

Abstract
Bioinformatics approaches to examine gene-gene models provide a means to discover interactions between multiple genes that underlie complex disease. Extensive computational demands and adjusting for multiple testing make uncovering genetic interactions a challenge. Here, we address these issues using our knowledge-driven filtering method, Biofilter, to identify putative single nucleotide polymorphism (SNP) interaction models for cataract susceptibility, thereby reducing the number of models for analysis. Models were evaluated in 3,377 European Americans (1,185 controls, 2,192 cases) from the Marshfield Clinic, a study site of the Electronic Medical Records and Genomics (eMERGE) Network, using logistic regression. All statistically significant models from the Marshfield Clinic were then evaluated in an independent dataset of 4,311 individuals (742 controls, 3,569 cases), using independent samples from additional study sites in the eMERGE Network: Mayo Clinic, Group Health/University of Washington, Vanderbilt University Medical Center, and Geisinger Health System. Eighty-three SNP-SNP models replicated in the independent dataset at likelihood ratio test P < 0.05. Among the most significant replicating models was rs12597188 (intron of CDH1)–rs11564445 (intron of CTNNB1). These genes are known to be involved in processes that include: cell-to-cell adhesion signaling, cell-cell junction organization, and cell-cell communication. Further Biofilter analysis of all replicating models revealed a number of common functions among the genes harboring the 83 replicating SNP-SNP models, which included signal transduction and PI3K-Akt signaling pathway. These findings demonstrate the utility of Biofilter as a biology-driven method, applicable for any genome-wide association study dataset.
Keywords: association, genetic interaction, complex disease
Introduction
Robust computational methods to explore gene-gene interactions are essential for elucidating the complex nature of common human traits. Genome-wide association study (GWAS) [Hindorff et al., 2009] has been the traditional paradigm for identifying main effects of genetic variants across the genome for one or more phenotypes and has yielded insufficient explanation about variation of common, complex traits [Eichler et al., 2010; Frazer et al., 2009; Maher, 2008; Manolio et al., 2009; Zuk et al., 2012]. Genetic interaction analysis offers an additional tool for exploring genetic association, and testing models that allow for interactions between genetic variants reflects the complex nature of biology [Cordell, 2009; Eichler et al., 2010; Frazer et al., 2009; Maher, 2008; Zuk et al., 2012]. Gene products do not function in isolation; rather, they physically interact with other proteins, perform regulatory roles, and operate dynamically in one or more pathway.
Bioinformatics methods have expanded in recent years to include searches for genetic interactions, yet many challenges remain in these analyses including extensive computational and time requirements as well as a high penalty for correction of multiple comparisons when exhaustively testing pairwise combinations of all genome-wide SNPs. One method for overcoming this challenge is to filter the number of loci investigated, thus reducing the number of tests. Two main strategies for filtering include: (1) limiting the interaction analyses to only include one or more variants that have demonstrated an association with the trait through GWAS or candidate gene studies; and (2) filtering SNPs based on biologically established gene-gene interactions [Sun et al., 2014].
Knowledge-based filtering decreases the investigation search space to biologically related gene pairs. Previous studies have shown success in applying prior knowledge to genetic interaction analyses [Kim et al., 2014; Ma et al., 2012; Wang et al., 2013]. Biofilter software [Bush et al., 2009; Pendergrass et al., 2013a; Ritchie, 2011] was developed to decrease the search space required for investigating genetic interactions using knowledge across numerous biological databases and has already been adopted for use in studies of complex diseases and traits such as multiple sclerosis [Bush et al., 2011; Ritchie, 2009], HIV pharmacogenetics [Grady et al., 2011], HDL cholesterol [Turner et al., 2014a], and other lipid traits (Holzinger et al., in preparation). Biofilter utilizes biologically validated knowledge of the relationships between sets of genes to build pairwise SNP-SNP models from functionally linked gene-gene pairs. This process takes advantage of biological knowledge of gene-gene relationships rather than requiring loci to have demonstrated a main effect, allowing for the detection of those variants that are only found to be associated with a given phenotype when acting in combination with another locus. In this study, we provided Biofilter with a set of genome-wide SNPs (∼500,000). By accessing biological knowledge available from open-access pathway, ontology, protein interaction, and gene function online databases, Biofilter identified 400 knowledge-driven gene-gene models with approximately 260,000 SNP-SNP models corresponding to the 400 gene pairs. This process reduced the search space from the over 100 billion SNP-SNP models required for an exhaustive pairwise analysis. Filtering based on knowledge decreases the investigation to only gene pairs that have established biological relationships with one another.
