

Abstract
Disease-associated BRCA2 mutations typically result in protein truncations that delete the phosphorylation-regulated S3291 BRCA2 domain that interacts with Rad51. BRCA2 hereditary breast cancers are usually ER+, differing from BRCA1 hereditary cancers, which are usually ER negative. We studied BRCA2 protein expression and S3291 phosphorylation in normal breast tissues and in sporadic breast cancers and observed that BRCA2 is expressed and phosphorylated in normal breast and 10 ER+ breast cancers but not in 10 ER negative breast cancers. In order to study this correlation between ER and BRCA2 expression we studied ER+ breast cancer cell lines. We found that a rapid increase in BRCA2 S3291 phosphorylation occurs following 17-beta-oestradiol (E2) treatment. This increase seen in BRCA2 total and phospho-S3291 protein levels was found to be unaffected with cycloheximide pre-treatment, but decreased following tamoxifen, ICI 182,780, or roscovitine treatment. This suggests a requirement for ER and cdk (cyclin-dependent kinase) in mediating the increased protein levels. MCF7 cell cycle distribution analysis following E2 both in the presence and absence of roscovitine (a cdk inhibitor) did not demonstrate any changes during an 8 hour period, which further supports our hypothesis that mitogenic effects of E2 are not predominant at early time points. Studies with MG132 proteasome inhibitor and siRNA to skp2 support a model in which skp2-mediated proteasomal degradation of BRCA2 rapidly degrades BRCA2 protein in the absence of hormone treatment which likely inhibits this pathway. E2 was shown to improve survival of MCF7 cells upon radiation treatment and roscovitine partially reversed this effect. We have demonstrated that BRCA2 protein is specifically expressed in ER+ breast cancers and are investigating a pathway that may show a link between E2 action and BRCA2 protein function in breast cancer.
Keywords: BRCA2, estrogen, breast cancer, phosphorylation
Introduction
Breast cancers are classified in a number of different ways, but the presence or absence of ER is key because ER+ and ER− cancers exhibit different biology and ER+ cancers preferentially respond to tamoxifen and aromatase inhibitors [1]. ER+ cancers usually occur in post-menopausal women while ER− cancers are more common in premenopausal women and generally have a poorer prognosis. Studies using microarrays have demonstrated several different breast cancer phenotypes that distinguish BRCA1 from BRCA2 hereditary cancers [2–4]. Large-scale population studies of BRCA1 mutation carriers indicate that only 4% of breast cancers in these patients are ER+ and this may statistically represent sporadic cancers occurring in patients harbouring BRCA1 mutations that did not produce the tumour [5]. Alternatively, the luminal phenotype expresses keratin 18, is ER+, and includes BRCA2 hereditary cancers and “BRCA2-like” sporadic cancers with similar molecular signatures [2,3]. These different phenotypes might result from different stem cell populations [1,6] or differences in ER expression in cells derived from a common stem cell. Interestingly, studies of mouse mammary stem cells indicate that mammary repopulating units are not exclusively basal or luminal and do not express ER, although the more differentiated Ma-CFCs do [7]. In addition to stem cell theories, several other hypotheses might explain why BRCA2 hereditary breast cancers are usually ER+, including: alteration of ER expression/function by BRCA proteins as shown for BRCA1 [8], specific molecular genetic interactions between E2 and BRCA2 protein, or selective expression of BRCA2 in ER+ breast cancer cells.
A single inherited BRCA2 mutation followed by LOH causes the BRCA2 hereditary cancer syndrome whereas inheritance of two BRCA2 mutations causes a subset of Fanconi’s anaemia [9]. Rad51 binds to BRCA2 at centrally located BRC repeats, and is dependent upon phosphorylation of serine 3291 which is located in the C-terminus of BRCA2 [10]. This C-terminal phosphorylated region of BRCA2 corresponds precisely to the area where BRCA2 truncations result in cancers, suggesting that a loss of S3291 phosphorylation might eliminate BRCA2 tumour suppressor function.
