

Abstract
Introduction
There is an unmet clinical need for the therapy of recurrent malignant gliomas. Novel therapies are developed in an attempt to target specific molecular mechanisms involved in abnormal signaling and resistance to apoptosis, and to overcome resistance to traditional chemotherapy drugs, such as temozolomide. The proteasome is a key regulator of multiple cellular functions, and proteasome inhibition in malignant astrocytic lines causes cell growth arrest and apoptotic cell death. Though the proteasome inhibitor bortezomib was reported to have very good in-vitro activity against malignant glioma cell lines, the drug was proven to have modest activity in animal models as well as in clinical trials as a single agent. Here, we describe the multiple effects of bortezomib in in-vitro and in-vivo glioma models, and we offer a novel explanation for its seeming lack of activity.
Materials and Methods
Brain tumors (glioblastoma multiforme) were collected by at surgery and then expanded. Temozolomide resistant cells were also cultured. The cells were treated with Bevacizumab and Bortezomib and XTT cell viability assays, apoptosis analysis, caspase -3 activity and proteasome activity was measured. Human glioma xenograft models were created in nude mice by subcutaneous injection. Bevacizumab was administered via intra-peritoneal (i.p.) injection at a dose of 5mg/kg daily. Bortezomib was given i.p. one hour after bevacizumab administration in doses of at a dose of 0.35 mg/kg on days 1, 4, 8 and 11 every 21 days. Tumors were measured twice weekly.
Results
We showed that bortezomib induced caspase-3 activation and apoptotic cell death in stable glioma cell lines and primary, stem-cell like glioma cultures (GSCs). Furthermore, temozolomide-resistant glioma cell lines retained susceptibility to the proteasome inhibition. The bortezomib activity in these cells was directly proportional with the base-line proteasome activity. However, the proteasome inhibition stimulated both Hypoxia Inducible Factor (HIF) 1α and the Vascular Endothelial Growth Factor (VEGF) production in the malignant GSCs. As such, the VEGF produced by the GSCs stimulated endothelial cell growth, an effect which could be reverted by the addition of bevacizumab (a VEGF antibody) to the media. Similarly, administration of bortezomib and bevacizumab to athymic mice carrying subcutaneous malignant glioma xenografts resulted in greater tumor inhibition and greater improvement in survival then either drug alone. These data indicate that simultaneous proteasome inhibition and VEGF blockade offer increased benefit as a strategy for malignant glioma therapy.
Conclusions
Our data indicate that combination therapies based on Bortezomib and Bevacizumab offer increased benefit. These drugs have a complementary mechanism of action and therefore can be used together to treat temozolomide resistant malignant gliomas.
INTRODUCTION
Malignant gliomas are among the most lethal tumors, highly resistant to chemotherapy and radiotherapy34. Recurrence following current “standard of care” surgery, radiation therapy and adjuvant chemotherapy is nearly universal27. The traditional therapies such as temozolomide rely on DNA damage and disruption of mitotic machinery, with limited effect in prolonging patient survival10. Thus, novel therapies which can overcome treatment resistance by targeting specific molecular mechanisms involved in abnormal signaling and resistance to apoptosis are needed.
Bortezomib (VelcadeR) is a novel therapy directed toward inducing apoptotic cell death in malignant cells by inhibiting the proteasome machinery2. The proteasome is one of the key regulators of cell function, being responsible for the degradation of intracellular proteins and preventing their accumulation as dysfunctional, misfolded adducts16. Thus it controls the cell cycle20, signal transduction 8, and response to oxidative stress and apoptosis 35 in the cell. Proteasome inhibition leads to apoptotic cell death in a number of malignant cell lines by inactivation of survival protein nuclear factor κB (NF-κB) 33, increased activity of p53 and Bax protein 11, and accumulation of cyclin-dependent kinase inhibitors such as p27 and p211,35. In malignant astrocytic stable cell lines proteasome inhibition causes cell growth arrest and apoptotic cell death by blocking the degradation of caspase -8, and -3, TRAIL activation, and mitochondrial dysfunction19,36. Also, we have recently reported that the proteasome inhibition is toxic only to malignant GSCs, and not to the human neural stem/precursor cells (NSC), suggesting that proteasome inhibition might have limited toxicities to the normal brain12. However, bortezomib activity in malignant glioma animal models is modest in spite of the fact that the drug is able to inhibit the proteasome activity in the xenografts, raising the question of possible alternative mechanisms counteracting the bortezomib efficacy21.
It has been previously shown that bortezomib can act as a double edge sword on endothelial cells: at low doses it allows cell proliferation and angiogenesis, whereas at high doses, it induces growth arrest and angiogenesis blockade29. The increased HUVEC proliferation and angiogenesis is explained by HIF-1α decreased degradation and subsequent nuclear accumulation in presence of proteasome inhibition29. HIF-1α levels directly correlates with the malignant glioma phenotype18, and directly regulate the expression of the major proangiogenic factor VEGF18.
Malignant GSCs drive tumor formation and treatment resistance22, but also enhance tumor angiogenesis through a VEGF-dependent mechanism3. We now propose that bortezomib has a similar effect of GSCs, causing increased HIF-1α levels and VEGF production, which in turn stimulates endothelial cell growth. We also show that the proangiogenic effects of bortezomib can be reversed by VEGF blockade in both the in-vitro and the in-vivo models, and suggest that simultaneous inhibition of proteolysis and angiogenesis offer increased benefit in the treatment of malignant gliomas.
METHODS
Materials
All chemicals and reagents were obtained from Sigma unless otherwise specified. Temozolomide (Merck) and O 6-benzylguanine (O 6-BG) were dissolved in dimethylsulfoxide and freshly prepared each time before use. Bortezomib was purchased from Millennium and bevacizumab from Roche.
