated whole muscles (excluding neuronal influences). Together these approaches can provide an understanding of overall muscle weakness in mice.
Sample preparation for cryosectioning—fresh frozen technique
Skeletal muscle cryosectioning can be extremely useful for a series of applications spanning from morphological analysis (to determine fiber CSA) to immunofluorescence and immunohistochemical stainings (for protein localization and expression) and to in situ hybridization.3
Prepare an insulated container with liquid nitrogen and a 100 ml Pyrex or a copper jar containing ∼30 ml isopentane (5-methylbutane).
Place the jar containing the isopentane into the liquid nitrogen, so it is partially submerged. It is critical that the isopentane reaches the correct temperature in order to perform a proper freezing of the muscle specimen. When the isopentane becomes slightly viscous and forms a solid white laminate lining the inside of the beaker (temperature: −160 °C), it is ready to use to freeze muscle tissue rapidly.
Skeletal muscles should be carefully removed from the hind limb (see Isolated whole-muscle contractility and Surgical instruments section below for a list of typical instruments used for dissection). In order to properly do so, extend a leg of the mouse by hanging it with the use of a buret clamp. Gently remove the skin and expose the underlying muscle tissues.
Remove the gastrocnemius (GSN), tibialis anterior (TA), extensor digitorum longus (EDL), soleus (SOL) and quadriceps muscles. Blot the muscles dry with filter paper and weigh the muscles on an analytical scale. Accuracy is extremely important, especially for small muscles, such as TA, EDL and SOL. Generally, any muscle can be frozen for histological examination. However, based on the purpose of the study, some muscles might be more suitable than others. For example, the EDL and the TA muscles contain predominantly fast-twitch, glycolytic and type II fibers, whereas the SOL muscle contains predominantly slow-twitch, oxidative and type I fibers (in rat, or a mix of type I and type II fibers in mouse). The muscle should be laid out straight and flat, and it should not be twisted or stretched.
Place a small amount of fresh embedding medium (for example, Tissue-Tek O.C.T. (optimal cutting temperautre) compound (Sakura Finetek, Torrance, CA, USA)) on a chuck (cork).
Handle the muscle by the tendon holding it vertically on the chuck, in order to maintain the orientation of the fibers and allow cross-sections. Gently immerse the base of the muscle into the embedding medium. It is important not to surround the muscle completely with the embedding medium, but it is critical to maintain the cross-sectional orientation.
Dip the chuck with the muscle (gently immersed into the embedding medium) into the isopentane bath (10 s). The usual freezing time is 7–15 s, depending on specimen size and composition. Immersion in the freezing solution should not last more than is needed to completely freeze the specimen. Freezing too long will fracture the tissue block, too short will cause ice crystal formation. A well-frozen specimen should be chalky white. Once the specimen is frozen, place it into a small plastic bag or a specimen tube (50 ml) and immediately store in a deep freezer at –80 °C or in liquid nitrogen.
Muscle sectioning and determination of CSA
Proper handling of skeletal muscle samples for histology is critical to perform accurate analysis of muscle fiber CSA. In order to do so, set the working temperature of the cryostat inner chamber at −23 °C and let your specimen come to working temperature for at least a couple of hours. Cut multiple 8-μm thick sections of the muscle specimen at its mid-belly region and collect them on Superfrost-Plus glass slides. For hematoxylin and eosin (H&E), let the sections dry at room temperature for at least 1 h. Otherwise, store the unstained slides at −80 °C for long-term or −20 °C for short-term storage.
For the evaluation of muscle morphology (CSA), perform H&E staining of muscle sections.4 H&E staining followed by a manual analysis of fiber size is commonly used to assess CSA in skeletal muscle, although some novel semi- or fully automated methods, associated with the immunohistochemical detection of the muscle fiber boundaries, are being actively tested.5,6 H&E is a charge-based staining. Hematoxylin acts as a basic dye, and therefore it mostly stains the cellular basophilic structures containing nucleic acids, such as ribosomes, cell nucleus and some RNA-rich cytoplasmic regions. Eosin acts as an acid dye and therefore stains most of the cytoplasm, as well as the intracellular or the extracellular protein components (Figure 1a).
Figure 1.

Histological analysis of skeletal muscle. (a) Photomicrograph of muscle cross-section stained by H&E with a schematic on how to use the ImgaeJ software to outline and measure the cross-sectional area of the fibers. Magnification 20X, scale bar: 50 μm. (b–c) Representative fiber typing. Consecutive 8-μm thick sections from liquid nitrogen-cooled isopentane frozen gastrocnemius muscle were stained for (b) Myosin Heavy Chain slow (BA-D5 mouse antibody diluted 1:50; green) or (c) Myosin Heavy Chain fast (SC-71 mouse antibody diluted 1:50; green) and visualized by anti-mouse-IgG Alexa Fluor-488 (Life Technologies) secondary antibody diluted 1:1000 in PBS. (d) Representative photomicrograph of C2C12 murine myotube cultures fixed in ice-cold 4% paraformaldehyde in PBS and subsequently incubated in anti-myosin heavy chain (MF-20 clone, Developmental Studies Hybridoma Bank) monoclonal antibody diluted 1:1000 in PBS overnight at 4 °C. Myotubes were then visualized using anti-mouse-IgG Alexa Fluor-488 (Life Technologies) secondary antibody diluted 1:1000 in PBS at room temperature for 30–45 min. Red bars indicate the minimum diameter for each fiber.
H&E staining procedure
Preparation
If frozen, bring the slides to room temperature and let them dry for at least 30 min.
Hematoxylin staining
Incubate the slides with Mayer's hematoxylin solution for 8–10 min. This will stain the nuclei dark blue/purple.