E2 has both genomic (transcriptional) and non-genomic effects. The non-genomic effects are ER-dependent, but occur even following inhibition of transcription or protein synthesis. Non-genomic hormonal signalling involves several pathways, including Src and MAP kinases [11,12]. Previous studies of BRCA gene transcription have shown that both BRCA1 and BRCA2 mRNA are expressed at high levels during pregnancy and that E2 increases BRCA1 mRNA levels in ovariectomized mice [13,14]. Despite the well-described examples of non-genomic effects of E2 [11,12], no published studies have evaluated whether E2 treatment alters protein levels or post-translational modifications of BRCA2. Oestrogens induce cell proliferation in part by stimulating transcription of cdk2 and cdk4 [15] {{{Ed Query check gene names and formatting}}}, activation of the cdk-activating phosphatase Cdc25A [15,16, as well as activation of both cyclin D1 and Rb [15]. Alterations in proteins such as cdk2 affect DNA repair since cdks also regulate key mediators such as chk1 [17]. The relationship of cdks and their inhibitors to E2 response is complex and bi-directional, as indicated by p21-mediated inhibition of selective ER transcriptional events [18].
Methods
Immunohistochemistry
Samples were stripped of identifiers and the research protocol was reviewed by the Colorado Institutional Review Board and determined to be exempt from review. After deparaffinization and rehydration of 4 μm sections of formalin fixed paraffin-embedded tissue, heat induced antigen retrieval was achieved in a 20mM citrate buffer at pH 6.0 in a decloaking chamber (Biocare Medical). Sections were incubated with the primary antibodies: pan BRCA2 (Calbiochem Ab-2) and S3291 phospho-specific BRCA2 at 3μg/ml and 4μg/ml in a humidified chamber overnight at 4°C, washed in buffer, and treated with a horseradish peroxidase labelled polymer detection system (DAKO Envision +, Dual Link) for 30 min at room temperature. Peptide blocking studies were performed by preincubating the phospho-specific antibody with a ten-fold excess of phosphorylated peptide antigen or an unphosphorylated peptide and staining as indicated. Chromagens used were Vector Nova Red (red), and DAKO DAB+ (brown), for the pan BRCA2 and phospho-specific BRCA2, respectively. The cytoplasmic yellow counterstain used was Metanil Yellow (ScyTek Laboratories).
Breast cancer cell culture
ER+ human breast cancer cells MCF7 and T47D cells were cultured in 10% charcoal-stripped serum/phenol free DMEM for two days prior to treatment and 10 nM E2 was added for 0.5 h (early), 4 h (intermediate), or 24 (extended) times. MCF-7 and T47D cells were grown in with 10 nM E2. Pathway inhibitors (U0126, roscovitine, Purvalanol A) were added to cells 1 h prior to addition of E2. Cells for colony formation assays were plated at 500 cells per plate and then scored microscopically after 14 days. After scoring, the plates were formaldehyde fixed, washed in phosphate buffered saline, and then stained with crystal violet. Statistical significance was determined on quadruplicate microscopic counts using Fisher’s exact test to determine 2 sided p values.
Generation of phospho-specific antibody
New Zealand White rabbits were immunized with the phosphorylated peptide TFVpSPAAKAGG and the resulting antisera were affinity purified by selective binding to phosphorylated and unphosphorylated peptides bound to Sepharose, elution of bound antibody, followed by gel filtration and dialysis against phosphate buffered saline.
Western blotting of proteins
Cell extracts in RIPA buffer, were standardized for equal protein then separated by either 1) Tris/glycine SDS-PAGE on 6% mini-gels for 2 hr at 125 V (Figure 2) then transferred to PVDF membranes at either 50 V constant for 200 Volt-hours, or 2) Tris/acetate SDS-PAGE on 3–8% Novex NuPage mini-gels for 1 h at 160 V then transferred in Tris/glycine transfer buffer containing 8% MeOH at 16°C (Figures 4–6). Membranes were blocked for 1 h at room temperature in 5% dried non-fat milk in PBS. The primary Ab BL320 (Bethyl) called pan-BRCA2, or the S3291 phospho-specific Ab were incubated overnight at 4°C diluted 1:500 in 0.5% dry non-fat milk/PBS-Tween 0.1%. After appropriate washes anti-rabbit-HRP (GE Healthcare/Amersham) diluted 1:5000 in 0.5% dried non-fat milk/PBS-Tween 0.1% was added and incubated for 1 h at room temperature. Detection was performed with ECL Plus reagents (GE Healthcare/Amersham).
Figure 2.