Standard Protocol Approvals, Registrations and Patient Consents
Institutional Review Board (IRB) approval was obtained at University of California Irvine Medical Center for the malignant glioma and human foreskins harvesting.
Isolation and Expansion of Cells
Fresh brain tumors (glioblastoma multiforme, GBM) were taken at surgery and dissociated, with neuropathological review completed by a specialty neuropathologist. Cells were cultured on matrigel-coated dishes in 1:1 DMEM:F12 medium (Irvine Scientific), containing 10% BIT9500 (Stem Cell Technologies), 292 μg/mL glutamine (Irvine Scientific), 40 ng/mL FGF, 20 ng/mL EGF, and 20 ng/mL PDGF. For expansion, one-half of this medium was replaced every other day, and the cultures were passaged every seven days or when confluent using Non-enzymatic Cell Dissociation Solution (Sigma). HuTuP01 glioblastoma multifome GSCs were a gift from Dr. David Panchision (Children’s National Medical Center) 25. D54-MG and U251- MG cell lines were a gift from Dr. Darrell Bigner, Duke. The lines are maintained in stationary monolayer culture at 37° C and 5% CO2, using DMEM-F12 media (Gibco) containing 10% fetal bovine serum (FBS). To create temozolomide-resistant cell lines, U251-MG and D54-MG cells were initially cultured in the presence of 10 μM of temozolomide (TMZ) and 25 μM O6-BG, as previously published5. After every three passages, the concentration of TMZ was increased 5–10 μM. The resulting resistant cell lines are denoted as TMZ resistance (TR) and O6-BG and TMZ resistance (OTR). Primary microvascular endothelial cells were a gift from Dr. Chung-Ho Sun (UC Irvine), and were obtained from fresh human foreskins as published7. The microvascular endothelial cells were maintained in Matrigel-coated 96 well plates.
XTT Cell Viability Assay
The malignant glioma cells were seeded at approximately 1×104/well in a final volume of 200 μl in 96-well microtiter plates, and treated with bortezomib, bevacizumab, temozolomide or the control vehicle, as indicated in the Results section. After 72 hours, 100 μl of XTT (2,3-bis (2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino) carbonyl]-2Htetrazolium hydroxide) (Roche Applied Science) was added to each well, and the plates were incubated at 37°C for another 4 hours. Absorbance was measured at 450 nm against a reference wavelength at 650 nm using a microplate reader (Beckman Coulter, DTX 880 Multimode Reader). To measure primary microvascular endothelial cells viability, media conditioned by GSCs for 72 hours were added to the Matrigel-coated 96 well plates already containing 1×104 cells per each well, and the cells were allowed to grow for an additional 72 hours. All the experiments were done in triplicate, and repeated for a minimum three separate times. The absorbance values are expressed reported to control cells.
Apoptosis Analysis by Flow Cytometry
U251-MG and D54-MG cells (106 per experiment) were treated with bortezomib (1, 5, and 10 nM). Two days later, the cells were collected and fixed in 70% ethanol for two hours. After fixation, the cells were pelleted and suspended in 1 ml Propidium Iodide (PI)/ Triton X staining solution containing 0.1 % Triton X-100, 20 μg/ml PI and 200 μg DNase-free RnaseA, and incubated for 30 min at room temperature. At the end of the incubation time, the cells were subjected to fluorescence-activated cell sorting analysis of DNA content. The percentage of cells with subdiploid DNA content was taken as a measure of the apoptotic rate of the cell population.
Caspase-3 Activation
U251-MG and D54-MG cells were treated with bortezomib or the vehicle control. Shortly after treatment, 50 μM of caspase-3 inhibitor Z-VAD-FMK was added (Enzyme Systems). Caspase-3 activity was measured using a Fluorometric Assay Kit (MBL). 106 cells were harvested 24 hours after treatment with bortezomib, pelleted, and resuspended in cell lysis buffer. 200 μg protein cell extract were incubated with 200 μM DEVD-pNA. Spectrophotometric detection of EVD-pNA was performed at 380 nm excitation and 460 nm emissions. The results are expressed as a percentage of control samples. The results are means +/- standard errors of six independent determinations.
Proteasome Activity Analysis
20S proteasome chymotrypsin-like activity was measured using the 20S Proteasome Activity Kit (Millipore). Briefly, malignant glioma cells (106 per experiment) were pelleted, and resuspended in cell lysis buffer. 200 μg protein cell extract (supernatant) were incubated with the proteasome substrate Suc-LLVY-AMC for two hours. The fluorescence was measured using a 380/460 nm filter set.
SDS-PAGE and Immunobloting
GSCs (DB17 and HuTuP01) were treated with bortezomib (10nM) or the vehicle control. Three days after treatment, the cells were harvested and lysed. After equalizing the protein content an SDS-PAGE was performed. The proteins separated on the SDS-PAGE were electroblotted onto a Millipore Immobilon PVDF membrane. The membranes were incubated over night in the cold room with primary antibody against HIF1-α (Novus) (1:500 dilution), then with the secondary antibody, horseradish peroxidase-labeled goat anti-mouse IgG (Millipore). The protein of interest was detected using an enhanced chemiluminescence immunochemical detection kit (Amersham). HIF1-α levels were quantified relative to actin expression.
VEGF ELISA
GSCs (DB17 and HuTuP01) were treated with bortezomib (10nM), bevacizumab (final concentration 0.5mg/mL) or the vehicle control. Three days after treatment, the conditioned media from the cells was harvested. Human VEGF Quantikine ELISA Kits (R&D Systems) were used according to the directions of the manufacturer. Two-hundred microliters of conditioned media were collected from triplicate samples.