Blueing reaction
Place the slides in a glass chamber and rinse under warm running tap water for up to 10 min. Hematoxylin coupling to the tissue occurs in combination with the metallic salt aluminum sulfate that, in an alkaline solution, combines with -OH groups of water to form aluminum hydroxide. Because most hematoxylin-metal formulae are fairly acid, the nuclei will at first be stained in purple. As tap water is considerably more alkaline compared with the pH of most hematoxylins (pH 2.6–2.9), washing the sections in tap water in order to neutralize the acid and free the -OH groups will change the color of the nuclei to blue.
Eosin staining
Add acetic acid (1:100) to the 0.1% eosin solution and incubate the slides for up to 2 min. The tissue will be stained in red-orange.
Washing
Put the slides in a glass chamber and wash three times for 1 min each with ddH2O to remove excess of eosin.
Dehydration
Perform the dehydration process by placing the slides in 70% ethanol for 1 min, 90% ethanol for 1 min, 100% ethanol for 2 min and xylene for 2 min.
Mounting
Mount the slides with 1–2 drops of a xylene-based mounting media (for example, Permount or similar) and cover with cover slides, carefully avoiding bubbles. Press the slides heavily to remove the excess medium and let dry at room temperature. Store the slides at room temperature.
The morphological features of the muscle are identified using digital images of the stained section. Images are acquired using a bright-light microscope (10 × or 20 × magnification objective is appropriate), digital camera and image capture software (such as ZEN Pro, Carl Zeiss Microscopy GmbH, Jena, Germany). The presence of a scale bar will be required to determine the CSA. It is recommended that at least 20–30 random fields be recorded for each muscle section.4 The images will then be analyzed for fiber size/diameter and the percentage of centralized nuclei. Centralized nuclei are a marker of muscle regeneration. In order to establish the muscle fiber size, determine the minimum ‘Feret's diameter' instead of the fiber area, as this is the most reliable tool due to the fact that this parameter is independent of the angle of sectioning.7 Other parameters, such as the ‘minimal inner diameter' and the ‘minimal outer diameter', also insensitive to the plane of sectioning, can be used as an indication of the fiber size instead of the Feret's diameter.
Other histological features (for example, fibrosis, myofiber necrosis) can also be identified manually by the researcher and quantified using image analysis software, such as ImageJ (NIH, U.S. National Institutes of Health, Bethesda, MD, USA). The data generated by doing so are then subjected to the appropriate statistical analyses.
Muscle fiber type specification
Mammalian skeletal muscle comprises different fiber types, whose identity is first determined during embryonic development and can be later modulated by several stimuli, among which are neural and hormonal factors. At the single fiber level, it is possible to distinguish four major fiber types, namely type 1, 2A, 2X and 2B, based on the presence of specific myosin heavy chain (MyHC) isoforms, whose complete inventory and expression pattern are now available: MyHC-1/slow, coded by the MYH7 gene, MyHC-2A, coded by MYH2, MyHC-2X, coded by MYH1, and MyHC-2B, coded by MYH4.8 It is also possible to differentiate these fiber types on the basis of their oxidative/glycolytic metabolism, type 1 and 2A fibers being more oxidative and type 2B fibers more glycolytic. These four fiber populations are present in mice, rats and many other mammalian species, although only type 1, 2A and 2X fibers are present in most human muscles.9 Moreover, intermediate hybrid fibers, containing type 1 and 2A, or 2A and 2X or 2X and 2B MyHCs, can be observed in normal muscles, as well as in conditions when fiber type shifts take place, either in response to exercise or electrical stimulation or during muscle wasting associated with atrophying conditions such as cancer, denervation or disuse.10,11,12,13
MyHC immunofluorescence staining
The easiest and the fastest method to determine the muscle fiber-type specification is based on the use of monoclonal antibodies specific for the different myosin isoforms MYHC1, MYHC2A and MYHC2B. These antibodies are produced from cultures of hybridoma cell lines BA-D5, SC-71 and BF-F3, respectively, and can be easily obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa.9 Briefly:
Prepare several consecutive muscle sections from muscle specimens, previously frozen in liquid nitrogen-cooled isopentane.
Let the sections dry at room temperature. For the purpose of fiber typing, no fixation is required.
Expose the muscle sections to blocking buffer (8% Bovine Serum Albumin in phosphate-buffered saline (PBS)) for at least 1 h.
Incubate the muscle sections with 10 μg ml−1 BA-D5, SC-71 and BF-F3 monoclonal antibodies for 1 h at room temperature (or 4 °C for overnight).
Wash the sections 3 times for 5 min each in PBS-Tween 0.1%.
Incubate the slides with goat anti-mouse secondary antibodies IgG2b Alexa Fluor 350, IgG1 Alexa Fluor 594 and IgM Alexa Fluor 488 (Life Technologies, Carlsbad, CA, USA) for 30–60 min.
Wash the slides three times for 5 min each in PBS-Tween 0.1% then perform a final PBS rinse.
Mount the slides in ProLong antifade reagent (Life Technologies) and image slides (Figure 1b and c).
Myosin ATPase staining
The histochemical assay for myofibrillar ATPase activity can be used to distinguish between fast- and slow-contracting muscle fibers. As previously stated, muscle fibers may be generally classified as type 1 (slow, oxidative fibers) and type 2 (fast, glycolytic fibers). Type 2 muscle fibers are further subdivided as 2A (glycolytic/oxidative) or 2B (glycolytic). 2C fibers are normally considered as fibers that are transitioning to other types in association with disease states.