E2 rapidly stabilizes BRCA2 protein levels in human breast cancer cells. a, MCF7 cells were treated with 10 nM E2 for 0.5, 4, or 24 h. CHX is 25μM cycloheximide. Cyclin D1 levels were analyzed to confirm inhibition of protein synthesis in CHX-treated groups. b, Inhibition of cyclin-dependent kinase (10 μM roscovitine) inhibits BRCA2 protein but MAP kinase inhibition (10μM U0126) does not. c, T47D cells show similar E2 stabilization of BRCA2 which is inhibited by roscovtine.
Figure 4.

Rapid BRCA2 protein stabilization occurs without increased transcription or cell cycle progression. a) MCF7 cells cultured for 48 hours in 10% charcoal stripped serum/phenol free DMEM were treated with 10 nM E2 for indicated times prior to RNA harvest for RT-PCR. pS2 (TFF1) is E2 induced control and 36B4 is a paired unaffected control [22] b) Data from RT-PCR experiments presented in a) which have been quantitated by fluorescent imaging in triplicate assays c) MCF7 cells were similarly cultured and treated with E2 prior to cell cycle analysis by Krishan’s stain.
Figure 6.

Time course of E2-mediated S3291 phosphorylation and BRCA2 protein stabilization in MCF7 cells. a. MCF-7 cell lysates blotted with C-terminal BRCA2 (pan BRCA2) or phosphospecific antibody (phospho) and following E2 treatment (top 2 panels), roscovitine pretreatment (3rd and 4th), 10 nM tamoxifen pretreatment (5th and 6th), and ICI182,780 pretreatment (7th and 8th). Lamin control is the bottom lane. b. T47D cell lysates treated with E2 (top 2) or 10 nM tamoxifen (3rd and 4th panels). Both Lamin isoforms runs as a single fused band under these gel electrophoretic conditions.
RT-PCR
Random hexamers, a first strand synthesis kit (Invitrogen) and mRNA from E2-and control-treated MCF-7 cells were used to generate cDNA for PCR with primers to: BRCA2: 5′AATTCATCTGCTTTCTCTGGA 3′ and 5′AGTGCTCTGGGTTTCTC TTATC3′; pS2 (TFF1): 5′ CGTGAAAGACAGAATTGTGGTT TT3′ and 5′CGTCGAAACAG CAGCCCTTA3′; 36B4; 5′CTCCAAGCAGATGCAGCA GA3′ and 5′TGCCCATCAGCA CCACAG3′. BRCA2 PCR conditions were 94° 1 min; 59° 2 min; 72° 1 min for 30 cycles and TFF1/36B4 PCR conditions were 95° 1 min; 51° 1 min; 72° 1 min for 25 cycles. PCR products were then resolved on 1.5% agarose gels in tris borate buffer containing ethidium bromide.
Flow cytometry
MCF7 cells were cultured for 48 h in 10% charcoal-stripped serum/phenol free DMEM prior to treatments. Roscovitine inhibitor was added to appropriate groups one hour prior to E2 treatment for the indicated times. Cells were released with trypsin, washed three times with cold PBS, and 1 × 106 cells were resuspended in Krishan’s stain and incubated overnight at 4°C prior to DNA content measurement by flow cytometry.
siRNA studies
The oligofectamine reagent was complexed with the siRNA and incubated for 20 min at room temperature. The complex was then added to the MCF7 cells and incubated for 6 hours. After the 6 hours had passed, DMEM phenol-red free/charcoal-stripped serum complete medium was added to the cells. An assay for gene activity was done at 48 hours post transfection. The Skp2 Smartpool and non-targeting siRNA #1: 5′UAGCGACUAAACACA UCAA-3′, were purchased from Dharmacon.
Results
Expression and Phosphorylation of BRCA2 in breast tissues
In order to understand the role of BRCA2 in the human breast in normal and malignant tissues, we studied BRCA2 protein expression and S3291 phosphorylation in breast tissue samples using immunohistochemistry (IHC). The phospho-specific antibody to S3291 BRCA2 clearly stains nuclei within a normal breast gland (Figure 1a, left panel) and this staining is inhibited by phosphopeptide (Figure 1a, middle panel) but not unphosphorylated peptide (Figure 1a, right panel), providing evidence for specificity of this reagent. Western blots using these antibodies in human cell lines are shown in Figures 2,4 and 5. As an additional test for antibody specificity, we tested breast cancer samples from patients with BRCA2 hereditary cancer (6174delT mutation) and showed a loss of nuclear immunostaining in the cancers but not the normal tissues from these patients (data not shown). These patients have a truncated BRCA2 protein that lacks this S3291 phosphorylation site in the cancers but retain the wild type allele in normal tissue; this expected result also confirms antibody specificity.