Human Glioma Xenografts
Athymic male BALB/c nu/nu mice were maintained in the Duke University Cancer Center Isolation Facility according to institutional policy. D-245 MG is a xenograft derived from an adult human malignant glioma, as described previously9. Xenografts were maintained in the flanks of athymic BALB/c nu/nu mice and early passages were used to minimize genetic drift. Subcutaneous tumor transplantation into the right flank of the animals was performed with inoculation volumes of 50 μl. 18 days after the malignant glioma cell inoculation, the animals (10 mice per group) were treated with bevacizumab (administered via intra-peritoneal (i.p.) injection at a dose of 5 mg/kg daily), or bortezomib (either single agent, or in combination with bevacizumab, given i.p. one hour after bevacizumab administration) at a dose of 0.35 mg/kg on days 1, 4, 8 and 11 every 21 days (mimicking human administration schedule). Tumors were measured twice weekly with hand-held vernier calipers (Scientific Products, McGraw, Ill.). Tumor volume was calculated according to the formula: [(width)2 × (length)]/2.
Statistical analysis
Graphs and statistical analyses were prepared using Prism 3.03 (Graph Pad). All values were presented as mean ± standard error of the mean (S.E.M.). Statistical significance was measured by simple paired t-tests or one-way ANOVA.
RESULTS
Bortezomib Kills Human Malignant Glioma Cell Lines and Glioma Stem-Like Cells by Caspase 3- Dependent Apoptotic Cell Death
We first examined the response of stable malignant gliomas cells lines (D-54 MG and U251-MG) and GSCs (HuTuP01 and DB17) to graded doses of bortezomib (Figure 1A). We found that low concentrations of bortezomib (1 nM) damages and kills the GSCs, but not the stable cell lines, while higher concentrations (10 nM) affects both the stem-like cells and the stable cell lines. In order to determine the observed decrease in cell numbers, we measured the activity of caspase-3, a reported mediator of bortezomib-induced apoptosis in cancer cells26. Twenty four hours after the treatment with bortezomib (10 nM and 100 nM), the levels of caspase-3 increased by two to three folds in D54-MG cells (Figure 1B). At the same timepoint, the population of cells with subdiploid content has also increased from less than 1% to 12% in the D54-MG cells treated with 10 nM of bortezomib as compared with control treated cells (representative results shown-Figure 1C).
Figure 1. Bortezomib Kills Human Malignant Glioma Stem-Like Cells and the Malignant Glioma Cell Lines U251-MG and D-54 MG by a Caspase 3- Dependent Apoptotic Cell Death.
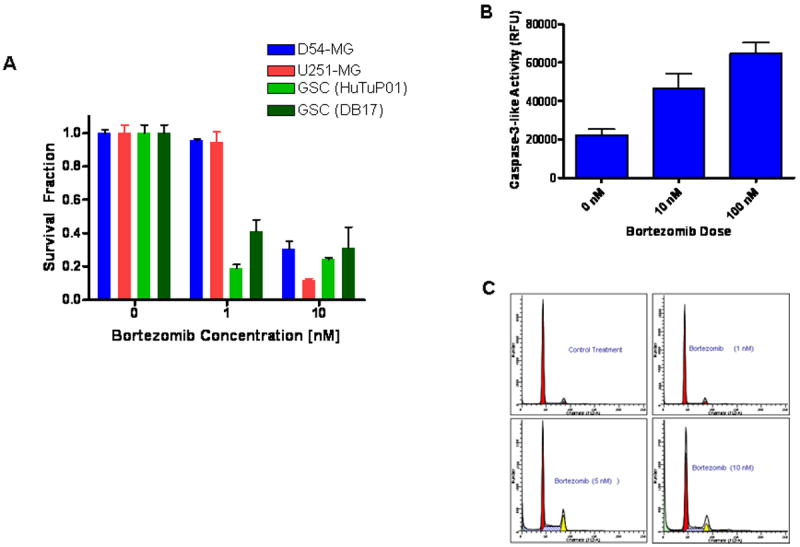
(A) In vitro treatment with 10 nM botezomib kills off the GSCs (HuTuP01 and DB17) as well as the stable glioma lines (D54-MG and U251-MG). (B) Caspase-3 activity was measured in cell extracts, 24 hours after treatment with bortezomib (10 nM and 100 nM). A doubling in aspase-3 activity was found in presence of 10 nM bortezomib. (C) 25% of the glioma cells treated with 10 nM bortezomib display subdiploid (SD) DNA content at 24 hours after treatment.
Bortezomib is Active against Temozolomide-Resistant Malignant Glioma Cell Lines
The temozolomomide (TMZ) and O6-BG resistant glioma lines were created in order to study resistance mechanisms and design new drug targets malignant gliomas. In patients, the most commonly described mechanism of TMZ resistance is MGMT and high MGMT levels predict poor response to TMZ14. The presence of the O6-BG precluded resistance caused by elevation of MGMT expression, as previously published4. Treatment with increasing doses of TMZ effectively kills the parent lines (U251 and D54), but has minimal effects on the survival of TMZ resistant (U251 TR and D54 TR) and TMZ and O6-BG resistant cells (U251 OTR and D54 OTR), as shown Figure 2A. However, both the TR and OTR lines retain sensitivity to proteasome inhibition (Figure 2B) at similar levels with the parent lines, suggesting that bortezomib is effective in TMZ resistant glioma cells.
Figure 2. Bortezomib is Active against Temozolomide-Resistant Malignant Glioma Cell Lines.