The myosin ATPase activity is indicative of contraction speed. This assay is based on a preincubation with either an acid (pH ∼4) or a basic (pH ∼10) solution. Acid preincubations inhibit the myosin ATPase activity in fast (mammalian type 2A, 2X and 2B) fiber types but not slow (mammalian type 1). Conversely, basic preincubations inhibit myosin ATPase in slow fiber types only. For this assay, ATP represents the reaction energy source and substrate, whereas inorganic phosphate (Pi) is the reaction product, and myosin is the enzyme. In order for the Pi to become histochemically visible, a chemical reaction with calcium (Ca2+) leads to the formation of a calcium phosphate (CaPO4) precipitate. Subsequently, CaPO4 is converted into cobalt sulfide (CoS2), a pH-dependent brown-black easily detectable precipitate. This assay is capable of discriminating between the different sets of fibers based on the fact that fast-contracting muscle fibers hydrolyze ATP faster than slow-contracting fibers; therefore, fast-contracting fibers will appear stained in dark, and slow-contracting fibers will appear light (see below for a detailed description of fiber typing based on the amount/intensity of the staining.
Several aspects of myosin ATPase staining are critical including the following: (1) the pH of all solutions, (2) the timing for each step, (3) the sodium hydroxide should be less than 2-week old (especially the 0.1 N) and (4) the stock of ammonium sulfide must still be yellow (as it oxidizes the color becomes red and does not react properly).
Pre-incubating solutions
For best fiber differentiation, prepare these solutions fresh!
(a) pH 9.4 ATP solution. This solution will reveal type 2 fibers as dark, type 1 fibers as light and type 2C fibers as dark intermediate. For 20 ml solution, prepare as follows: 4.0 ml 0.1 M sodium barbital, 4.0 ml 0.18 M calcium chloride and 12.0 ml deionised water. Adjust pH to 9.7–9.8 just prior to use with a few drops of 0.1 N NaOH (muscle other than human usually requires a pH of ∼10.2)
(b) pH 4.6 ATP solution. This solution will identify type 1 fibers as the darkest, type 2B and 2C fibers as intermediate and type 2 A fibers as very light. For 20 ml solution, prepare as follows: 5.0 ml Barbital Acetate Solution, 10.0 ml 0.1 N HCl and 5.0 ml deionized water. Adjust pH between 4.60 and 4.62 just prior to use with a few drops of 1 N HCl.
(c) pH 4.3 ATP solution. This solution shows type 1 fibers as the darkest, type 2C fibers as intermediate and type 2A and 2B as the lightest. For 20 ml solution, prepare as follows: 5.0 ml Barbital Acetate Solution, 10.0 ml 0.1 N HCl and 5.0 ml deionized water. Adjust pH between 4.25 and 4.3 just prior to use with a few drops of 1 HCl.
(d) ATP incubating solution (volume reported is sufficient for three staining jars). ATP powder (60 mg), 6.0 ml 0.1 M Sodium Barbital, 21.0 ml deionized water and 3.0 ml 0.18 M Calcium Chloride (added as the last in order to prevent precipitation of ATP). Prepare fresh prior to use and adjust pH to 9.4 (9.40–9.45) with a few drops of 1 N NaOH. Remember: do not allow the pH to become excessively alkaline, as this will cause precipitation of the ATP.
Staining procedure
Cut three consecutive (adjacent) sections from fresh frozen muscle samples.
Place the pre-incubating solutions (pH 4.6, pH 4.3 and pH 9.4) in separate glass jars.
Incubate the muscle sections in the pre-incubating solutions (start incubations at the same time).
In particular, incubate the sections in the pH 4.6 and pH 4.3 ATP solutions for no more than 5 min at room temperature. Incubate in the pH 9.4 ATP solution for 15 min at room temperature.
Rinse each section one time with deionized water.
Add the ATP solution to each section and incubate for 25 min for the pH 4.6 and pH 4.3 ATP solutions or 15 min for the pH 9.4 ATP solution.
Wash each section with three changes of 1% calcium chloride (3 min each).
Expose each section to 2% cobalt chloride solution for 10 min.
Wash with 3–5 changes of 0.1 M sodium barbital (diluted 1:20 in deionized water).
Rinse in deionized water for at least five times.
In a fume hood, prepare a 2% v/v solution of ammonium sulfide and add to each section for 20–30 s (sections will turn very dark).
Rinse in the fume hood in tap water for at least five times.
Perform the dehydration process by placing the slides in 70% ethanol for 1 min, 90% ethanol for 1 min, 100% ethanol for 2 min and xylene for 2 min.
Mount the glass slides with Canada Balsam.
C2C12 myoblasts as a model of muscle atrophy in vitro
The cell line C2C12 is an immortal line of mouse skeletal myoblasts originally derived from satellite cells isolated from the muscle of a C3H mouse donor.14 From the C2 cell line, the immortal subline C2C12 was originated.15 These cells differentiate well into myotubes under appropriate culture conditions. In particular, myogenic differentiation occurs upon reaching 90% confluence by switching the myoblasts to a medium containing 2% horse serum. Alternatively, it is feasible to use satellite cells from a donor biopsy and use these to produce myotubes.
During the differentiation process, the general monolayer architecture changes from fusiform or star-shaped cells to elongated confluent cells, thus forming long, multinucleated myotubes that can be used to study aspects of muscle differentiation and muscle homeostasis in vitro.16 During differentiation, the originating myotubes progressively express actin and myosin, whereas myoblasts do not normally show expression of these proteins. Here below is a brief protocol for the use of C2C12 as a model for the study of muscle atrophy in vitro3,17 (Figure 1d). These cells require passage every 1–2 days.
Cell growth in vitro
Thaw a vial of C2C12 cells in water bath at 37 °C. Transfer cells to a proper culture dish with 20 ml fresh complete medium (Dulbecco's Modified Eagle Medium (DMEM), high glucose, 10% fetal bovine serum, 1% sodium pyruvate, 1% penicillin-streptomycin and 1% glutamine). Place in incubator. After one day, remove the medium and add fresh media.
When cells are 50–60% confluent (very few cells are physically touching each other), split 1:4. Please note: it is very important to not let the cells become fully confluent. Indeed, the cell–cell contact may partially begin the differentiation process.
To passage, remove and discard the culture medium.
Rinse twice with sterile PBS.