Figure 1.

BRCA2 phosphorylation and expression in breast cancers a. IHC on normal breast with phospho-antibody. Brown chromogen shows S3291 phosphoBRCA2 with yellow counterstain. Upper panels are 40X; lower panels 100X. Phospho-antibody staining (left panels), is inhibited by phosphorylated peptide TFVpSPAAQKAGG (central panels), but not by unphosphorylated peptide (right panels). b. IHC on sporadic breast cancers. Left panels ER+, right panels ER−. All 40X magnification. Left panels employ phosphoBRCA2 (brown chromagen) and right panels employ pan BRCA2 (red chromagen) for each ER tumour type. Note that virtually all the immunostaining in ER− breast cancers appears in lymphocytes (marked by arrows) or stromal cells but not in tumour nuclei.
Figure 5.
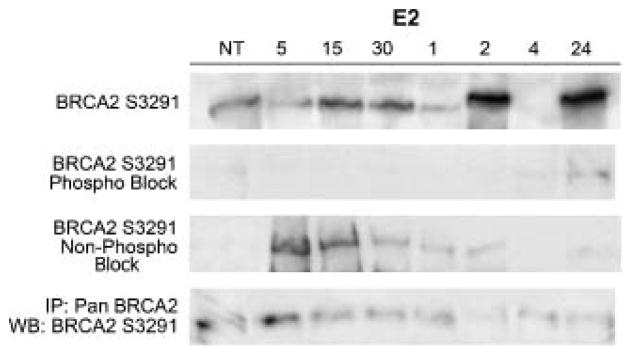
Assessment of phosphoS3291 antiserum specificity. Time course studies showing protein levels in indicated number of min after 10 nM E2 treatment (E2). Western blotting with phosphospecific BRCA2 antibody 1st panel incubated with either the non-phosphorylated peptide TFVSPAAQKAGG (2nd panel) or the phosphopeptide TFVSpPAAQKAGG (3rd panel). Bottom panel shows results of immunoprecipitation with pan-BRCA2 antibody followed by western blotting with S3291 phosphospecific antibody.
We next analyzed whether there is a correlation between ER status and the expression and phosphorylation of BRCA2 at S3291 in sporadic breast cancers. IHC results show that BRCA2 is present and phosphorylated at increased protein levels in ten ER+ cancers but not in ten ER negative cancers. Note that virtually all the immunostaining from either the pan-BRCA2 or phosphospecific BRCA2 antibodies in ER negative breast cancers appears in lymphocytes or stromal cells but not in tumour nuclei (arrows in Figure 1b). These results indicate that BRCA2 protein expression and phosphorylation is highly correlated with expression of oestrogen receptor and is present in normal tissues and in ER positive breast cancers but not in ER negative breast cancers.
Having observed this strong correlation between ER and the expression and phosphorylation of BRCA2 protein, we decide to test ER+ cell lines to see if we could use these cells as a model system to understand the relationship between oestrogen, ER, and BRCA2 protein
E2 non-genomically stabilizes BRCA2 protein in ER+ cell lines
E2 treated MCF7 cells exhibited a rapid increase in BRCA2 protein levels, suggesting a non-genomic mechanism involving protein stabilization. Indeed, increased protein levels were still observed at early (30 min) and intermediate (4 h) time points in cells pre-treated with 25 μM cycloheximide to inhibit protein synthesis (Figure 2a). Showing maximal stabilization of BRCA2 protein by E2 requires culturing in phenol red free media and charcoal-stripped serum for 48 h since MCF7 cells cultured in full serum and phenol red containing media express high levels of BRCA2 protein (data not shown). The increased BRCA2 protein observed at 24 hours was blocked by cycloheximide indicating that the later effects of E2 on BRCA2 protein are likely genomic effects.