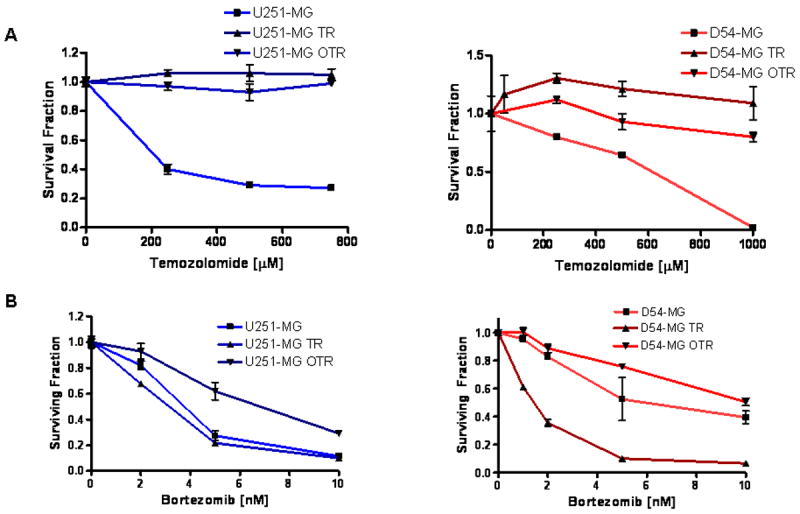
(A) In vitro treatment with graded doses of temozolomide leads to a decreased number of U251-MG and D54-MG parent lines, while minimally affecting the temozolomide resistant derived glioma lines (U215-TR and U251-OTR, D54-TR and D54-OTR). (B) In vitro treatment with graded doses of temozolomide is similarly effective in killing of U251-MG and D54-MG parent lines as well as the temozolomide resistant derived glioma lines (U215-TR and U251-OTR, D54-TR and D54-OTR).
Bortezomib Activity is Directly Proportional with the Baseline Proteasome Levels in Temozolomide-Resistant and Parent Malignant Glioma Cell Lines
The proteasome chymotrypsin-like base-line activity has been reported to correlate with bortezomib activity in multiple myeloma cells6. We found that in our malignant glioma cell lines the cells with the lowest chymotrypsin-like activity (U251-MG) were the most vulnerable to the toxic effects of bortezomib (10 nM), while the D54 OTR cells which had the highest chymotrypsin levels were the most resistant (Figure 3A and 2 C and D). All the other cell lines (U251 TR, U251 OTR, D54 MG and D54 TR) sensitivity to proteasome inhibition is also directly proportional with the proteasome activity levels.
Figure 3. Bortezomib Sensitivity is Directly Proportional with the Baseline Proteasome Chymotrypsin-Like Activity Levels in Temozolomide-Resistant and Parent Malignant Glioma Cell Lines.
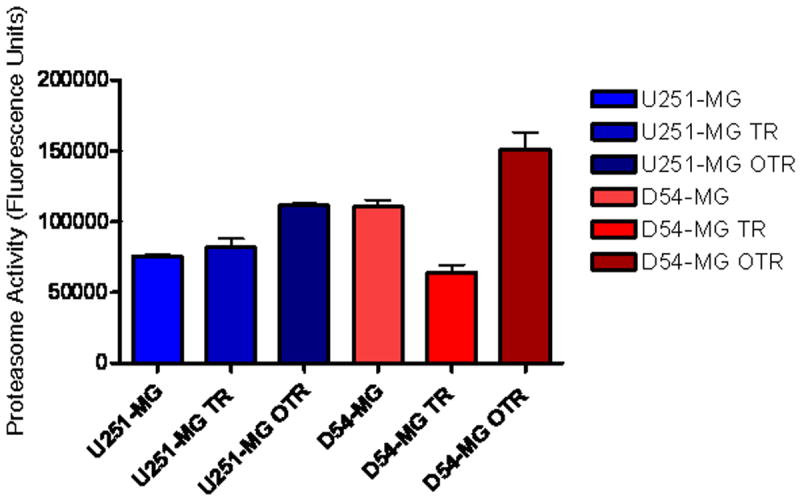
Proteasome chymotrypsin-like activity was measured in cell extracts. The two cell lines that displayed the highest resistance to bortezomib (U251-OTR and D54-OTR) also had the highest baseline proteasome levels, while the most sensitive line to bortezomib (D54-TR) had the lowest baseline proteasome activity.
Bortezomib Treatment Induces HIF1-α and VEGF Levels in Malignant Glioma Stem-Like Cells
To evaluate for the effect of bortezomib (10 nM) on HIF1-α, we compared HIF1-α protein levels on multiple GSCs (representative results with HuTuP01 and DB17 shown). Thought bortezomib treatment (10 nM) potentially kills up to 70% of the GSCs (Figure 1A), it also significantly increases the HIF1-α protein expression (Figure 4A). Also, the VEGF levels in the GSCs conditioned media were consistently increased up to five times after bortezomib treatment compared with the vehicle-treated cell (Figure 4A). The addition of the neutralizing anti-VEGF antibody bevacizumab (final concentration 0.5mg/mL) lowered the VEGF detection in conditioned media collected from both bevacizumab and bortezomib-bevacizumab treated GSCs.
Figure 4. Bortezomib Treatment Induces HIF1-a and VEGF Levels in Malignant Glioma Stem-Like Cells.