We applied this method to age-related cataract, which is the leading cause of blindness worldwide [Black and Wood, 2005] and is responsible for approximately 60% of Medicare costs related to vision [Ellwein and Urato, 2002]. Summary prevalence estimates indicate that 17.2% of Americans that are 40 years and older have cataract in either eye and 5.1% have had pseudophakia/aphakia (previous cataract surgery). Several loci have previously been found to be associated with age-related cataract, and it has been suggested that as many as 40 genes may be involved [Hejtmancik and Kantorow, 2004]. Our recent GWAS of age-related cataract revealed novel loci associated with this trait using electronic medical record (EMR) data [Ritchie et al., 2014]. Despite the identification of numerous associated loci, the molecular mechanisms that lead to age-related cataract remain unclear [Asbell et al., 2005]. However, many genes that have been implicated function together in pathways and interact with one another [Asbell et al., 2005; Bao et al., 2012; Cho et al., 2007; Chong et al., 2009; Martinez and de Iongh, 2010]. Investigation of SNP-SNP interactions for age-related cataract is relevant, given the molecular complexity involved in lens development and maintenance. A recent exploratory gene-gene interaction analysis of age-related cataract implicated genetic interactions in the genetic etiology of the complex trait [Pendergrass et al., 2013b]. However, no replication in a separate dataset was performed in those analyses for validation of results. Here, we present findings of the first genetic interaction study for age-related cataract with replication across two separate studies.
Using PLATO software [Grady et al., 2010], we tested the Biofilter-generated SNP-SNP models for association with age-related cataracts in discovery and replication datasets as part of the NHGRI-funded electronic MEdical Records & GEnomics (eMERGE) Network [Crawford et al., 2014; Gottesman et al., 2013; McCarty et al., 2011]. We identified 83 SNP-SNP models that replicated across the discovery and replication datasets. The results discussed herein demonstrate the utility of Biofilter as a robust method for elucidating the genetic interactions underlying complex traits such as age-related cataract.
Methods
Phenotypic Data
The eMERGE Network implemented an electronic phenotype algorithm to select cataract cases and controls [McCarty et al., 2011]. Age-related cataract as a phenotype was selected by Marshfield Personalized Medicine Research Project (PMRP) as its primary phenotype. The algorithm, which uses diagnostic (ICD-9) and procedure codes (CPT) as well as natural language processing (NLP), was developed by the Marshfield PMRP investigators [Peissig et al., 2012]. The five participating study sites from eMERGE included in this study are Marshfield PMRP [McCarty et al., 2008], Group Health/University of Washington, Vanderbilt University [Roden et al., 2008], Mayo Clinic from eMERGE I, and Geisinger Health System from eMERGE II. In eMERGE I, each of the participating studies applied electronic phenotyping algorithms to identify cases and controls of a specific disease or individuals with a specific phenotype based on their respective EMRs [McCarty et al., 2011]. DNA samples from individuals selected for study were then genotyped for the original phenotype of interest, and these same individuals were available for additional electronic phenotyping with new algorithms including age-related cataract. No additional GWAS-level genotyping was performed in eMERGE II; thus, additional eMERGE study sites joined the network with existing GWAS data available on study participants linked to EMRs which enabled additional electronic phenotyping [Gottesman et al., 2013].
Cataract cases and controls had to meet the following inclusion criteria: cases—aged 50 years and older at the time of diagnosis or surgery, and controls—ages 50 years or older at the time of most recent eye exam and had an eye exam in the previous 5 years. Controls had no diagnostic codes for cataract or evidence of cataract surgery. Cases were identified as “surgical” or “diagnosis-only.” Surgical cases had undergone a cataract extraction in at least one eye. Diagnosis-only cases were required to have either cataract diagnoses on two or more dates, or have one diagnosis date and one or more mention of cataracts identified by NLP of electronic chart notes or paper records converted to electronic using optical character recognition [Peissig et al., 2012; Rasmussen et al., 2012]. Validation of case/control status was conducted at each site through manual abstraction of random samples of patient charts.
All participants were collected at their respective eMERGE site with appropriate patient protections and IRB protocols in place.
Genotypic Data
Genome-wide genotyping was been performed on approximately 18,000 samples across the eMERGE I study sites at the Broad Institute and at the Center for Inherited Disease Research (CIDR) using the Illumina 660W-Quad or 1M-Duo BeadChips. DNA samples from Marshfield Clinic, Group Health/University of Washington, Mayo Clinic, and Vanderbilt University were genotyped using the Illumina 660W-Quad array as previously described [Turner et al., 2011b]. The eMERGE discovery dataset prequality control (QC) included 3,912 (1,356 controls, 2,556 cases) from the Marshfield Clinic. The pre-QC replicating dataset included 2,345 samples (110 controls, 2,193 cases) from Group Health/University of Washington, 952 (346 controls, 606 cases) from Mayo Clinic, and 185 (80 controls, 105 cases) from Vanderbilt University. Added to the replication dataset were samples from eMERGE II by the Geisinger Health System, genotyped on Illumina Human Omni Express (875 pre-QC samples: 221 controls, 654 cases). Due to incomplete overlap of SNPs genotyped on the Illumina 660W Quad platform and the Omni HumanExpress platform, we used imputed data for the Geisinger samples and genotype data for all other sites. In eMERGE, genetic data are imputed to 1,000 genomes reference panel (March 2012 release) [Abecasis et al., 2012]. Imputation for all eMERGE sites was performed on datasets separated by site and platform [Verma, 2014] using IMPUTE2 software [Howie et al., 2009] on the phased genotyped data (SHAPEIT2 was used for phasing) [Delaneau et al., 2013]. For the purpose of this study, we used hard calls derived from imputed data where the genotypes with a probability score >0.9 were reported in PLINK [Purcell et al., 2007] binary files.
