

Abstract
Langerhans cell histiocytosis (LCH) is a group of idiopathic disorders characterized by proliferation of bone marrow derived Langerhans cells and mature eosinophils. Their clinical features simulate common oral findings such as gingival enlargement, oral ulcers, and mobility of teeth, along with nonspecific radiographic features; hence, diagnosing such lesions becomes difficult for the oral physicians. These lesions are commonly seen in childhood; however, we are reporting a case of LCH in 29-year-old adult male. A provisional diagnosis of giant cell granuloma was considered based on history and examination, although the lesion was histologically proven to be LCH and was confirmed with immunohistochemical staining of S100 protein and CD1a antigen. The purpose of this paper is to enhance the understanding of diverse, nonpathognomical oral presentation of LCH that is easily misdiagnosed and overlooked by dentist.
Keywords: Eosinophilic granuloma, immunohistochemistry, Langerhans cell histiocytosis, management, radiographic features
Introduction
Formerly known as histiocytosis X, the term Langerhans cell histiocytosis (LCH), is now preferred, as it emphasizes the histogenesis of the condition, clarifying the type of cellular basis of lesional cell and removing the connotation of the unknown X. LCH demonstrates neoplastic proliferation of histiocytes and other inflammatory cells leading to accumulation and pathological dissemination of histiocytes resulting in destruction of hard and soft tissues. Though the etiology is controversial, various theories have suggested contribution of environment, infections, immunology, genetics, and neoplastic process. Limited data are available regarding the epidemiology of LCH, mainly, because of its relatively low incidence, with estimation of 2–5 cases per million inhabitants per year in the year 2009.[1] The diagnosis of LCH is solely based on microscopic examination and the disease is broadly categorized into three disorders based on the clinical presentations, each of which is staged clinically according to Greenberg et al. clinical staging system.[2] Oral involvements are commonly the first manifestation in LCH but the initial symptoms are generally nonspecific, causing misdiagnoses. The purpose of reporting this article is to discuss clinical and radiological features of LCH and role of dentists in diagnosing and managing such lesions. Management and prognosis of LCH are based on the clinical presentation of the lesion.
Case Report
A 29-year-old male patient, well-built, and nourished reported to our department with a history of swelling in right lower half of the face since 2 months. The swelling was gradual in onset and was not associated with any symptoms. No significant history of trauma or medical history was provided by the patient. General physical examination revealed all vital signs within normal limits, although, a single right submandibular lymph node was palpable, nontender, spherical in shape, soft in consistency, and freely movable.
No gross asymmetry or surface changes was noticed on clinical examination of the face, however, on palpation, an ill-defined solitary swelling measuring approximately 2.5 cm × 3 cm was present on the right parasymphyseal region which was 2 cm above the inferior border of the mandible. The swelling was nontender, bony hard in consistency with smooth diffuse border. No step deformity, crepitus, paresthesia, or local rise in temperature was appreciated on palpation. No significant inspectory and palpatory findings were noticed intraorally. Vitality tests for all associated teeth were positive. Hence, based on history and clinical features, a provisional diagnosis of central giant cell granuloma on the right mandibular region was considered, along with keratocystic odontogenic tumor and ameloblastoma as differential diagnosis.