Add 0.25% trypsin-EDTA and as soon as the cells start to detach from the culture dish transfer them to a new one (typically 6- or 12-well plates work fine) and add fresh complete medium.
Differentiation treatment
When the cells are fully confluent, initiate the differentiation process by rinsing the cells once with PBS and by adding freshly prepared low-serum differentiation medium (DMEM high glucose, 2% Horse Serum, 1% sodium pyruvate, 1% penicillin-streptomycin and 1% glutamine).
Replace the medium every 24 h up to the 72 h time point and, after that, every 12 h (as the differentiation progresses, the cells acidify the medium more quickly).
The cells are fully differentiated and ready to be used for experiments after 4–5 days in the differentiation medium.
We previously took advantage of this model to investigate the mechanisms that cause myotube atrophy in the presence of pro-inflammatory cytokines, such as interleukin-6 or tumor necrosis factor, previously shown to be causative of muscle wasting in both in vitro and in vivo conditions.3,17,18 In order to do so, the cell layer is generally exposed to pro-catabolic cytokines (100 ng ml−1), with or without specific inhibitors/antagonists. Typically, muscle atrophy in the myotubes can be assessed at several time points, early (30 min, 1, 2, 6 h), as well as late ones (24, 48, 72 h). Quite often, it is hard to detect modulation of fiber size at early time points, although the activation of signaling pathways responsible for muscle atrophy might be already evident (Figure 1d 3,17,18).
Assessment of myotubes size
Prepare ice-cold 4% paraformaldehyde in PBS.
Aspirate the medium and rinse the 6- or 12-well plates with ice-cold 1 × PBS.
Fix the cells in ice-cold 4% paraformaldehyde in PBS for 5 min at room temperature.
Wash the cell monolayers twice with ice-cold PBS for 5 min each. Alternatively, cells can be kept in 4 °C for several days before proceeding to further steps.
Add 8% bovine serum albumin/PBS and incubate for 1 h at room temperature to avoid the nonspecific binding of the antibodies.
Wash three times in PBS, 5 min each.
Add the mouse anti-MyHC monoclonal antibody (MF-20 clone, Developmental Studies Hybridoma Bank). This antibody recognizes light meromyosin, the rod-like tail region of MyHC protein). This antibody is generally diluted 1:1000 in PBS. Incubate overnight at 4 °C.
Wash three times in PBS, 5 min each.
Apply goat anti-mouse-IgG Alexa Fluor-488 (Life Technologies) secondary antibody diluted 1:1000 in PBS and incubate at room temperature for 30–45 min.
Wash three times in PBS, 5 min each.
Incubate each well with 1 μg ml−1 DAPI at room temperature for up to 5 min.
Wash twice in PBS, 7 min each.
Acquire the images as previously described for muscle. Normally, 15 fields per well, with a 10 × or 20 × magnification, are enough to get a statistically relevant analysis of myotube size.
Proceed to assess the myotube size (for example, the myotubes minimum diameter) by using ImageJ (or similar software; Figure 1d).3,17
In vivo assessment of muscle strength
The quantitative assessment of muscle strength in rodents has been the object of investigation for several purposes, due to the fact that these animals can be used to model many human pathologic conditions, among which neurodegenerative diseases, cancer, aging and drug toxicity. For all of these models, a full assessment of skeletal muscle strength and of any motor deficit is required in order to fully characterize the effects on skeletal muscle. Here we present some of the most commonly used techniques to assess the strength in laboratory mice.
Forelimb grip strength
The assessment of limb strength in experimental mice can be performed by means of a commercially available automatic grip strength meter (for example, Bioseb grip strength meter). This test is a widely used and a noninvasive method, particularly designed to evaluate mouse forelimb and/or hind limb strength and to assess the effects of a disease (for example, cancer, dystrophy) or a drug treatment on skeletal muscles.4,19 It is based on the natural tendency of the mouse to grasp a bar or a grid when it is suspended by the tail and gently pulled backward (Figure 2a). Forelimb grip strength can be used to evaluate in vivo neuromuscular performance. The procedure works by recording the peak resistance force when the experimental animal loses grip from the grid as it is being pulled away from the device. To reduce procedure-related variability, an average from several repeated peak force measurements in the same animal is recommended.
Figure 2.
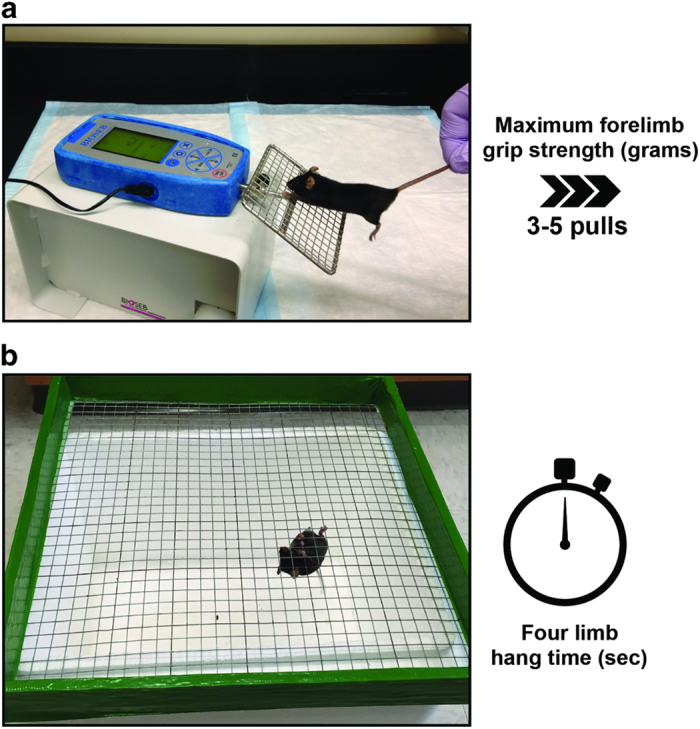
In vivo assessment of muscle strength. (a) Forelimb grip strength test of mouse grasping wire screen as it is being pulled by the tail horizontally relative to the force meter. The mouse is pulled steadily until it loses grip. The peak force is measured by the force meter. (b) Four limb-hanging test (Kondziella's inverted screen test) of mouse grasping wire screen as it is inverted. The time of sustained limb tension to oppose their weight is measured (hang time and minimal holding impulse).