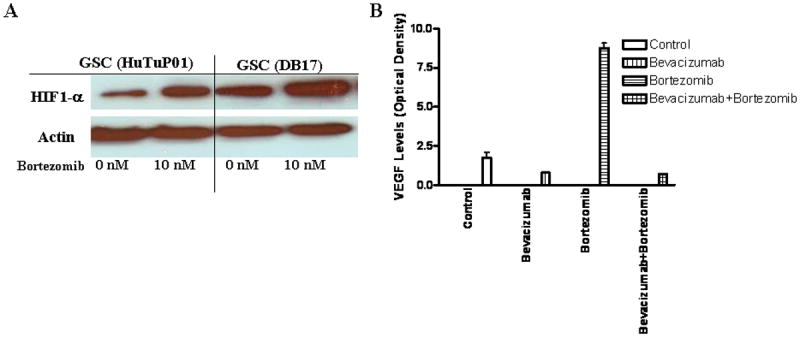
(A) 72 hours after treatment with bortezomib (10nM), HIF1-α protein levels (as determined by Western blot analysis) are increased in GSCs (HuTuP01 and DB17). (B) VEGF expression levels in media conditioned for 72 hours from GSCs treated with either bortezomib (10nM), bevacizumab (0.5mg/mL), or the combination were measured using a Human VEGF Quantikine ELISA Kits (R&D Systems). Bortezomib induced a four-time increase in the VEGF levels as compared with the control treatment, while bevacizumab was able to block the VEGF increase.
Conditioned Media (CM) from Malignant GSCs Treated with Bortezomib induces Microvascular Endothelial Cell Growth due to VEGF Over-expression
Two main cellular components are found in astrocytic tumors: glioma cells and endothelial cells. As such, the effects of chemotherapy depend not only on the sensitivity of the glioma cells to the therapeutic agent, but also on the sensitivity of the endothelial component. In addition, one of the most important mechanisms that govern GBM treatment and resistance to treatment is the formation of VEGF-regulated endothelial cell growth and new vasculature formation- a process controlled by the VEGF production of GSCs. In order to test if bortezomib treatment can potentially induce angiogenesis in our glioma models, we collected conditioned media from GSCs (treated with bortezomib and/or bevacizumab and the control vehicle) and added to primary human microvascular endothelial cells cultures (Figure 5 A). We found that the conditioned media from the GSCs treated with bortezomib strongly induce the human microvascular endothelial cells growth (twice as many cells were found on the bortezomib treated cells than in the control cells). This is consistent with the data presented in Figure 4B, which show that the conditioned media from GSCs treated with bortezomib has very high VEGF levels. Furthermore, the conditioned media from the GSCs treated with bevacizumab alone or with bortezomib in combination with the bevacizumab (and having low VEGF levels) do not induce microvascular endothelial cells growth.
Figure 5. Conditioned Media (CM) from Malignant Glioma Stem-Like Cells Treated with Bortezomib induces Microvascular Endothelial Cell Growth due to VEGF Over-expression.
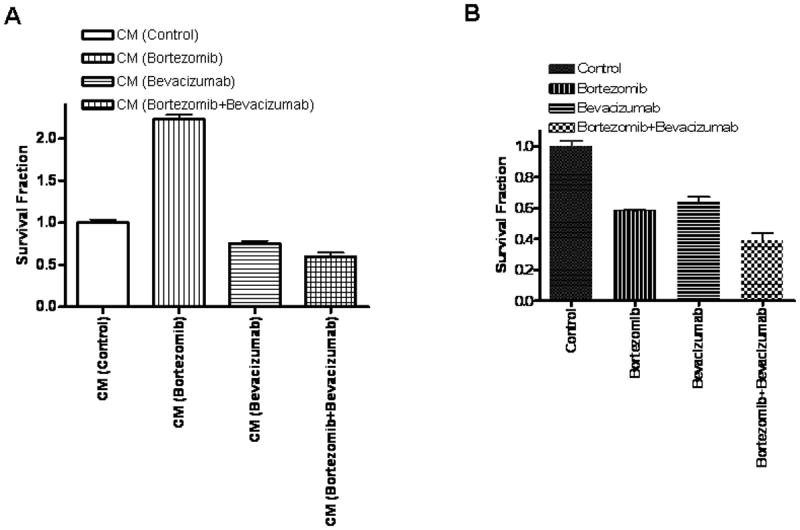
(A) Media conditioned for 72 hours by GSCs treated with bortezomib stimulates microvascular endothelial cell growth, while the conditioned media from the GSCs treated with both bortezomib and the VEGF neutralizing antibody, bevacizumab doesn’t increase endothelial cell growth. (B) Direct treatment of microvacular endothelial cells with with bortezomib (10 nM) as well as with bevacizumab (0.5mg/mL) caused a 40% reduction in microvascular endothelial cell viability, while the combined treatment caused a 60% decrease in cell viability, statistically superior to either drug alone.
In a separate set of experiments, we added bortezomib (10 nM) and bevacizumab (final concentration 0.5mg/mL) directly to the primary human microvascular endothelial cells cultures, in order to determine the direct effect of the drugs on the endothelial cells (Figure 5 B). We found that both drugs decrease endothelial cells viability by 30-40%, while the combination of the two drugs causes a 60% reduction in viability (p=0.02 for bortezomib versus combination treatment, and p=0.014 for bevacizumab versus combination treatment), suggesting that the two drugs might have an additive effect on the endothelial cells (Figure 5 B).
In vivo Treatment with Simultaneous Proteasome Inhibition (Bortezomib) and VEGF Blockade (Bevacizumab) Leads to Reduced Tumor Growth and Extended Survival
We also studied the effectiveness of bortezomib and bevacizumab, and the combination in athymic mice carrying subcutaneous malignant glioma xenografts (D245-MG). Intraperitoneal administration bortezomib (5 mg/kg daily), bevacizumab (0.35 mg/kg on days 1, 4, 8 and 11 every 21 days, mimicking human administration schedule) and the combination was well-tolerated in the tumor-bearing, atymic mice, with no difference in weight gain and no toxic deaths. After 16 days of treatment, the vehicle-treated mice group had an average tumor volume of 1155 μl, and the bortezomib-treatment very modestly decreased the tumor volume (Figure 6 A). As previously published, bevacizumab treatment significantly slowed D245-MG xenograft growth (mean tumor volume was 530 μl). However, the bortezomib-bevacizumab treated mice group was found to have the smallest tumor volume (465 μl, p=0.01).