Hematological and biochemical investigation demonstrated patient was anemic with the increase level of erythrocyte sedimentation rate (ESR), white blood cell (WBC) count and eosinophilic count. Serum alkaline phosphatase levels were increased to 145 IU/L. The patient was referred for medical and endocrinological assessment and found to be was normal. Liver and renal function tests were within the normal range. Mandibular true occlusal radiographic analysis revealed, an ill-defined radiolucency at premolar region bilaterally and also on right parasymphyseal region [Figure 1]. No cortical plate perforation, expansion, or displacements of teeth were noticed on occlusal radiograph. Panoramic radiograph [Figure 2] revealed mandibular cortical border to be scalloped and uninterrupted. Multiple radiolucencies were present on ramus and basal bone of mandible bilaterally with normal coronoid and condylar process. Large radiolucencies appear to be well-defined and nonsclerotic; however, smaller lesions were poorly defined. The internal structure of radiolucencies appeared clear with no trabeculae or calcifications. No alveolar bone resorption was noticed although lamina dura appeared to be discontinued at the periapical region of all the teeth without root resorption. Mandibular canal and mandibular teeth appeared to be undisplaced. No abnormalities detected in the maxilla except for missing 21. PA mandible radiograph [Figure 3] showed similar mandibular multiple radiolucencies. A complete skeletal radiographic survey was also performed on patient revealing no abnormalities. Computed tomography guided fine needle aspiration cytology of mandible was performed suggestive of a giant cell tumor due to the presence of osteoclast-like giant cells with scanty inflammatory cells and no atypical features. Trephination was performed at 46, 47, 48 region and obtained specimen was submitted for histopathological investigation. The specimen revealed, proliferation of large mononuclear pale staining cells with ill-defined cellular margins, coalition of Langerhans cells interspersed with inflammatory cells, histopathologically suggestive of LCH [Figure 4a]. To confirm the lesion, immunohistochemical staining was performed; the result was positive for S100 [Figure 4b] and CD1a [Figure 4c] marker. Based on the age of the patient, clinical presentation of the lesion, involving single bone with altered hemogram values and positive microscopic examination a final diagnosis of eosinophilic granuloma type of LCH was made. Based on the clinical findings, a grading of type 1a LCH was given based on Greenberger's criteria.[2] After we had confirmed the diagnosis of single bone involvement and no lesions in other organs, surgical curettage with chemotherapy was recommended protocol. The patient was unwilling for surgery due to esthetic reasons, hence, was referred to the nearby cancer institute for chemotherapy. He is currently on chemotherapy with treatment protocol of administration of Vincristine 1.5 mg/m2 intravenous on the 1st day with repeated dose 9 times every 4–6 weeks, and prednisolone 40 mg/m2 daily initially for 4 weeks followed by 20 mg/m2 daily for a whole year. No further enlargement of swelling or any associated symptoms is reported by the patient as on date, and the patient is on regular follow-up.
Figure 1.
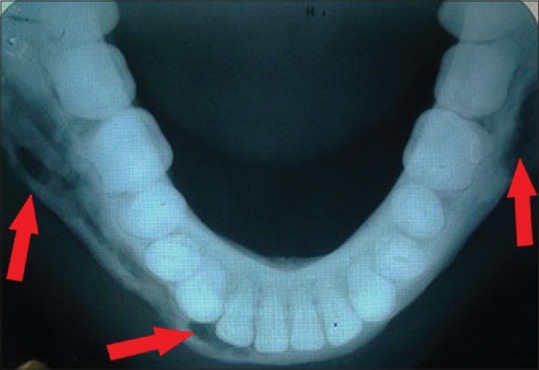
Mandibular true occlusal radiograph
Figure 2.
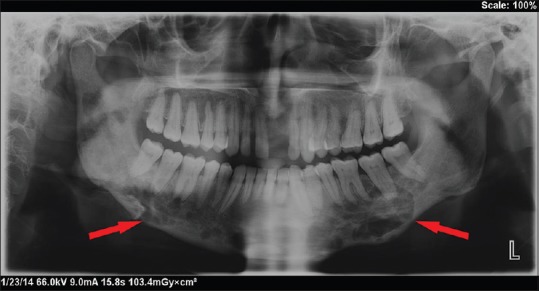
Panoramic radiograph with irregular radiolucencies of entire body of mandible on both sides with irregular inferior cortex
Figure 3.