The grip strength method is not a pure readout of muscle strength, as it is dependent upon neurological, anatomical and behavioral factors. Nonetheless, grip strength measurements can be useful for phenotype screening in conjunction with more comprehensive methods for evaluation of the neuromuscular function. In addition, forelimb grip strength would be amenable to studies of age-related neuromuscular decline. Generally speaking, increases in grip strength have been interpreted as an evidence of increased muscle strength, whereas, on the contrary, decreases in grip strength have been considered indicative of weakness and augmented fatigability. In order to accurately perform this test, the operator must take into consideration the following: (1) the test cannot be used in mice that are very young (that is, generally less than 2-week old); (2) this method allows determination of the absolute grip strength (expressed in grams) or the normalized grip strength (obtained by dividing the force value by the body weight of every single mouse); (3) some mice will not be willing to cooperate with the study, and therefore they will need to be excluded; (4) multiple trials3,4,5 per animal are required to generate reliable data that can be statistically validated; (5) this test is an in vivo behavioral test, as such significant variability can affect the study; and 6) mice may get used to the test quickly and lose their interest for the task, especially if the test is repeated too frequently. To avoid bias of habituation, the number of trials in each session should be limited (usually <5).
In carefully controlled settings, the variability within a group of experimental animals (assuming the animals are age-matched, same gender and genotype) should stand in the 10–25% range. Analogous variability should be observed among repeated measures performed in the same animal during the trial. In case greater variability is reported, this might be due to several conditions (such as animal fatigue, different operator performing the test and different environmental conditions) that will need to be addressed. Generally, in order to reduce variability in performing the test, it is imperative that the same operator performs the test for all the animals involved in the study.
This test also allows one to calculate the degree of fatigue by simply comparing the first two pulls with the last two pulls. In case 5 pulls are determined, the measure of fatigue obtained by the formula (4+5)/(1+2) will theoretically be 1 in mice that show no fatigue.
Four limb-hanging test
The four limb-hanging test (also known as Kondziella's inverted screen test) represents a method to assess muscle strength using all four limbs and to determine the general condition over time.20,21
This test makes use of a wire grid system to noninvasively measure the ability of mice to exhibit sustained limb tension to oppose their weight (Figure 2b). As a result, the test can provide important information, such as the ‘four limb hang time' (in seconds), as well as the ‘minimal holding impulse' (body mass x hang time). In particular, this test has been widely utilized to determine the natural course of neuromuscular disease, to demonstrate neuromuscular impairment and the lack of coordination or to study the effects of genetic or pharmacologic treatments on skeletal muscle functionality.22 Four limb hang testing would also be amenable to studies of age-related neuromuscular decline.
The test is easy to perform and relatively inexpensive. However, a few considerations must be taken into account:
This test does not measure the maintenance of a minimum force required to oppose the mouse gravitational force.
Variability can be challenging. Therefore, it is necessary to test a considerable number of mice in order to detect statistically significant differences.
Animal condition, as well as sex, age and, especially, body weight, can influence the outcome of the test. In particular, whenever it is clear that body weight can influence the result of the test, determining the holding impulse will correct the negative effects of body mass on the hang time.
The grid should be set at a height of about 35 cm, and soft bedding should be placed underneath to protect the mice from harm should they fall off the grid.
Every measurement may be repeated several times for each mouse with a rest interval between each trial of about 2 min.
If a fixed maximum holding time (for example, 10 min) is established, mice that fall off the grid before the time limit will be tested for 2 more tries, whereas mice that hang for the entire duration of the trial (10 min) will be placed back in the cage. In this case, either the maximum hang time observed or the mean of the observed hang times can be used as the dependent variable. Please note: when applying a fixed maximum time, the holding impulse cannot be used, as the unlimited maximum hanging time is not known.
If mice are capable of hanging for shorter periods (for example, <10 min), typically each mouse should be given several attempts to hold to the grid with suitable rest periods between each trial. In this case, either the maximum hang time observed or the mean of the observed hang times can be used as the dependent variable. The holding impulse should be assessed as well.
Mice that jump off the grid should be immediately retested. If this event is recurrent, these mice should be excluded from the study.
Isolated whole-muscle contractility
Skeletal muscle force production can readily be measured in isolated whole-muscle or single myofiber preparations. Pioneer studies on muscle function dates back to the 18th century when Caldani and Galvani discovered that muscle contraction could be triggered by electrical stimulation.23 The methods for force measurement in isolated skeletal muscles, which are still used, were delineated in the early-mid 20th century.23,24,25,26 The EDL and SOL muscles are typically used for contractility measurements, as these muscles are relatively easily dissected with intact tendons for the purpose of attachment to a force transducer, and the small size allows diffusion of oxygen to the interior of the muscle during testing. In the mouse, the EDL is dominated by type 2 fibers and is used as a model for studies on fast-twitch muscle function.8 The SOL has more type 1 fibers and is commonly used as a representative slow-twitch muscle.
It is also possible to dissect single intact mammalian muscle fibers with intact tendons.27 The use of dissected muscle fibers in conjunction with fluorescent indicators provides a powerful way to analyze muscle excitation–contraction coupling at the single cell level as it enables measurement of force production simultaneously with cytoplasmic-free [Ca2+] ([Ca2+]i).28 However, force measurement in single fibers is less widespread compared with whole muscle likely due to the meticulous dissection skill needed.