Figure 6. In vivo Treatment with Simultaneous Proteasome Inhibition (Bortezomib) and VEGF Blockade (Bevacizumab) Leads to Reduced Tumor Growth and Extended Survival.
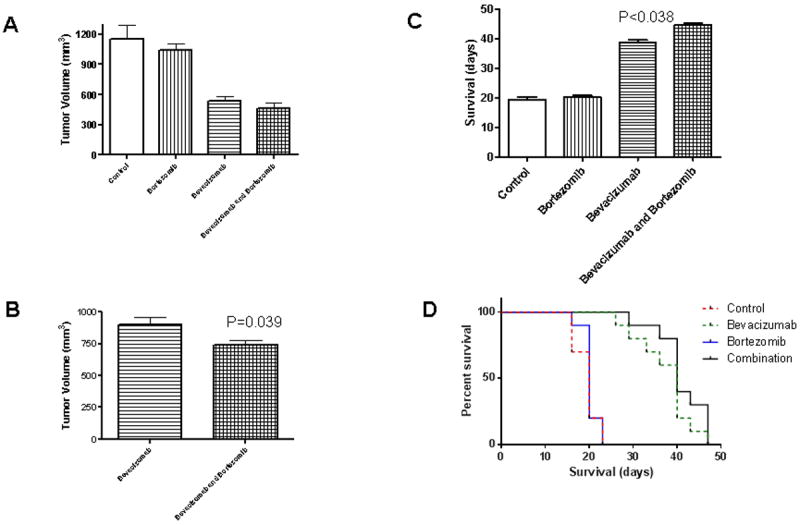
D245-MG xenografts were grown in the flanks of atymic mice. 18 days after the malignant glioma cell inoculation, the animals (10 mice per group) were treated with bortezomib (0.35 mg/kg on days 1, 4, 8 and 11 every 21 days) and/or bevacizumab (5 mg/kg daily). Tumor volume was calculated according to the formula: [(width)2 × (length)]/2. The animals that received the bortezomib-bevacizumab combination had smaller tumors both at day 16 and day 27 (p= 0.039) than the bevacizumab-only treated group (A, B). The combination also increased animal survival with 6 days (16%, p<0.038) as compared with the bevacizumab-only treated group (C).
After 27 days of treatment, only the bevacizumab and the combination (bevacizumab-bortezomib) mice were still alive (Figure 6 C). At that time, the average tumor volume was 900 μl for bevacizumab treated-mice, and 740 μl for the combination-treated group (p=0.04, Figure 6 B).
In our model, bortezomib monotherapy did not increase survival, while bevacizumab lead to a significant increase in median animal lifespan. The combination yielded further increases in lifespan, with a median lifespan of 19.4 days for control mice, 20.2 days for bortezomib-treated animals, 38.7 days for bevacizumab-treated animals, and 44.7 days for the combination-treated animals (p=0.038 against bevacizumab monotherapy).
DISCUSSION
By inhibiting a single molecular target, the proteasome, bortezomib affects multiple signaling pathways. The anti-neoplastic effect of bortezomib in glioma likely involves several distinct mechanisms, including inhibition of cell growth and survival pathways and induction of apoptosis32,36. The mechanisms by which bortezomib elicits its antitumor activity may vary among tumor types, and the extent to which each affected pathway is critical to the inhibition of tumor growth could also differ1.
Recent publications from our group12 as well as from other groups have suggested that tumor stem-like cells derived from malignant glioma tumor specimens have low proteasome activity as compared with the neural stem cells30. Here, we show that GSCs are more sensitive to the proteasome inhibition when very low dose of borteasome is used, but equally sensitive with the more differentiated stable glioma cell lines D54-MG and U251-MG when a higher dose of bortezomib is used (still nanomolar levels). This data confirm that glioma cells (both stem-like and more differentiated) are very sensitive to proteasome inhibition, to a level similar with the previously published data for meyloma and leukemia cell lines17. We also investigated what is the mechanism of proteasome activity in our glioma cell lines, confirming that proteasome inhibition leads to major caspase-3 activation and subsequently apoptotic cell death.
A major clinical problem in neuro-oncology is that 40% of more of the GBM tumors are resistant to temozolomide from the time of the initial diagnosis13. Notably, these patients do not benefit from the temozolomide treatment, and their prognosis remains poor 14. In prostate cancer models, bortezomib induces apoptosis in cells that over express bcl-2, a genetic trait that confers unregulated growth and resistance to conventional chemotherapeutics 23. Presently, we show that bortezomib has activity against both temozolomide sensitive glioma cell lines (U251-MG and D54-MG) as well as against the lines selected for temozolomide resistance (U251 TR and OTR and D54-TR and OTR)7.
Increased proteasome expression and activity has reported to establish apoptosis resistance and to confere decreased sensitivity to bortezomib in lymphoma cells, but no such correlation was previously established for gliomas. We now show that the bortezomib activity in malignant glioma cell lines is proportional with the proteasome chymotrypsin-like activity. In our system, the glioma cells with the lowest chymotrypsin-like activity (U251-MG) were the most vulnerable to the toxic effects of bortezomib, while the D54-OTR cells which had the highest chymotrypsin levels were the most resistant. This finding opens a potential avenue for using base-line (pretreatment) proteasome activity as a biomarker to stratify the malignant glioma patients currently receiving proteasome inhibitors in clinical trials.