Data were cleaned using the eMERGE QC pipeline developed by the eMERGE Genomics Working Group [Zuvich et al., 2011]. This process includes evaluation of sample and marker call rate, sex mismatch, duplicate and HapMap concordance, batch effects, Hardy-Weinberg equilibrium, sample relatedness, and population stratification. For the discovery dataset, QC thresholds included: marker call rate > 99%, sample call rate > 99%, and minor allele frequency (MAF) > 5%. For the replication dataset, QC thresholds included: marker call rate > 98%, sample call rate > 99%, and there was no MAF threshold so as to allow for testing of the highest number of quality variants in the replication analysis. After QC, 3,377 samples and 499,456 SNPs were used for discovery analysis, and 4,311 samples and 1,930 SNPs were included for replication (Table1). The SNPs included for replication analysis were those found in significant models among the discovery dataset only. The 3,377 samples from the Marshfield PMRP included: 3,350 European Americans, one African American, eight Hispanic Americans, and 18 samples of other descent. The 4,211 samples in the replication dataset included: 2,330 samples from Group Health (2,143 European Americans, 81 African Americans, 11 Hispanic Americans, and 95 samples of other descent), 923 samples from Mayo Clinic (894 European Americans, seven African Americans, two Hispanic Americans, and 20 samples of other descent), 183 samples from Vanderbilt University (158 European Americans, 22 African Americans, and three samples of other descent), and 875 samples from Geisinger Health System (866 European Americans, five African Americans, and four Hispanic Americans). All genotype data and a detailed QC report for each individual study site, as well as the merged eMERGE dataset, can be found on dbGaP and the detailed eMERGE QC pipeline can be found in Turner et al. [2011b] and Zuvich et al. [2011].
Table 1.
Study population characteristics
| No. of cases | No. of controls | Total | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| eMERGE study site | Male | Female | All | Male | Female | All | Male | Female | All | |
| Discovery | Marshfield | 934 | 1,258 | 2,192 | 474 | 711 | 1,185 | 1,408 | 1,969 | 3,377 |
| Replication | Mayo, Group Health/University of Washington, Vanderbilt, Geisinger | 1,726 | 1,843 | 3,569 | 400 | 342 | 742 | 2,126 | 2,185 | 4,311 |
| Total | All | 2,600 | 3,101 | 5,761 | 874 | 1,053 | 1,927 | 3,534 | 4,154 | 7,688 |
Sample sizes are given for cataract cases, controls, and total population for the discovery and replication datasets. Sample information for discovery and replication samples after quality control.
Biofilter
Biofilter [Bush et al., 2009; Pendergrass et al., 2013a] was developed for high-throughput annotation, model building, and filtering of genetic data through automated access to multiple biological databases. Biofilter software is open source and freely available for noncommercial research institutions. For more information, see: http://ritchielab.psu.edu/ritchielab/software/.
Biofilter accesses several publicly available biological knowledge databases through the external database compiler called the Library of Knowledge Integration (LOKI) [Pendergrass et al., 2013a]. Data sources utilized by Biofilter, and compiled through LOKI, include information about biological networks, connections, and/or pathways for determining relationships between genes. Sources compiled within LOKI include: the Kyoto Encyclopedia of Genes and Genomes (KEGG) [Ogata et al., 1999], Reactome [Matthews et al., 2009], Gene Ontology (GO) [Ashburner et al., 2000], protein families database [Punta et al., 2012], NetPath [Kandasamy et al., 2010], Biological General Repository for Interaction Databases (BioGrid) [Stark et al., 2011], and the Molecular INTeraction Database (MINT) [Licata et al., 2012]. Additionally, Biofilter maps SNPs to genes using knowledge from the National Center for Biotechnology (NCBI) dbSNP [Sherry et al., 2001] database.
Building SNP-SNP models with Biofilter for our analyses involved several steps. First, QC-filtered SNPs were mapped to genes using Biofilter with a 50 kb window upstream and downstream of each gene to encompass potential regulatory regions close to the genes. Gene-gene pairs were established by Biofilter using knowledge from the databases within LOKI (Fig. 1A and B). It was required that a link between a gene-gene pair be validated in five or more separate databases (implication index ≥ 5) within LOKI for a gene-gene model to be considered for analysis. All gene-gene models were generated because of their connections to one another, independent of phenotype. Once gene-gene models were verified by five or more databases, pairwise SNP-SNP combinations of all loci within each gene-gene model were created (Fig. 1C) and output for regression using PLATO software [Grady et al., 2010]. Because we allowed for a 50 kb gene boundary, it was possible for a given SNP to map to more than one gene, and thus, to be paired with SNPs within the same gene. Therefore, models containing two SNPs within the same gene were dropped from our results.