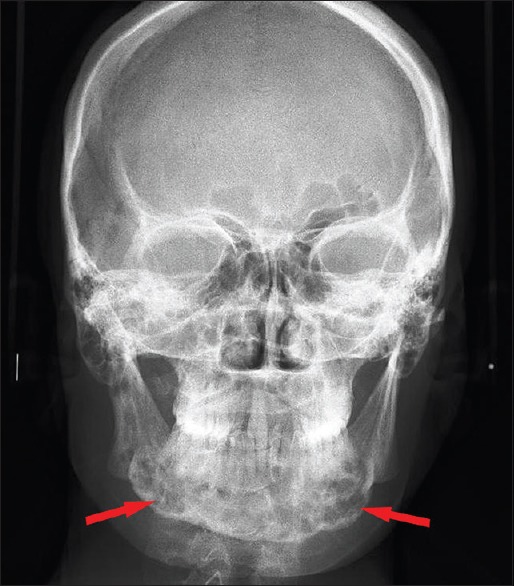
PA mandible view with multiple radiolucencies of mandible
Figure 4.
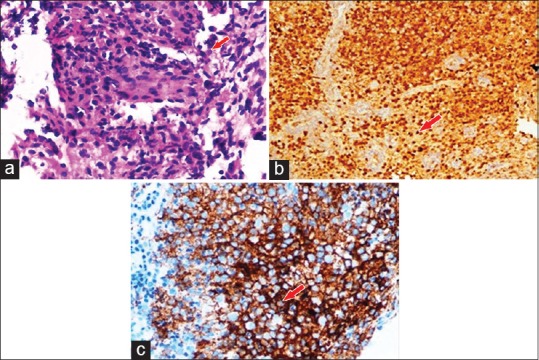
(a) Large mononuclear pale staining cells with ill-defined cellular margins, Langerhans cells interspersed with inflammatory cells are seen on microscopic examination. (b) Immunohistochemistry showing presence of Langerhans cell S100 protein. (c) Immunohistochemistry showing presence of Langerhans cell CD1a antigen
Discussion
Proposed by Lichtenstein in 1953 as histiocytosis X, the term LCH was introduced as a collective designation for a spectrum of clinicopathologic disorders characterized by proliferation of histiocyte such as cells accompanied by eosinophils, lymphocytes, plasma cells, and multinucleated giant cells. The destructive histiocytes are Langerhans cells, dendritic mononuclear cells normally found in the epidermis, mucosa, lymph nodes, and bone marrow. Their normal function is to process and present antigens to T-lymphocytes, however, in LCH, literature review suggests a monoclonal proliferation of langerhan cells leading to hard and soft tissue destruction. Although, etiology and pathogenesis of LCH is unclear, reviewing of litrature suggested two widly accepted hypothesis, one being the disorder due to immune regulation and other being neoplastic proliferation.[3]
LCH may be encountered over a wide age range, although, cases are frequently reported under the age of 15 years, with a peak incidence of 2–4 years with a male predilection, twice that of the females. The age of onset varies depending upon the variety of LCH and are rarely seen in adults with incidence around 1–2 cases/million persons per year.[4] The present case was reported in adult male of 29-year-old. Solitary or multiple bone lesions are the most common clinical presentations seen in the majority of cases, with a most frequent sites involving skull, ribs, vertebrae, and jaws. Among jaws in particular, posterior aspect of the mandible is more commonly involved especially the region distal to canine;[5] however, in present the case, the entire mandible was involved which could be due to extension of posterior lesions to involve anterior aspect during the process of osteolysis.
This distinct group of disease that is collectively referred as LCH is categorized into three variants based on the age and clinical presentation; clear separation is difficult on the basis of histological manifestations. These variants include (1) Acute disseminated form with multiple system involvement often occurring mainly in infants (Letterer-Siwe disease). (2) Chronic disseminated form with osseous lesions which are frequently multiple and with extraskeletal lesions (Hand-Schuller-Christian disease) (3) Chronic localized form with solitary or multiple skeletal lesions and occasionally extraskeletal involvement mainly seen in adult (Eosinophilic granuloma). Hashimoto Pritzker syndrome is a congenital form of LCH presenting with a deep subcutaneous skin lesions.[4,6] Solitary type of eosinophilic granuloma is twice as common as multiple variants and is most common among other three variants. In the present case, the patient exhibited a solitary skeletal lesion with no extraskeletal involvement hence was considered as eosinophilic granuloma variant. A clinical staging system was proposed by Greenberg et al. in 1981,[2] [Table 1], wherein all cases of LCH were categorized under five stages based on the clinical presentation, age of the patient at the time of diagnosis and the number of bone lesions. Based on same criteria, the present case was considered under stage 1a due to the involvement of single bone with no extraskeletal features.