Protein phosphorylation is a common mechanism for rapid changes in protein stability or function. As Cdk-2 is known to phosphorylate BRCA2 at S329110 we pre-treated cells with the cyclin-dependent kinase inhibitor roscovitine which repressed the E2-induced increase in BRCA2 protein levels at early and intermediate time points (30 min and 4 h) but not later time points (24 h) in both MCF7 (Figure 2b) and T47D cells (Figure 2c). Note that treatment of cells with cycloheximide and E2 eventually inhibits BRCA2 protein levels at 24 h, presumably because the half life of E2-stabilized BRCA2 protein is still less than 12 h (Figure 2a, right panel). In contrast, inhibition of the MAPK pathway, a common mediator of non-genomic E2 effects [11, 12], with U0126 had little or no effect on the increase in BRCA2 protein levels in MCF7 cells (Figure 2b). In order to determine whether cdk-dependent stabilization of BRCA2 protein was specific to MCF7 cells we also tested ER+ T47D cells and showed a similar result (Figure 2c).
In order to analyze the potential mechanism for BRCA2 protein stabilization we treated cells with the proteasome inhibitor MG132 in the presence of absence of E2. The results show both a rapid and sustained increase in BRCA2 protein levels within one hour of MG132 treatment in the absence of E2 but no increase in BRCA2 protein levels during the first two hours in the presence of E2 (Figure 3a). Once we had demonstrated that proteasomal degradation was a key regulator of BRCA2 protein levels, we tested whether skp2 was a likely E3 ligase mediating BRCA2 protein stability, as had been previously published in prostate cells [19]. Figure 3b shows that inhibition of skp2 by siRNA results in a 5-fold increase in BRCA2 protein levels while controls including mock transfection, no treatment, and a negative control siRNA sequence show no effect.
Figure 3.

BRCA2 protein stabilization by inhibitors of proteasomal degradation A, MCF7 cells were treated in the presence of the proteasomal inhibitor MG132 (20 μM) for 1, 2, 4 and 24 h or vehicle alone: 0 h (Left panels). Cell lysates were analyzed for the levels of total BRCA2 and Lamin by immunoblotting. B, MCF7 cells were transfected with 100 nM siRNA targeted to the E3 ligase Skp2 (Skp2), non-targeting siRNA (Neg), and mock transfected with the transfection reagent alone (Mock). The cells were allowed to adhere for 48 h before {{Ed Query please check this sentence is as meant}} transfection and collected at 48 h post-transfection. Similar results were obtained from two independent experiments.
E2 does not rapidly induce BRCA2 mRNA or change cell cycle distribution
Because E2 also regulates transcription and promotes cell cycle traverse in hormone responsive cells, we tested whether E2 treatment altered either BRCA2 mRNA levels or altered cell cycle distribution during the first few hours following hormone addition. The initial effects of E2 are predominantly post-transcriptional since RT-PCR analysis of E2 treated samples indicates that E2 only slightly induces BRCA2 mRNA levels at 24 hours (Figure 4a,b). To demonstrate that E2 did activate transcription in our cells we analyzed the pS2 (TFF1) gene and this showed a 6-fold mRNA induction 4 h after E2 and a 10-fold induction 24 h after E2 administration. A cell cycle analysis demonstrated that E2 clearly alters cell cycle distribution of cells 24 h after its addition, but has minimal effects during the initial 8 h period following hormone administration (Figure 4c).
E2 rapidly phosphorylates BRCA2 at serine 3291 in ER+ cultured cells
To demonstrate antibody specificity we performed peptide competition experiments to inhibit phospho-specific antibody binding with the phosphorylated (TFV-phosphoS-PAAQKAGG) and unphosphorylated (TFVSPAAQKAGG) peptides. These studies showed that the phosphorylated BRCA2 protein was inhibited by phosphorylated but not by unphosphorylated peptides (compare 1st through 3rd panel in Figure 5). As an additional test for specificity of the phospho-specific antibody, we immunoprecipitated with a pan-BRCA2 antibody and then western blotted with the S3291 phosphospecific antisera and showed the antisera identified a single E2-regulated protein of 440 kDa (Figure 5, bottom panel).