Figure 1.
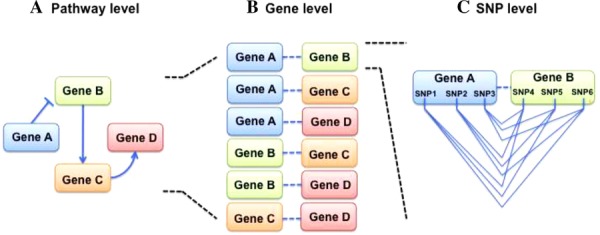
Steps involved in generating Biofilter SNP-SNP models. (A) Biofilter accessed LOKI-compiled databases with information about connections between genes (for the example shown here: connections within a pathway). (B) Biofilter-generated gene-gene models based on connections between genes that were validated by five or more databases. (C) For each gene-gene model, pairwise SNP-SNP models were created for each unique combination of loci across a gene pair.
Statistical Analyses
Pairwise SNP-SNP model tests of association were performed using logistic regression with PLATO assuming an additive genetic model for 259,845 Biofilter-generated SNP-SNP models in the Marshfield Clinic discovery dataset. To determine the significance of the interaction term, we performed a likelihood ratio test (LRT) between the full (Y = β0 + β1SNP1 + β2SNP2 + β3SNP1×SNP2) and reduced (Y = β0 + β1SNP1 + β2SNP2) models. We calculated principal components (PCs) using program Eigenstrat [Price et al., 2006] to identify any potential population substructure and adjusted our analyses for the first three PCs, sex, and year of birth.
The independent replication dataset included samples from Group Health/University of Washington, Vanderbilt University, Mayo Clinic, and Geisinger Health System. We targeted an initial set of 2,452 SNP-SNP models that passed an LRT P-value threshold of P < 0.01 in the Marshfield discovery dataset for replication. There were 2,149 unique SNPs in the 2,452 discovery-significant SNP-SNP models. After applying a QC filter (marker call rate: 98%) in the replication set, 1,930 SNPs remained, and thus, 2,092 SNP-SNP models (of the 2,452 discovery-significant models) were available for testing in the replication dataset. All methods and adjustments used for the discovery dataset were applied for the replication analyses in addition to adjusting for study site. The pipeline used for the discovery and replication analysis is shown in Figure 2. Permutation tests were performed for all 2,092 SNP-SNP models in the replication dataset. For permutation, the phenotype was randomly shuffled 1,000 times and an LRT P-value was calculated for each model per 1,000 permutations. The permuted P-value for each SNP-SNP model was determined as the fraction of times any permuted P-value had a lower P-value than the LRT P-value derived from the natural phenotype.
Figure 2.
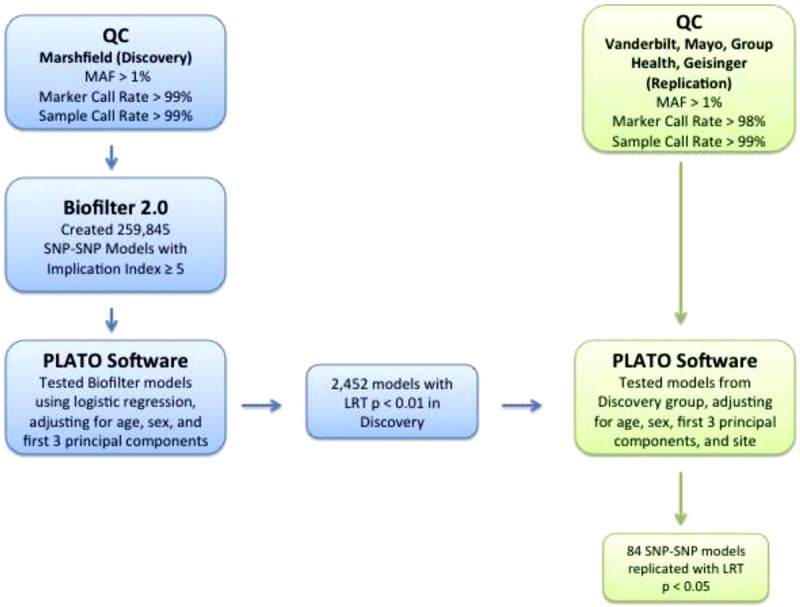
Flow chart of steps in the discovery and replication analyses.
Results
In the discovery dataset, 2,452 SNP-SNP models were significant with an LRT P-value < 0.01, and these were tested in the independent replication dataset. Of these, 83 models were significant in the replication dataset with an LRT P-value < 0.05 (supplementary Table S1). Additionally, for all of the 83 models, the permuted P-value was ≤ 0.05. There were 22 SNP-SNP models that replicated with an LRT P-value < 0.01 (Fig. 3).
Figure 3.