Here we summarize a protocol for measurement of isometric force in whole EDL and SOL muscle preparations.
Dissection of EDL and SOL muscle
Careful dissection is the most critical step for accurate measurement of force in an isolated skeletal muscle preparation; to dissect out an undamaged muscle takes practice. Therefore, perfecting the isolation of muscles is essential before performing any functional testing. Factors that will negatively influence the muscle function include overstretching, cutting or pinching the muscle and drying of the muscle.
The procedure for isolating EDL and SOL muscles from rodents can vary between dissectors, but the following scheme should be followed: 1) euthanize the animal by CO2 asphyxiation and confirm by cervical dislocation; 2) remove the hind limbs and ensure that the musculature is entirely free of the skin; 3) maintain the limbs in physiological salt solution (for example, Tyrode's solution, see Physiological solutions) at all times to ensure viability; and 4) using a dissection microscope and limbs fixed to the bottom of the chamber with pins, free the whole muscle using micro-dissection. An alternate procedure step is possible whereby the EDL and SOL are affixed to stainless steel hooks prior to freeing the muscle from their insertion points.29,30 Extreme care must be taken using this method to ensure that the muscle is not stretched and fibers are not damaged during manipulations (see Surgical instruments section below for a list of typical instruments used for dissection).
EDL dissection—anterior side of the hind limb
After removing the TA muscle, the EDL can be visualized as the relatively pale elongated muscle lateral to the tibia.
Cut the tendons as far from the muscle and as close to the bone insertion point as possible—at the distal end of the EDL, the tendon could be cut where it branches out into the toes, and at the proximal end of the muscle the tendon originates at the knee joint level.
To get good exposure of the insertion point of tendon, the knee joint can be dissected open before attempting to cut the tendon. The goal is to get as much of the tendon stump as possible as this will make it easier to attach the muscle to the hooks in the experimental chamber.
SOL dissection—posterior side of the hind limb
Dissection of the SOL muscle starts by cutting the Achilles tendon and thereafter lifting up the GSN muscle in proximal direction.
The red SOL muscle will be exposed beneath the GSN muscle. The distal tendon SOL is part of the Achilles tendon, and the proximal tendon inserts close to the posterior side of the knee joint.
Start by cutting the tendon at the knee joint, and then carefully dissect out the distal SOL tendon from the Achilles tendon bundle.
Care should be taken to free the muscles with a large portion of the tendons intact.
Muscle force measurement
The setup for isometric force measurement could be a commercial (for example, Aurora Scientific, Aurora, ON, Canada or World Precision Instruments, Sarasota, FL, USA) or a laboratory built system. The essential components of such a system include the following:
Chamber to hold the muscle bathed in physiological solution. The solution is normally bubbled with oxygen or a gas mixture of CO2/O2 (5/95%) when using a bicarbonate-buffered solution.
Force transducer
Bi-phasic stimulator
A/D converter and computerized acquisition and analysis of data.
Isolated muscles are tied to stainless steel hooks very tightly with 4–0 silk suture. If the attachments are not secured, the contracting muscle can slip away and cause loss of data. Both during and after dissection, the muscles should be stored in physiological solution. We have noted that the viability is best preserved when the muscle is hooked up in the muscle bath, bubbled and slightly stretched. Under these conditions, the muscle can be kept for several hours with fully preserved force generating capacity.
The muscle is placed in the chamber with one tendon attached to the force transducer and the other tendon attached to an adjustable hook, so that the muscle can be adjusted to its optimal length (L0). The muscle is electrically stimulated via electrodes made of platinum or some other inert conducting material that are placed along the long axis of the muscle. It is important to note that the electrodes should not touch the muscle tissue. Many muscle baths have a jacketed construct that allows the temperature of the physiological solution to be controlled by circulating pre-heated water into the jacket (Figure 3a).
Figure 3.

Isolated whole muscle contractility of mouse skeletal muscle. (a) Schematic illustration of the steps to determine isolated whole muscle contractility of skeletal muscle: (i), the muscle is excised from the lower hind limb; (ii), stainless steel hooks are attached via silk suture to the tendons; (iii), the muscle is bathed in physiological solution (Tyrode's buffer) bubbled with oxygen and stimulated to contract via platinum electrodes, and force is recorded by the force transducer; (iv), the optimal length (L0) is measured using vernier style calipers; (v), the semi-dry weight of the muscle is measured using an analytical balance (mg). (b) Representative single muscle data at various frequencies using the EDL excised from 28-week-old C57BL/6 mice. (c) Representative absolute force and specific force of the EDL from 28-week-old C57BL/6 mice (n=10±s.e.m.).
Electrical stimulation of contraction
Muscle contraction is triggered by electrically stimulating the muscle in the bath, which will activate voltage sensitive channels in the sarcolemma and start the excitation–contraction coupling process.23,25 To measure maximum isometric force generation, all muscle fibers in the muscle must be stimulated. Therefore, the supramaximal stimulation must be determined for each test system employed. Triggering pulses are generated by an electrical stimulator (for example, Aurora 701C stimulator, Grass Technologies Square Pulse Stimulator (Warwick, RI, USA) or WPI Pulsemaster (World Precision Instruments)). The electrical signal is transferred to the platinum electrodes in the muscle bath that generates an electrical field over the muscle. The output signal can be increased by incorporating a power amplifier to generate a sufficiently large field strength in the muscle bath.
The protocol used to stimulate the muscle varies depending on the muscle type and the scientific question. The basic stimulation is an electrical pulse that mimics the action potential, for example, a brief pulse, 0.5–1 ms in duration at a voltage that exceeds the activation threshold. Twitch contraction is achieved by electrically stimulating the muscle with a single electrical pulse. As the stimulation frequency increases, the twitches start to fuse, and tetanic contractions occur with a resultant increase in tension (Figure 3b). A train of repeated pulses (for example, 1 ms pulses at 1–200 Hz for 300–600 ms) is used to increase the muscle tension. Fast-twitch muscle (for example, EDL) needs a higher stimulation frequency to reach the maximum tetanic contraction than does slow-twitch muscle (for example, SOL).31
The stimulation voltage should be sufficient to exceed the activation threshold for triggering action potentials throughout the muscle preparation.