We next analyzed the kinetics of S3291 phosphorylation and learned that phosphorylation increased within 15–30 min following E2 treatment (Figure 6a). This rapid phosphorylation precedes the stabilization of BRCA2 protein since S3291 phosphorylation of BRCA2 is maximal at 15 min while overall BRCA2 protein stability (determined by pan-BRCA2 antibody) continues over 24 h. Although BRCA2 phosphorylation at serine 3291 is maximal at 15 min, there is also a biphasic response with significant S3291 phosphorylated BRCA2 present at 24 hours after E2 (Figure 6a, second panel). We used the cdk inhibitor roscovitine to compare effects of cdk inhibition on phosphorylation and protein stability and demonstrated that roscovitine inhibits both phosphorylation and protein stability at early and intermediate time points (Figure 5a, compare upper 4 panels). Roscovitine appears to inhibit S3291 phosphorylated BRCA2 protein at 24 h but does not completely inhibit total BRCA2 protein at 24 h, as determined with the pan-BRCA2 antibody (Figure 6a, panels 3 and 4). This suggests that the S3291 phosphorylated protein is not completely identical with the total BRCA2 protein identified by the pan-BRCA2 antibody since some non-phosphorylated protein is likely present.
To determine whether the ER mediated this effect we investigated whether the antiestrogen, 4-hydroxytamoxifen, could eradicate the increase in BRCA2 serine 3291 expression following E2 treatment. We found that pretreatment with 4-hydroxytamoxifen inhibits the E2 mediated increase in both BRCA2 protein and phosphorylation in MCF-7 cells (Figure 6a, panels 5, 6) and T47D cells (Figure 6b, panels 3, 4). Pretreatment with the absolute E2 antagonist ICI 182,780 [20] also inhibits the E2-mediated increase in BRCA2 S3291 phosphorylation and protein levels in MCF-7 cells (Figure 6b, panels 7&, 8).
E2 increases radiation response by a cdk dependent mechanism
We next examined whether the cdk-mediated E2 effect has biological significance for radiation survival. Because cdk inhibitors can have complex effects in cultured cells, we used both roscovitine and Purvalanol A for these experiments. Both of these small molecules inhibit cdk activity by competition for ATP at the catalytic kinase site [21]. Pretreatment of MCF7 cells with 10 nM E2 prior to irradiation with 1 Gy resulted in greater numbers of colonies that were larger in size (Figure 7a, compare panels 1 and 2) whereas treatment with the E2 plus the cdk inhibitors roscovitine or purvanalol A treatment resulted in smaller microscopic colonies (Figure 7a). Figure 6b shows that E2 treatment significantly increases the total number of MCF7 colonies (visible and microscopic) which survive ionizing radiation but the cdk2 inhibitor roscovitine inhibits this effect of E2; purvalanol A inhibits to a lesser extent. This effect of E2 on radiation survival is more significant at 1 and 2 Gy of ionizing radiation then at 4 Gy (Figure 7b).
Figure 7.

Effects of E2 and cdk inhibitors on radiation sensitivity. a. MCF7 cell colonies present after pretreatment with E2 and/or purvalanol A followed by radiation. Plates are labelled as 1. 1 Gy radiation; 2. 1 Gy radiation following 1 h pretreatment with 10 nM E2. 3. 1 Gy radiation following 1 h pretreatment with 400 nM purvalanol A. 4. 1 Gy radiation following 1 h pretreatment with 10 nM E2 and 400 nM purvanalol A. b. Graph showing results of quadruplicate colony counts of MCF7 cells treated with 1–4 Gy radiation and/or 10 μM roscovitine, 400 nM purvalanol and/or 10 nM E2. Bars are mean +/− SE. * denotes p<0.5 and ** denotes p< 0.2 by Fisher’s exact test.
Discussion
In this study, we show that 1) BRCA2 protein is expressed and phosphorylated in normal breast tissues and in ER+ breast cancers but not in ER negative breast cancers; 2) E2 phosphorylates and stabilizes BRCA2 protein by a cdk dependent mechanism that likely involves inhibition of proteasomal degradation, 2) BRCA2 is specifically phosphorylated and expressed in ER+ but not ER− breast cancers, and 3) E2 enhances radiation survival in a cdk-dependent manner. These results support a model that E2 activates BRCA2 function to increase DNA repair responses in ER+ breast cancer cells.
Previous research showed that cdk2-dependent phosphorylation of serine 3291 BRCA2 regulates its association with Rad51 and function in DNA repair [10], but did not demonstrate a link to tumour cell biology or demonstrate that hormones phosphorylated or regulated BRCA2. We now report that E2 rapidly phosphorylates S3291 BRCA2 protein and also stabilizes the protein leading to a 10-fold increase in BRCA2 protein levels. The initial increased expression appears to be largely non-genomic at early time points since it is unaffected by protein synthesis inhibitors and not observed at the mRNA level. In order to investigate the mechanism of protein stabilization of BRCA2 we inhibited proteasomal degradation with MG132 and skp2-mediated proteasomal degradation with skp2 siRNA. The results appear to show that skp2-mediated proteasomal degradation is responsible for BRCA2 protein stability as has been described for prostate cells previously [19]. This protein stabilization is inhibited by tamoxifen, ICI-182,780, and roscovitine indicating it requires ER and cyclin-dependent kinases.