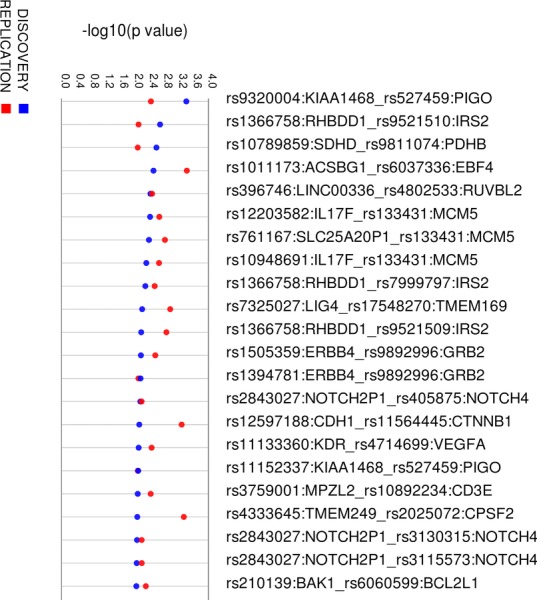
All replicating SNP-SNP models with LRT P < 0.01 in both the replication and discovery datasets. SNP-SNP models are shown above with the –log10 of the P-value in the track directly beneath (discovery values are in blue and replication values are in red). Visualization was performed using Synthesis View software [Pendergrass et al., 2010].
Thirteen replicating models were significant with an LRT P-value < 0.001 in the discovery sample and three models with LRT P < 0.001 in the replication sample. Figure 4 shows the replicating SNP-SNP models with the 10 lowest LRT P-values for the discovery (Fig. 4A) and replication (Fig. 4B) datasets.
Figure 4.

Ten most significant replicating SNP-SNP models, ranked by significance level in the discovery (A) and replication (B) samples. For both figures, the SNP-SNP models and their nearest genes are listed to the left. The track to the right of each displays the –log10 of the P-value for the discovery (blue) and replication (red) groups. Figures were made using Synthesis View.
The SNPs within the model with the lowest LRT P-value in the discovery group were rs2303436 (a missense SNP in DLAT) and rs9811074 (near PDHB; discovery LRT P = 2.9 × 10−4, replication LRT P = 0.013; Fig. 4A). Other significant models in the discovery group included intronic SNP rs9320004 (KIAA1468) and rs527459 (542 bp 3′ of PIGO) as well as rs10789856 (intron of DIXDC1) and rs9811074 (near PDHB).
The replicating SNP-SNP model with the lowest LRT P-value in the replication sample was rs1011173 (intron of ACSBG1) and rs6037336 (near EBF4; discovery LRT P = 0.0031, replication LRT P = 3.9 × 10−4; Fig. 4B). Other top SNP-SNP models were rs4333645 (near TMEM249) and rs2025072 (intron of CPSF2) as well as rs12597188 and rs11564445 in CDH1 and CTNNB1, respectively.
In order to identify common function across genes in all replicating models, SNPs in every replicating SNP-SNP model were mapped to their closest gene. Ninety unique genes were identified as harboring SNPs in the 83 SNP-SNP models. These 90 genes were subsequently annotated with all group information (such as pathway) using Biofilter. Groups linked to the largest number of genes included: signal transduction (8 genes), adaptive immune system (12 genes), pathways in cancer (12 genes), innate immune response (11 genes), apoptosis (10 genes), DNA replication (10 genes), extracellular vesicular exosome (9 genes), microRNAs in cancer (9 genes), positive regulation of cell proliferation (9 genes), proteoglycans in cancer (9 genes), PI3K-Akt signaling pathway (8 genes), EGFR signaling pathway (8 genes), focal adhesion (8 genes). Figure 5 shows two examples of these common groups and the genes with which they were annotated.
Figure 5.

Common groups relating to genes in replicating SNP-SNP models. Figures display two of the most common groups (yellow) and the genes that Biofilter annotated with that group (blue): (A) signal transduction and (B) PI3K-Akt signaling pathway. Solid lines indicate group-gene connection, dotted line indicates gene-gene connection from the interaction analysis. Plots were generated using Cytoscape software [Saito et al., 2012].
Finally, we used the Tissue-specific Gene Expression and Regulation (TiGER) database [Liu et al., 2008; Yu et al., 2006] to determine how many of the genes in our models are expressed in the eye. Though cataracts develop in the lens specifically, no comprehensive analysis of gene expression in the lens has been published, to the authors’ knowledge, so we focused on gene expression in the eye. We found that, of the identified 90 genes, 61 (∼68%) are expressed in the human eye. This is a far greater percentage than the fraction of all genes that are expressed in the eye (289) out of all the genes that were analyzed for the compilation of the database (∼20,000), which is ∼1%. Thus, we see a greater proportion of genes expressed in the eye represented in our final set of genes.
Discussion
In this first replication study of gene-gene interactions associated with age-related cataract, we found 83 SNP-SNP models that replicated across two independent datasets with an LRT P-value less than 0.01 in the discovery sample and 0.05 in the replication sample. Many of the replicating SNP-SNP models were in or near genes expressed in the eye and/or relating to lens development and maintenance as well as the development of cataracts, as further described below.