Table 1.
The clinical staging proposed by Greenberg et al.[2]
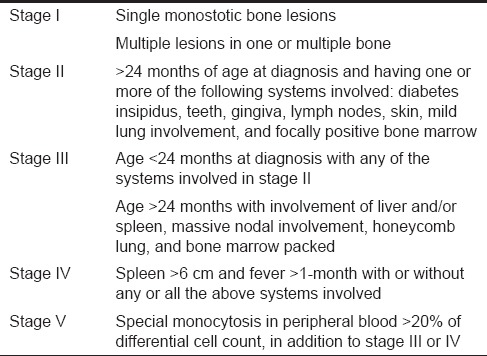
LCH has a wide clinical spectrum wherein clinical course of LCH depends upon the age of the patient and pathological infiltration of langerhans cells in various system such as bone, skin, lymph nodes, bone marrow, liver, spleen, lung, endocrine system, ear, and brain. Adults and older children present single system disease commonly affecting bone, however, young babies demonstrate multisystem involvement with initial general clinical presentation includes skin rash, otitis media, fever, organomegaly, anemia, pain and pathological fracture of involved bone, and diabetes insipidus.[7] As the adult cases are rare, clinical features are poorly described in literature, although in most cases, first manifestation of LCH occur in oral cavity with nonspecific multiple presentations such as gingival hypertrophy, oral ulceration, mobility of teeth with alveolar expansion, jaw pain, facial swelling, and mental nerve anesthesia.[5,8] Intraoral involvement being the first sign and in some occasion being the only manifestation of LCH, as seen in the present reported case, hence, leading the patient to consult a dentist.
As LCH does not give any pathognomonic clinical and radiographic features, the diagnosis of LCH is based on microscopic examination of obtained biopsy specimen from the bony lesions. The histopathological pattern demonstrates a diffuse infiltration of large pale staining mononuclear cells that resemble histiocytes with indistinct cytoplasmic borders and rounded or indented vesicular nuclei. The number of eosinophils vary and are typically interspersed among the histiocytes along with the presence of Plasma cells, lymphocytes, and multinucleated giant cells. The histopathology is similar in all LCH variants except in acute disseminated form as they also demonstrate acute form of lymphomas.[9] The presence of Langerhans cells is necessary for diagnosis, which require additional diagnostic methods. Electron microscopic evaluation is the gold standard that shows rod-shaped cytoplasmic structures known as Birbeck granules within Langerhans cells, differentiating them from other mononuclear phagocytes. Confirmatory diagnosis is by immunohistochemical procedures because of their immunoreactivity with antibodies directed against CD1a. Lesional cells have S100 protein immunoreactivity.[10] In the present case, the biopsy specimen was positive for CD1a marker and S100 protein, thus, confirming the diagnosis.
The radiological features are present due to the destruction of bone by infiltration of Langerhans cells that are similar in all three forms of LCH. This process of osteolysis may involve any part of any bone, however, in head and neck region, it is commonly seen in the posterior region of mandible. In the disseminated form of the disease, multiple areas of bone destructions are involved, of varying size leading to perforation of the cortical plate and pathological fracture. Osseous expansions are usually not observed in the case of LCH and were not observed in the current case also. At basal bone of mandible, lesions are often present punched out radiolucencies without a corticated rimming suggestive of active disease, occasionally giving an ill-defined pattern, probably due to confluence of many smaller lesions[11] which was observed in this case also. Involvement of superficial alveolar bone shows characteristic scooped out appearance with floating of teeth, displacement, periodontitis, and often loss of teeth at a very early age. The mucosal lesion may develop as gingival mass if disease breaks out of bone[10] although, in the present case, no intraoral or dental findings were appreciated.