The modest bortezomib activity in malignant glioma animal models in spite of the fact that the drug is able to inhibit the proteasome activity in the subcutaneous xenografts21 was long postulated to be causes by the reduced (but not absent) penetrance of bortezomib in brain15. However, bortezomib is able to reverse in vivo the hedgehog signaling pathway activation by blocking the degradation of Patched 1 protein in the cerebellar tumor cells of medulloblastoma-carrying Pax7ICNm /WTPtc1F1-2m/WTp53F2-10/F2-10 mice28, suggesting that even small concentrations of bortezomib might the able to exercise an effect on the brain tumors which have an abnormal blood-brain barrier. We now show that bortezomib has a dual effect on the glioma stem cells-which are postulated to drive tumor formation and treatment resistance22. We demonstrate that GSCs are very sensitive to low-concentrations of bortezomib, but the surviving GSCs have increased HIF1-α protein expression, and release they release large amounts of VEGF in the media. Furthermore, when human primary endothelial cells are exposed to the conditioned media from the GSCs treated with bortezomib, they are driven into increased proliferation. The addition of bevacizumab- a VEGF blocking antibody which is now used as second-line GBM treatment31 is able to reverse endothelial cell proliferation.
Our xenograft results are consistent with our in-vitro findings. As previously discussed, bortezomib alone is not able to affect malignant glioma xenograft outcomes, while, as expected, bevacizumab is a potent suppressor of tumor growth3. However, the bortezomib-bevacizumab combination was more active than both agents alone, and slowed tumor growth while prolonging animal survival.
Our results suggest that combination therapies based on two drugs with different, but complementary mechanisms of action which act simultaneously on the glial cell component (including the glioma stem cells) and on the tumor vascular endothelium might bring the next advance in the treatment of malignant gliomas. The data for the single bortezomib, phase I trial in recurrent malignant gliomas were recently presented, and bortezomib was well tolerated in this patient populations24. We now conclude a phase II trial of the bortezomib-bevacizumab combination, and will publish our results in the immediate future.
Acknowledgments
This work was supported by University of California, Irvine start-up funds to D. Bota. We thank Dr. Ronald C. Kim for the neuropathologic diagnosis of our tumor samples, Drs. David Panchision (Children’s National Medical Center), and Chung-Ho Sun (UC Irvine) for their gifts of cells, and Ms. Madelaine Afable for her technical help with the manuscript.
References
- 1.Adams J. Potential for proteasome inhibition in the treatment of cancer. Drug Discov Today. 2003;8:307–315. doi: 10.1016/s1359-6446(03)02647-3. [DOI] [PubMed] [Google Scholar]
- 2.Adams J, Palombella VJ, Elliott PJ. Proteasome inhibition: a new strategy in cancer treatment. Invest New Drugs. 2000;18:109–121. doi: 10.1023/a:1006321828515. [DOI] [PubMed] [Google Scholar]
- 3.Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, et al. Stem Cell-like Glioma Cells Promote Tumor Angiogenesis through Vascular Endothelial Growth Factor. Cancer Res. 2006;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 4.Bektas M, Johnson S, Poe W, Bigner D, Friedman H. A sphingosine kinase inhibitor induces cell death in temozolomide resistant glioblastoma cells. Cancer Chemotherapy and Pharmacology. 2009;64:1053–1058. doi: 10.1007/s00280-009-1063-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bektas M, Johnson SP, Poe WE, Bigner DD, Friedman HS. A sphingosine kinase inhibitor induces cell death in temozolomide resistant glioblastoma cells. Cancer Chemother Pharmacol. 2009;64:1053–1058. doi: 10.1007/s00280-009-1063-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bianchi G, Oliva L, Cascio P, Pengo N, Fontana F, Cerruti F, et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood. 2009;113:3040–3049. doi: 10.1182/blood-2008-08-172734. [DOI] [PubMed] [Google Scholar]
- 7.Chang C-J, Sun C-H, Liaw L-HL, Berns MW, Nelson JS. In vitro and in vivo photosensitizing capabilities of 5-ALA versus Photofrin® in vascular endothelial cells. Lasers in Surgery and Medicine. 1999;24:178–186. doi: 10.1002/(sici)1096-9101(1999)24:3<178::aid-lsm2>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 8.Ciechanover A, Orian A, Schwartz AL. The ubiquitin-mediated proteolytic pathway: mode of action and clinical implications. J Cell Biochem Suppl. 2000;34:40–51. doi: 10.1002/(sici)1097-4644(2000)77:34+<40::aid-jcb9>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 9.Friedman HS, Houghton PJ, Schold SC, Keir S, Bigner DD. Activity of 9-dimethylaminomethyl-10-hydroxycamptothecin against pediatric and adult central nervous system tumor xenografts. Cancer Chemotherapy and Pharmacology. 1994;34:171–174. doi: 10.1007/BF00685936. [DOI] [PubMed] [Google Scholar]
- 10.Friedman HS, Kerby T, Calvert H. Temozolomide and treatment of malignant glioma. Clin Cancer Res. 2000;6:2585–2597. [PubMed] [Google Scholar]
- 11.Fujioka S, Schmidt C, Sclabas GM, Li Z, Pelicano H, Peng B, et al. Stabilization of p53 is a novel mechanism for proapoptotic function of NF-kappaB. J Biol Chem. 2004;279:27549–27559. doi: 10.1074/jbc.M313435200. [DOI] [PubMed] [Google Scholar]