Our studies also indicate that E2-mediated phosphorylation at S3291 does not completely explain BRCA2 protein stabilization. Side by side comparison of S3291 phosphorylated BRCA2 protein and total BRCA2 protein (Pan-BRCA2) shows that the phosphorylated BRCA2 and total BRCA2 have somewhat different time courses in response to E2 treatment. Phosphorylation of BRCA2 is more rapid and is biphasic with a maximal increase at 15 min and a second later peak of phosphorylated protein at 24 hours. We do not understand the basis of the biphasic response observed but have considered the possibility that the initial response my be due to protein kinase and dephosphorylase effects, and the later time points may be mediated by more complex cellular events. Total levels of BRCA2 protein increase more slowly but also show elevated levels at 24 h. It is possible that S3291 phosphorylation in cultured cells alters BRCA2 protein sublocalization or protein-interactions which ultimately lead to protein stabilization even after S3291 is no longer phosphorylated. However, we note that in human tissue samples BRCA2 phosphorylation and total BRCA2 protein levels appear highly correlated.
Our finding that E2 regulates BRCA2 provides a simple hypothesis why BRCA2 hereditary cancers are usually ER+: because E2 is required for BRCA2 function in breast cells. BRCA2 is phosphorylated in normal breast acini (Figure 1a) supporting a physiological function for S3291 BRCA2 in the normal breast. Analysis of breast cancer specimens by both IHC and western blotting showed that BRCA2 is phosphorylated and present at increased protein levels in ER+ cancers compared to ER− cancers. The expression of phosphorylated BRCA2 in ER+ cancers may provide enhanced radiation survival and DNA repair as suggested by our data showing that E2 enhances radiation survival in a cdk-dependent manner. Future work should address whether S3291 phosphorylation of BRCA2 affects Rad51 binding or localization and whether this affects the DNA repair pathway in breast cells.
More generally, this study provides a conceptual framework for the tissue specificity of BRCA hereditary cancers. Mutations in the BRCA genes most commonly result in breast or ovarian cancer and this tissue specificity has remained largely unexplained. Our study indicates that BRCA2 mutations lead to hormone related malignancies because the normal BRCA2 function is itself hormone regulated. Understanding the relationship between hereditary cancers and hormone signalling may provide new strategies for chemotherapy or radiation therapy which involve combination of hormone therapy with agents targeting specific DNA repair pathways such as those mediated by BRCA2.
Acknowledgments
Grant support: Grants from Public Health Service, National Cancer Institute and DOD: NCI R01CA85269 (JTH); DOD BC051061 (ACN); NIH PO1-HD38129 and RO1-DK063674 (SMA)
References
- 1.Kurbel S. Selective reduction of ER positive breast cancer occurrence by estrogen receptor modulators supports etiological distinction between ER positive and ER negative breast cancers. Med Hypothesis. 2005;64:1182–1187. doi: 10.1016/j.mehy.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 2.Hedenfalk L, Duggan D, Chen Y. Gene expression profiles in hereditary breast cancer. N Engl J Med. 2001;344:539–548. doi: 10.1056/NEJM200102223440801. [DOI] [PubMed] [Google Scholar]
- 3.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nature Rev Cancer. 2004;4:814–819. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 4.Richardson AL, Wang ZC, De Nicolo A, Lu X, Brown M, Miron A, et al. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006;9:121–132. doi: 10.1016/j.ccr.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 5.Foulkes WD, Metcalfe K, Sun P, Hanna WM, Lynch HT. Estrogen receptor status in BRCA1- and BRCA2-related breast cancer: the influence of age, grade, and histological type. Clin Cancer Res. 2004;10:2029–2034. doi: 10.1158/1078-0432.ccr-03-1061. [DOI] [PubMed] [Google Scholar]