Based on clinical and radiological findings of the present case, possible differential diagnosis was central giant cell granuloma; however, they are more commonly seen in female patients exhibiting aggressive clinical pattern with root resorption and expansion of cortical plate. The radiolucency of central giant cell granuloma also shows internal wispy septa with sclerotic margins. Giant cell lesions of hyperthyroidism were ruled out due to negative serum chemistry test results. Aneurysmal bony cyst was considered in differential diagnosis based on age of the patient and the location of the lesion but no cortical expansion, internal septa, or sclerotic margins were observed in present case neither any history of trauma was reported by the patient. The absence of any symptoms, cortical expansion, impacted tooth, and internal septas excluded ameloblastoma and KOT from differential diagnosis although the age of the patient and location of the lesion was in favor. Multiple myeloma and metastatic carcinoma were also ruled out due to no disclosed general systemic manifestations or any other skeletal abnormalities.
The histiocyte society has established a set of guidelines to assist in the diagnosis, and study of LCH includes complete physical and hematological examination along with liver function tests, urine osmolality, and bone marrow examination. For a definitive diagnosis, a complete skeletal radiographic survey and chest radiography is also necessary.[12] In the present case, the blood investigation showed increased ESR rate and reduced hemoglobin level along with altered WBC count, eosinophilic count and increased serum alkaline phosphatase level, hence, the patient was referred for complete medical evaluation. Patient with any abnormalities requires additional assessments such as pulmonary function tests, lung biopsy, small bowel series, liver biopsy, endocrine evaluation, and otolaryngoscopy.[12] These investigations were not carried out in the present case due to no reported abnormalities on general physical examination. Since LCH patients have a recurrence to this disease, follow-up studies are required every month to 6 months. In the present case, the patient is on a regular follow-up.
Optimal treatment of LCH remains controversial due to high clinical variability and absence of standard diagnostic and evaluation criteria; also most of the treatment related data are confined to the juvenile cases as the disease is common in children. The treatment of LCH depends upon pathogenesis of the disease, age of the patient and dissemination of the lesion, to some extent. Usually, course of the disease is unpredictable wherein majority show self-limiting process along with alternating phase of relapse and remission, hence, multidisciplinary evaluation is necessary for diagnosing and treating such patients. With respect to management of LCH, the main aim is to resolve the signs and symptoms of the disease and to prevent the permanent consequences. Diverse therapeutic options are available, still progress is required in the field of management to achieve higher cured rate. Treatment of LCH is reserved for symptomatic patients as asymptomatic patients are kept under observation because some lesions resolve spontaneously. Currently, single system disease exhibiting benign course or solitary small bone lesions are usually treated with surgical curettage followed by local injection of steroids for remission of symptoms.[1,3,5,13] Treatment protocol for disseminated or multisystem disease involves two major approaches; a conservative approach with minimal chemotherapy along with steroids for low-risk diseases and an intensive chemotherapy complimented with low dose radiotherapy or surgery for high-risk diseases.[1,5,13,14] Radiotherapy is an effective adjunct in management of widespread bony or soft tissue lesions, although, their use is decreased considerably due to the risk of secondary tumor occurrences, a threat to the function of the involved organs and nonfeasibility of local infiltration of corticosteroids.[13] In the present case, though the patient showed single bone involvement of the mandible with no other systemic involvement, surgical curettage was an optimum treatment modality; however, a conservative chemotherapeutic approach was initiated due to unwillingness of the patient. Prognosis is unpredictable and poor prognostic factors that are accepted universally are age <2 years and in patients with disseminated disease. The ten-year survival rate for a single bone involvement has been reported to be 100% while in a multisystem involvement, it is reported to be 77%.[15] Recurrence rate depends upon degree of tissue involvement and the treatment method opted, hence, the patient should be closely monitored for a longer period of time.[8]