The anterior surface of the lens is made up of a single layer of epithelial cells that divide and differentiate throughout life into fiber cells, which make up the largest part of the lens [Goodenough, 1992]. As differentiation occurs, fiber cells experience unique loss of the nucleus and other organelles in addition to high expression of crystallin proteins, both of which are essential for transparency of the lens as well as a high refractory index [Benedek, 1971]. Because the lens is an avascular tissue and fiber cells lack organelles, cell-to-cell junctions, both among-fiber cells as well as between-fiber cells and lens epithelial cells, are crucial for cell maintenance and survival including nutrient delivery and metabolic waste removal [Donaldson et al., 2001]. Both gap junctions and adherens junctions are present in lens cells [Cooper et al., 2008]. Studies have shown that mutations in genes encoding gap junction connexin (Cx) proteins have led to cataracts in mice [Gong et al., 1997; White et al., 1998] and are associated with cataract development in humans [Wei et al., 2004; White and Paul, 1999]. Adherens junctions and their components, classical cadherins and interacting β-catenin, play a crucial part in lens development and maintenance as well [Cooper et al., 2008; Martinez and de Iongh, 2010; Pontoriero et al., 2009].
Among the replicating model with the lowest LRT P-value in the replication dataset were two intronic SNPs, rs12597188 and rs11564445 which are in cadherin 1, type 1, E-cadherin (CDH1), and catenin (cadherin-associated protein) beta 1 (CTNNB1), respectively (Fig. 3B). CDH1 encodes E-cadherin, a calcium-dependent glycoprotein that maintains epithelial cell-cell adhesion at adherens junctions [Perez-Moreno et al., 2003], and CTNNB1 encodes β-catenin, which acts as an anchor protein for E-cadherin so as to maintain a connection to intracellular actin. In addition to its role in adherens junction formation, β-catenin also has known signaling functions [Martinez and de Iongh, 2010]. β-catenin has been shown to translocate to the nucleus and activate transcription in complex with lymphoid enhancer-binding/T-cell factor in response to Wnt signaling [Nusse, 2005]. The Wnt/β-catenin pathway is known for regulating cell proliferation, differentiation, as well as migration [Logan and Nusse, 2004]. Normal Wnt/β-catenin signaling is thought to be essential in the formation and maintenance of the lens epithelium [Martinez and de Iongh, 2010]. The pathway’s response to transforming growth factor beta (TGFβ) induction has been implicated in epithelial-mesenchymal transition (EMT) [Bao et al., 2012; Guarino et al., 2009], an event that has been shown to lead to posterior capsular opacification, also known as secondary cataracts, in humans [Apple et al., 1992; Awasthi et al., 2009]. This process includes loss of cell polarity and cell-cell adhesion, which involves downregulation of E-cadherin, transcriptional reprograming, and migration.
Another pathway involved in induction of EMT by TGFβ is the phosphatidylinositol-3-kinase (PI3K)/Akt pathway, which has demonstrated importance in downregulation of connexin-43 [Yao et al., 2008]. We found eight genes that harbor the replicating SNP-SNP models that were annotated with the PI3K/Akt pathway group (Fig. 5B). These results reinforce previous findings on the importance of typical function of E-cadherin, β-catenin, and the PI3K/Akt pathway in lens maintenance.
Additional growth factors are crucial for lens development and maintenance. The aqueous humor provides lens cells with growth factors including FGF, IGF, PDGF, and epidermal growth factor (EGF), and these are important for lens structure and polarity [Martinez and de Iongh, 2010]. Further, it is thought that these factors regulate cell proliferation via the MAPK/Erk and PI3K/Akt pathways. “Signal Transduction” was among the two most common groups, with 15 genes relating to it (Fig. 5A). Signal transduction can be considered a somewhat generic group into which a large number of proteins fall. Nonetheless, the genes found to relate to this group in our study are involved in specific transduction events known to be related to cataract. Gene-gene models found to relate to signal transduction here were NOTCH1, which has demonstrated involvement in lens development [Rowan et al., 2008] and NOTCH4 as well as EGF and EGFR. The intronic SNPs of EGF and EGFR, rs3796947 and rs6954351, respectively, were among the five most significant replicating models in the discovery dataset. EGF encodes a mitogenic factor that acts by binding to the EGFR, encoded by EGFR. EGF and EGFR are part of both the MAPK/Erk and PI3K/Akt signaling pathways. Both factors are important for epithelial cell proliferation, and previous findings have demonstrated that EGFR RNAi treatment suppresses proliferation of lens epithelial cells following cataract surgery in rats [Huang et al., 2011].
Some limitations to our method may have decreased our ability to identify additional genetic interactions predictive of age-related cataract. The current application of Biofilter focuses on building models from protein-coding gene regions. Future additions to the software, including incorporation of regulatory regions, will allow identification of loci that fall outside of the 50 kb gene window that may still be involved in the expression of a trait. The challenge of genetic heterogeneity has yet to be addressed with this method as well. If there are multiple disease loci spread across subsets of cases, we would have had little power to detect them. Methods for binning variants in genes and/or pathways may increase our ability to identify more genetic interactions. Additionally, the current approach considered genetic variation without allowing for interactions with the environment. Incorporating exposure data with this approach will further elucidate the complex underpinnings of age-related cataract.