At the start of each experiment, the muscle force-length relationship should be optimized for each muscle such that the muscle length (L0) is set to yield the maximum force for a given stimulation.32
L0 is determined by activating the muscle to twitch (or tetanic) contractions and incrementally altering the muscle length until maximal force production is achieved.
To avoid fatiguing the muscle due to repeated stimulation, a rest period should be present between each contraction (for example, >30 s between twitch contractions and >1 min or more between tetani).
EDL force-frequency response is determined using the following parameters: 0.5 ms pulses; 1, 10, 20, 30, 50, 70, 100, 120, 150 and 200 Hz; pulse train duration 350 ms.
SOL force-frequency response is determined using the following parameters: 0.5 ms pulses; 1, 10, 20, 30, 50, 70, 100, 120 and 150 Hz; pulse train duration 600 ms.
Repeated contractions cause a reduction in muscle force that can be recovered after a period of rest. This physiological process is known as muscle fatigue.24 Isolated skeletal muscles can be used to study the mechanisms of fatigue.24 However, it is important to be aware that, during intense repeated contractions in large isolated whole muscles, the diffusion of oxygen into the interior of the muscle is limited and can accelerate development of muscle fatigue in a non-physiological way.33,34 To study fatigue, isolated muscle fiber is a better suited preparation where oxygen diffusion distance is small and does not cause a limitation.28
Quantification of specific force
The force production will depend on the size of the muscle (the CSA, the number of myofilaments in parallel) and the intrinsic properties of the contractile machinery. Thus, muscle weakness could be due to a reduced muscle mass and/or an impaired excitation–contraction coupling.23,24 To differentiate between these mechanisms, it is recommended to measure the specific muscle force (sP0), that is, the absolute force (P0) normalized to the whole-muscle CSA. The CSA can be approximated from the mass (m) and length (L0) of the muscle together with the muscle density (∼1.06 mg mm−3).25 The following equation is typically used to compute the specific force: sP0=P0 × (muscle mass/1.06 × L0); Figure 3c). The optimal length (L0) should be measured with the use of a caliper when the muscle is mounted in the bath. Muscle mass is measured using an analytical balance at the end of the experiment, after the tendons and other non-muscle tissues have been removed and after brief contact with absorbent material such as filter paper to remove excess solution (Figure 3a,v).
Physiological solutions
The muscle must be kept in physiological salt solution to buffer pH, preserve osmolality and to maintain the excitation properties of the membrane and muscle fiber integrity. The choice of solution varies with local laboratory traditions. In our hands, a bicarbonate-buffered Tyrode's solution is well suited for muscle experiments. The following recipe can be used (this solution yields a physiological pH 7.4 if constantly bubbled with O2/CO2 (95/5%)). Physiological buffer should be used for all dissection steps. Tyrode's solution: 121 mM NaCl, 5.0 mM KCl, 1.8 mM CaCl2, 0.5 mM MgCl2, 0.4 mM NaH2PO4, 24 mM NaHCO3, 0.1 mM EDTA and 5.5 mM glucose.
Surgical instruments
Equipment necessary to cleanly dissect skeletal muscle from mice will vary according to individual needs. Representative equipment includes the following:
dissecting microscope (e.g. Leica S6).
10 mm spring scissors (Fine Science Tools, cat. no. 15006-09).
Dumont #5 standard tip forceps (Fine Science Tools, cat. no. 11295-00).
dissection platform (any suitable material, for example, paraffin wax in shallow dish).
4–0 non-absorbable silk suture.
hand-crafted hooks made from 23-guage stainless steel wire.
Discussion
Evaluation of the strength of skeletal muscles can provide direction for therapeutic candidates for muscle diseases. In this report, we have provided basic protocols that can be used for assessment of physiological strength. Representative data for these methods will vary greatly depending on age, strain and sex of mice under evaluation. It is advised that group size to obtain statistically significant results be determined by power analysis using pilot data. When combined with imaging and biochemical methods (for example, calcium handling and myofibrillar protein function), these techniques provide an understanding of the overall muscle strength in mice. These techniques can be adapted to the study of many disease states that display a muscle weakness component (for example, cachexia/sarcopenia, cancer, dystrophy, etc).
Acknowledgments
AB is supported by the National Cancer Institute (Grant R21-CA190028-01). DCA is supported by the Swedish Heart Lung Foundation, Åke Wiberg Foundation, French Muscular Dystrophy Association and a grant from the Stockholm County Council. DLW is partially supported by the American Cancer Society and IU Simon Cancer Center (grant IRG-84-002-28 and IU Health Strategic Research Initiative in Oncology). AB and DLW thank Dr Teresa A Zimmers and Dr Theresa A Guise for supporting their work. Supported by the IBMS-ECTS Young Investigators.
Footnotes
The authors declare no conflict of interest.