The present case tells us that LCH should not be confined as a disease of children as cases are also reported in adults. Besides following a benign course, considerable morbidity is seen secondary to both its treatment and disease itself; hence, long-term follow-up for a minimum of one year is essential in each patient to ensure remission.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Madrigal-Martínez-Pereda C, Guerrero-Rodríguez V, Guisado-Moya B, Meniz-García C. Langerhans cell histiocytosis: Literature review and descriptive analysis of oral manifestations. Med Oral Patol Oral Cir Bucal. 2009;14:E222–8. [PubMed] [Google Scholar]
- 2.Greenberger JS, Crocker AC, Vawter G, Jaffe N, Cassady JR. Results of treatment of 127 patients with systemic histiocytosis. Medicine (Baltimore) 1981;60:311–38. doi: 10.1097/00005792-198109000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Jonas N, Mulwafu W, Khosa SA, Hendricks M. Case study: Langerhan's cell histiocytosis (LCH) Int J Pediatr Otorhinolaryngol. 2008;3:61–5. [Google Scholar]
- 4.Yashoda-Devi B, Rakesh N, Agarwal M. Langerhans cell histiocytosis with oral manifestations: A rare and unusual case report. J Clin Exp Dent. 2012;4:e252–5. doi: 10.4317/jced.50728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shevale VV, Ekta K, Snehal T, Geetanjal M. A rare occurrence of Langerhans cell histiocytosis in an adult. J Oral Maxillofac Pathol. 2014;18:415–9. doi: 10.4103/0973-029X.151335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aruna DR, Pushpalatha G, Galgali S, Prashanthy Langerhans cell histiocytosis. J Indian Soc Periodontol. 2011;15:276–9. doi: 10.4103/0972-124X.85675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tesluk EW, Szutkowski Z, Kawecki A. Langerhans cell histiocytosis of bone - A case report and review of literature. J Oncol. 2003;53:161–4. [Google Scholar]
- 8.Divya KS. Oral manifestion of Langerhans cell histiocytosis mimicking inflammation. Indian J Dent Res. 2014;25:228–30. doi: 10.4103/0970-9290.135930. [DOI] [PubMed] [Google Scholar]
- 9.Neville BW, Damm DD, Allen CM, Bouquot JE. Oral and Maxillofacial Pathology. Ch. 13. Vol. 2. New Delhi: Reed Elsevier India Pvt. Ltd; 2004. Langerhans cell histiocytosis. Hematological disorders; pp. 513–4. [Google Scholar]
- 10.Lajolo C, Campisi G, Deli G, Littarru C, Guiglia R, Giuliani M. Langerhans's cell histiocytosis in old subjects: Two rare case reports and review of the literature. Gerodontology. 2012;29:e1207–14. doi: 10.1111/j.1741-2358.2012.00629.x. [DOI] [PubMed] [Google Scholar]
- 11.Langlais RP, Langland OE, Nortje CJ. Ch. 11. Vol. 1. Malvern, PA, USA: Williams & wilkins; 1995. Poorly defined radiolucencies. Diagnostic Imaging of the Jaws; pp. 417–23. [Google Scholar]
- 12.Jindal M, Sharma VK, Ahmed I, Agrawal A. Langerhans cell histiocytosis of maxilla and mandible in 6 years old child: A case report. Int J Clin Pediatr Dent. 2009;2:45–9. doi: 10.5005/jp-journals-10005-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ladisch S, Gadner H. Treatment of Langerhans cell histiocytosis – Evolution and current approaches. Br J Cancer Suppl. 1994;23:S41–6. [PMC free article] [PubMed] [Google Scholar]
- 14.Desai VD, Priyadarshinni SR, Varma B, Sharma R. Langerhans cell histiocytosis: An illusion of hope. Int J Clin Pediatr Dent. 2013;6:66–70. doi: 10.5005/jp-journals-10005-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernstrand C, Sandstedt B, Ahström L, Henter JI. Long-term follow-up of Langerhans cell histiocytosis: 39 years’ experience at a single centre. Acta Paediatr. 2005;94:1073–84. doi: 10.1111/j.1651-2227.2005.tb02048.x. [DOI] [PubMed] [Google Scholar]