- 12.Gong X, Schwartz PH, Linskey ME, Bota DA. Neural stem/progenitors and glioma stem-like cells have differential sensitivity to chemotherapy. Neurology. 76:1126–1134. doi: 10.1212/WNL.0b013e318212a89f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hegi ME, Diserens AC, Godard S, Dietrich PY, Regli L, Ostermann S, et al. Clinical trial substantiates the predictive value of O-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res. 2004;10:1871–1874. doi: 10.1158/1078-0432.ccr-03-0384. [DOI] [PubMed] [Google Scholar]
- 14.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 15.Hemeryck A, Geerts R, Monbaliu J, Hassler S, Verhaeghe T, Diels L, et al. Tissue distribution and depletion kinetics of bortezomib and bortezomib-related radioactivity in male rats after single and repeated intravenous injection of 14C-bortezomib. Cancer Chemotherapy and Pharmacology. 2007;60:777–787. doi: 10.1007/s00280-007-0424-9. [DOI] [PubMed] [Google Scholar]
- 16.Hough R, Pratt G, Rechsteiner M. Ubiquitin-lysozyme conjugates. Identification and characterization of an ATP-dependent protease from rabbit reticulocyte lysates. J Biol Chem. 1986;261:2400–2408. [PubMed] [Google Scholar]
- 17.Houghton PJ, Morton CL, Kolb EA, Lock R, Carol H, Reynolds CP, et al. Initial testing (stage 1) of the proteasome inhibitor bortezomib by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;50:37–45. doi: 10.1002/pbc.21214. [DOI] [PubMed] [Google Scholar]
- 18.Jensen R, Ragel B, Whang K, Gillespie D. Inhibition of hypoxia inducible factor-1α (HIF-1α) decreases vascular endothelial growth factor (VEGF) secretion and tumor growth in malignant gliomas. Journal of Neuro-Oncology. 2006;78:233–247. doi: 10.1007/s11060-005-9103-z. [DOI] [PubMed] [Google Scholar]
- 19.Kim S, Choi K, Kwon D, Benveniste EN, Choi C. Ubiquitin-proteasome pathway as a primary defender against TRAIL-mediated cell death. Cell Mol Life Sci. 2004;61:1075–1081. doi: 10.1007/s00018-004-3477-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koepp DM, Harper JW, Elledge SJ. How the cyclin became a cyclin: regulated proteolysis in the cell cycle. Cell. 1999;97:431–434. doi: 10.1016/s0092-8674(00)80753-9. [DOI] [PubMed] [Google Scholar]
- 21.Labussiere M, Pinel S, Delfortrie S, Plenat F, Chastagner P. Proteasome inhibition by bortezomib does not translate into efficacy on two malignant glioma xenografts. Oncol Rep. 2008;20:1283–1287. [PubMed] [Google Scholar]
- 22.Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir I, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Molecular Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McConkey DJ, Pettaway C, Elliott P, Adams J, Papandreou C, Herrmann JL. The proteasome as a new drug target in metastatic prostate cancer. Proceedings of 7th Annual Genitourinary Oncology Conference; Houston, TX. 1999. [Google Scholar]
- 24.Phuphanich S, Supko J, Carson K, Grossman S, Burt Nabors L, Mikkelsen T, et al. Phase 1 clinical trial of bortezomib in adults with recurrent malignant glioma. Journal of Neuro-Oncology. 100:95–103. doi: 10.1007/s11060-010-0143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pistollato F, Chen H-L, Rood BR, Zhang H-Z, D’Avella D, Denaro L, et al. Hypoxia and HIF1alpha Repress the Differentiative Effects of BMPs in High-Grade Glioma. Stem Cells. 2009;27:7–17. doi: 10.1634/stemcells.2008-0402. [DOI] [PubMed] [Google Scholar]
- 26.Ri M, Iida S, Nakashima T, Miyazaki H, Mori F, Ito A, et al. Bortezomib-resistant myeloma cell lines: a role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia. doi: 10.1038/leu.2010.137. [DOI] [PubMed] [Google Scholar]
- 27.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 28.Taniguchi E, Cho MJ, Arenkiel BR, Hansen MS, Rivera OJ, McCleish AT, et al. Bortezomib reverses a post-translational mechanism of tumorigenesis for patched1 haploinsufficiency in medulloblastoma. Pediatric Blood & Cancer. 2009;53:136–144. doi: 10.1002/pbc.21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Veschini L, Belloni D, Foglieni C, Cangi MG, Ferrarini M, Caligaris-Cappio F, et al. Hypoxia-inducible transcription factor-1 alpha determines sensitivity of endothelial cells to the proteosome inhibitor bortezomib. Blood. 2007;109:2565–2570. doi: 10.1182/blood-2006-06-032664. [DOI] [PubMed] [Google Scholar]
- 30.Vlashi E, Pajonk F. Targeted cancer stem cell therapies start with proper identification of the target. Mol Cancer Res. 8:291. doi: 10.1158/1541-7786.MCR-09-0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, Reardon DA, Quinn JA, Sathornsumetee S, et al. Bevacizumab and Irinotecan is an Effective Treatment for Malignant Gliomas. Neuro-oncol. 2006;8:454. [Google Scholar]
- 32.Wagenknecht B, Hermisson M, Groscurth P, Liston P, Krammer PH, Weller M. Proteasome inhibitor-induced apoptosis of glioma cells involves the processing of multiple caspases and cytochrome c release. J Neurochem. 2000;75:2288–2297. doi: 10.1046/j.1471-4159.2000.0752288.x. [DOI] [PubMed] [Google Scholar]
- 33.Wang CY, Mayo MW, Baldwin AS., Jr TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 34.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 35.Yan Y, Yu X. Regulation of apoptosis: the ubiquitous way. FASEB J. 2003;17:790–799. doi: 10.1096/fj.02-0654rev. [DOI] [PubMed] [Google Scholar]
- 36.Yin D, Zhou H, Kumagai T, Liu G, Ong JM, Black KL, et al. Proteasome inhibitor PS-341 causes cell growth arrest and apoptosis in human glioblastoma multiforme (GBM) Oncogene. 2005;24:344–354. doi: 10.1038/sj.onc.1208225. [DOI] [PubMed] [Google Scholar]