- 6.Clarke RB, Spence K, Anderson E, Howell A, Okana H, Potten CS. A putative human breast stem cell population is enriched for steroid receptor-positive cells. Dev Biol. 2005;277:443–456. doi: 10.1016/j.ydbio.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 7.Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, et al. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- 8.Razandi M, Pedram A, Rosen EM, Levin ER. BRCA1 inhibits membrane estrogen and growth factor receptor signaling to cell proliferation in breast cancer. Mol Cell Biol. 2004;24:5900–5913. doi: 10.1128/MCB.24.13.5900-5913.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.King MC, Marks JH, Mandell JB The New York Breast Cancer Study Group. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 10.Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin, et al. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005;434:598–604. doi: 10.1038/nature03404. [DOI] [PubMed] [Google Scholar]
- 11.Boonyarantanakornit V, Scott MP, Ribon V, Sherman L, Anderson SM, Maller JM, et al. Progesterone receptor contains a praline-rich motif that directly interacts with SH3 domains and activates c-scr family tyrosine kinases. Molecular Cell. 2001;8:269–280. doi: 10.1016/s1097-2765(01)00304-5. [DOI] [PubMed] [Google Scholar]
- 12.Improta-Brears T, Whorton AR, Codazzi F, York JD, Meyer T, McDonnell DP. Estrogen-induced activation of mitogen-activated protein kinase requires mobilization of intracellular calcium. Proc Natl Acad Sci USA. 1999;96:4686–4691. doi: 10.1073/pnas.96.8.4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajan JV, Marquis ST, Gardner HP, Chodosh LA. Developmental expression of Brca2 colocalizes with Brca1 and is associated with proliferation and differentiation in multiple tissues. Dev Biol. 1997;184:385–401. doi: 10.1006/dbio.1997.8526. [DOI] [PubMed] [Google Scholar]
- 14.Rajan JV, Wang M, Marquis ST, Chodosh LA. Brca2 is coordinately regulated with Brca1 during proliferation and differentiation in mammary epithelial cells. Proc Natl Acad Sci USA. 1996;93:13078–13083. doi: 10.1073/pnas.93.23.13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foster JS, Henley DC, Bukovsky A, Seth P, Wimalasena J. Multifaceted regulation of cell cycle progression by estrogen: regulation of CDK inhibitors and cdc25A independent of cyclin D1-cdk4 function. Mol Cell Biol. 2001;21:794–810. doi: 10.1128/MCB.21.3.794-810.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster JS, Fernando IR, Ishida N, Nakayama KI, Wimalasena J. Estrogens down-regulate p27Kip1 in breast cancer cells through Skp2 and through nuclear export mediated by the ERK pathway. J Biol Chem. 2003;278:41355–41366. doi: 10.1074/jbc.M302830200. [DOI] [PubMed] [Google Scholar]
- 17.Maude SL, Enders GH. Cdk inhibition in human cells compromises chk1 function and activates a DNA damage response. Cancer Res. 2005;65:780–786. [PubMed] [Google Scholar]
- 18.Fritah A, Saucier C, Mester J, Redeuilh G, Sabbah M. p21WAF1/CIP1 selectively controls the transcriptional activity of estrogen receptor alpha. Mol Cell Biol. 2005;25:2419–2430. doi: 10.1128/MCB.25.6.2419-2430.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moro L, Arbini AA, Marra E, Greco M. Upregulation of Skp2 after prostate cancer cell adhesion to basement membranes results in BRCA2 degradation and cell proliferation. J Biol Chem. 2006;281:22100–22107. doi: 10.1074/jbc.M604636200. [DOI] [PubMed] [Google Scholar]
- 20.Long X, Nephew KP. Fulvestrant (ICI 182,780)-dependent interacting proteins mediate immobilization and degradation of estrogen receptor-alpha. J Biol Chem. 2006;281:9607–9615. doi: 10.1074/jbc.M510809200. [DOI] [PubMed] [Google Scholar]
- 21.Senderowicz AM. Small-molecule cyclin-dependent kinase modulators. Oncogene. 2003;22:6609–6620. doi: 10.1038/sj.onc.1206954. [DOI] [PubMed] [Google Scholar]
- 22.Katzenellenbogen BS, Kendra, Norman MJ, Berthois Y. Proliferation, hormonal responsiveness, and estrogen receptor content of MCF-7 human breast cancer cells grown in the short-term and long-term absence of estrogens. Cancer Res. 1987;47:4355–4360. [PubMed] [Google Scholar]