References
- Call JA, McKeehen JN, Novotny SA, Lowe DA. Progressive resistance voluntary wheel running in the mdx mouse. Muscle Nerve 2010; 42: 871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim CH, Wasala NB, Duan D. Evaluation of muscle function of the extensor digitorum longus muscle ex vivo and tibialis anterior muscle in situ in mice. J Vis Exp 2013; 72: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab 2012; 303: E410–E421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetto A, Penna F, Minero VG, Reffo P, Bonelli G, Baccino FM et al. Deacetylase inhibitors modulate the myostatin/follistatin axis without improving cachexia in tumor-bearing mice. Curr Cancer Drug Targets 2009; 9: 608–616. [DOI] [PubMed] [Google Scholar]
- Mula J, Lee JD, Liu F, Yang L, Peterson CA. Automated image analysis of skeletal muscle fiber cross-sectional area. J Appl Physiol 2013; 114: 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Fry CS, Mula J, Jackson JR, Lee JD, Peterson CA et al. Automated fiber-type-specific cross-sectional area assessment and myonuclei counting in skeletal muscle. J Appl Physiol 2013; 115: 1714–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briguet A, Courdier-Fruh I, Foster M, Meier T, Magyar JP. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul Disord 2004; 14: 675–682. [DOI] [PubMed] [Google Scholar]
- Schiaffino S, Reggiani C. Fiber types in mammalian skeletal muscles. Physiol Rev 2011; 91: 1447–1531. [DOI] [PubMed] [Google Scholar]
- Smerdu V, Karsch-Mizrachi I, Campione M, Leinwand L, Schiaffino S. Type IIx myosin heavy chain transcripts are expressed in type IIb fibers of human skeletal muscle. Am J Physiol 1994; 267: (6 Pt 1): C1723–C1728. [DOI] [PubMed] [Google Scholar]
- Klitgaard H, Bergman O, Betto R, Salviati G, Schiaffino S, Clausen T et al. Co-existence of myosin heavy chain I and IIa isoforms in human skeletal muscle fibres with endurance training. Pflugers Arch 1990; 416: 470–472. [DOI] [PubMed] [Google Scholar]
- Maier A, Gorza L, Schiaffino S, Pette D. A combined histochemical and immunohistochemical study on the dynamics of fast-to-slow fiber transformation in chronically stimulated rabbit muscle. Cell Tissue Res 1988; 254: 59–68. [DOI] [PubMed] [Google Scholar]
- DeNardi C, Ausoni S, Moretti P, Gorza L, Velleca M, Buckingham M et al. Type 2X-myosin heavy chain is coded by a muscle fiber type-specific and developmentally regulated gene. J Cell Biol 1993; 123: 823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson MF, Stephenson GM, Stephenson DG. Denervation produces different single fiber phenotypes in fast- and slow-twitch hindlimb muscles of the rat. Am J Physiol Cell Physiol 2006; 291: C518–C528. [DOI] [PubMed] [Google Scholar]
- Yaffe D, Saxel O. A myogenic cell line with altered serum requirements for differentiation. Differentiation 1977; 7: 159–166. [DOI] [PubMed] [Google Scholar]
- Blau HM, Pavlath GK, Hardeman EC, Chiu CP, Silberstein L, Webster SG et al. Plasticity of the differentiated state. Science 1985; 230: 758–766. [DOI] [PubMed] [Google Scholar]
- Burattini S, Ferri P, Battistelli M, Curci R, Luchetti F, Falcieri E. C2C12 murine myoblasts as a model of skeletal muscle development: morpho-functional characterization. Eur J Histochem 2004; 48: 223–233. [PubMed] [Google Scholar]
- Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG et al. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS ONE 2011; 6: e22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetto A, Penna F, Minero VG, Reffo P, Costamagna D, Bonelli G et al. Glutamine prevents myostatin hyperexpression and protein hypercatabolism induced in C2C12 myotubes by tumor necrosis factor-alpha. Amino Acids 2011; 40: 585–594. [DOI] [PubMed] [Google Scholar]
- Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med 2009; 15: 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon RM. Measuring the strength of mice. J Vis Exp 2013; 76: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondziella W. [A new method for the measurement of muscle relaxation in white mice]. Arch Int Pharmacodyn Ther 1964; 152: 277–284. [PubMed] [Google Scholar]
- Carlson CG, Rutter J, Bledsoe C, Singh R, Hoff H, Bruemmer K et al. A simple protocol for assessing inter-trial and inter-examiner reliability for two noninvasive measures of limb muscle strength. J Neurosci Methods 2010; 186: 226–230. [DOI] [PubMed] [Google Scholar]
- Hill JA, Olson EN. Muscle: Fundamental Biology and Mechanisms of Disease Academic Press: Burlington, VT, USA, 2012; . [Google Scholar]
- Allen DG, Lamb GD, Westerblad H. Skeletal muscle fatigue: cellular mechanisms. Physiol Rev 2008; 88: 287–332. [DOI] [PubMed] [Google Scholar]
- Close RI. Dynamic properties of mammalian skeletal muscles. Physiol Rev 1972; 52: 129–197. [DOI] [PubMed] [Google Scholar]
- Hill AV. The position occupied by the production of heat, in the chain of processes constituting a muscular contraction. J Physiol 1911; 42: 1–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannergren J, Westerblad H. The temperature dependence of isometric contractions of single, intact fibres dissected from a mouse foot muscle. J Physiol 1987; 390: 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerblad H, Allen DG. Changes of myoplasmic calcium concentration during fatigue in single mouse muscle fibers. J Gen Physiol 1991; 98: 615–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waning DL. Soleus dissection (http://youtu.be/IG7H7bQdA94)2014; Available from https://www.youtube.com/user/IUBoneandCancer.
- Waning DL. EDL dissection (http://youtu.be/8qwZXkaooRQ)2014; Available from https://www.youtube.com/user/IUBoneandCancer.
- Ranatunga KW. Temperature-dependence of shortening velocity and rate of isometric tension development in rat skeletal muscle. J Physiol 1982; 329: 465–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon AM, Huxley AF, Julian FJ. The variation in isometric tension with sarcomere length in vertebrate muscle fibres. J Physiol 1966; 184: 170–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay CJ. Modelling diffusive O(2) supply to isolated preparations of mammalian skeletal and cardiac muscle. J Muscle Res Cell Motil 2005; 26: 225–235. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Bruton JD, Katz A, Westerblad H. Limited oxygen diffusion accelerates fatigue development in mouse skeletal muscle. J Physiol 2006; 572: (Pt 2): 551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]