The results described in this study are consistent with previous findings relating to lens cell maintenance and structure as well as cataract development. Use of Biofilter decreased the search space to identify and replicate putative SNP-SNP combinations. These results demonstrate the role of genetic interactions in the development of complex phenotypes like age-related cataract. Other genetic epidemiology studies would benefit from the annotation, filtering, and model-building functions of Biofilter.
Acknowledgments
This study was supported by the following U01 grants from the National Human Genome Research Institute (NHGRI), a component of the National Institutes of Health (NIH), Bethesda, MD, USA: U01HG004608 (Marshfield Clinic); U01HG006389 (Essentia Institute of Rural Health & Marshfield Clinic Research Foundation); U01HG004610 and U01HG006375 (Group Health/University of Washington); U01HG04599 and U01HG006379 (Mayo Clinic); U01HG04603 and U01HG006378 (Vanderbilt University); U01HG006385 (Vanderbilt University); U01HG004438 (CIDR, Johns Hopkins University); U01HG004424 (The Broad Institute); and U01HG006382 (Geisinger Health System). Additional support was provided by UO1AG06781, HG006828 and F31 HG008588. We would like to thank Dr. Dokyoon Kim for his assistance in generating Figure 1.
Supporting Information
Appendix
References
- Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apple DJ, Solomon KD, Tetz MR, Assia EI, Holland EY, Legler UF, Tsai JC, Castaneda VE, Hoggatt JP, Kostick AM. Posterior capsule opacification. Surv Ophthalmol. 1992;37(2):73–116. doi: 10.1016/0039-6257(92)90073-3. [DOI] [PubMed] [Google Scholar]
- Asbell PA, Dualan I, Mindel J, Brocks D, Ahmad M, Epstein S. Age-related cataract. Lancet. 2005;365(9459):599–609. doi: 10.1016/S0140-6736(05)17911-2. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi N, Guo S, Wagner BJ. Posterior capsular opacification: a problem reduced but not yet eradicated. Arch Ophthalmol. 2009;127(4):555–562. doi: 10.1001/archophthalmol.2009.3. [DOI] [PubMed] [Google Scholar]
- Bao XL, Song H, Chen Z, Tang X. Wnt3a promotes epithelial-mesenchymal transition, migration, and proliferation of lens epithelial cells. Mol Vis. 2012;18:1983–1990. [PMC free article] [PubMed] [Google Scholar]
- Benedek GB. Theory of transparency of the eye. Appl Opt. 1971;10(3):459–473. doi: 10.1364/AO.10.000459. [DOI] [PubMed] [Google Scholar]
- Black A, Wood J. Vision and falls. Clin Exp Optom. 2005;88(4):212–222. doi: 10.1111/j.1444-0938.2005.tb06699.x. [DOI] [PubMed] [Google Scholar]
- Bush WS, Dudek SM, Ritchie MD. Biofilter: a knowledge-integration system for the multi-locus analysis of genome-wide association studies. Pac Symp Biocomput. 2009:368–379. [PMC free article] [PubMed] [Google Scholar]
- Bush WS, McCauley JL, DeJager PL, Dudek SM, Hafler DA, Gibson RA, Matthews PM, Kappos L, Naegelin Y, Polman CH. A knowledge-driven interaction analysis reveals potential neurodegenerative mechanism of multiple sclerosis susceptibility. Genes Immun. 2011;12(5):335–340. doi: 10.1038/gene.2011.3. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HJ, Baek KE, Saika S, Jeong MJ, Yoo J. Snail is required for transforming growth factor-beta-induced epithelial-mesenchymal transition by activating PI3 kinase/Akt signal pathway. Biochem Biophys Res Commun. 2007;353(2):337–343. doi: 10.1016/j.bbrc.2006.12.035. [DOI] [PubMed] [Google Scholar]
- Chong CC, Stump RJ, Lovicu FJ, McAvoy JW. TGFbeta promotes Wnt expression during cataract development. Exp Eye Res. 2009;88(2):307–313. doi: 10.1016/j.exer.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MA, Son AI, Komlos D, Sun Y, Kleiman NJ, Zhou R. Loss of ephrin-A5 function disrupts lens fiber cell packing and leads to cataract. Proc Natl Acad Sci USA. 2008;105(43):16620–16625. doi: 10.1073/pnas.0808987105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell HJ. Detecting gene-gene interactions that underlie human diseases. Nat Rev Genet. 2009;10(6):392–404. doi: 10.1038/nrg2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DC, Crosslin DR, Tromp G, Kullo IJ, Kuivaniemi H, Hayes MG, Denny JC, Bush WS, Haines JL, Roden DM. eMERGEing progress in genomics-the first seven years. Front Genet. 2014;5:184. doi: 10.3389/fgene.2014.00184. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods. 2013;10(1):5–6. doi: 10.1038/nmeth.2307. [DOI] [PubMed] [Google Scholar]
- Donaldson P, Kistler J, Mathias RT. Molecular solutions to mammalian lens transparency. News Physiol Sci. 2001;16:118–123. doi: 10.1152/physiologyonline.2001.16.3.118. [DOI] [PubMed] [Google Scholar]
- Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11(6):446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellwein LB, Urato CJ. Use of eye care and associated charges among the Medicare population: 1991–1998. Arch Ophthalmol. 2002;120(6):804–811. doi: 10.1001/archopht.120.6.804. [DOI] [PubMed] [Google Scholar]
- Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nat Rev Genet. 2009;10(4):241–251. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- Goodenough DA. The crystalline lens. A system networked by gap junctional intercellular communication. Semin Cell Biol. 1992;3(1):49–58. doi: 10.1016/s1043-4682(10)80007-8. [DOI] [PubMed] [Google Scholar]
- Gong X, Li E, Klier G, Huang Q, Wu Y, Lei H, Kumar NM, Horwitz J, Gilula NB. Disruption of alpha3 connexin gene leads to proteolysis and cataractogenesis in mice. Cell. 1997;91(6):833–843. doi: 10.1016/s0092-8674(00)80471-7. [DOI] [PubMed] [Google Scholar]
- Gottesman O, Kuivaniemi H, Tromp G, Faucett WA, Li R, Manolio TA, Sanderson SC, Kannry J, Zinberg R, Basford MA. The Electronic Medical Records and Genomics (eMERGE) Network: past, present, and future. Genet Med. 2013;15(10):761–771. doi: 10.1038/gim.2013.72. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady BJ, Torstenson E, Dudek SM, Giles J, Sexton D, Ritchie MD. Finding unique filter sets in PLATO: a precursor to efficient interaction analysis in GWAS data. Pac Symp Biocomput. 2010:315–326. [PMC free article] [PubMed] [Google Scholar]
- Grady BJ, Torstenson ES, McLaren PJ, PI DEB, Haas DW, Robbins GK, Gulick RM, Haubrich R, Ribaudo H, Ritchie MD. Use of biological knowledge to inform the analysis of gene-gene interactions involved in modulating virologic failure with efavirenz-containing treatment regimens in ART-naive ACTG clinical trials participants. Pac Symp Biocomput. 2011:253–264. doi: 10.1142/9789814335058_0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarino M, Tosoni A, Nebuloni M. Direct contribution of epithelium to organ fibrosis: epithelial-mesenchymal transition. Hum Pathol. 2009;40(10):1365–1376. doi: 10.1016/j.humpath.2009.02.020. [DOI] [PubMed] [Google Scholar]
- Hejtmancik JF, Kantorow M. Molecular genetics of age-related cataract. Exp Eye Res. 2004;79(1):3–9. doi: 10.1016/j.exer.2004.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106(23):9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5(6):e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WR, Fan XX, Tang X. SiRNA targeting EGFR effectively prevents posterior capsular opacification after cataract surgery. Mol Vis. 2011;17:2349–2355. [PMC free article] [PubMed] [Google Scholar]
- Kandasamy K, Mohan SS, Raju R, Keerthikumar S, Kumar GS, Venugopal AK, Telikicherla D, Navarro JD, Mathivanan S, Pecquet C. NetPath: a public resource of curated signal transduction pathways. Genome Biol. 2010;11(1):R3. doi: 10.1186/gb-2010-11-1-r3. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Li R, Dudek SM, Frase AT, Pendergrass SA, Ritchie MD. Knowledge-driven genomic interactions: an application in ovarian cancer. BioData Min. 2014;7:20. doi: 10.1186/1756-0381-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata L, Briganti L, Peluso D, Perfetto L, Iannuccelli M, Galeota E, Sacco F, Palma A, Nardozza AP, Santonico E. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012;40(Database issue):D857–D861. doi: 10.1093/nar/gkr930. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Yu X, Zack DJ, Zhu H, Qian J. TiGER: a database for tissue-specific gene expression and regulation. BMC Bioinform. 2008;9:271. doi: 10.1186/1471-2105-9-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- Ma L, Brautbar A, Boerwinkle E, Sing CF, Clark AG, Keinan A. Knowledge-driven analysis identifies a gene-gene interaction affecting high-density lipoprotein cholesterol levels in multi-ethnic populations. PLoS Genet. 2012;8(5):e1002714. doi: 10.1371/journal.pgen.1002714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher B. Personal genomes: the case of the missing heritability. Nature. 2008;456(7218):18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez G, de Iongh RU. The lens epithelium in ocular health and disease. Int J Biochem Cell Biol. 2010;42(12):1945–1963. doi: 10.1016/j.biocel.2010.09.012. [DOI] [PubMed] [Google Scholar]
- Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, de Bono B, Garapati P, Hemish J, Hermjakob H, Jassal B. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 2009;37(Database issue):D619–D622. doi: 10.1093/nar/gkn863. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty CA, Chapman-Stone D, Derfus T, Giampietro PF, Fost N. Community consultation and communication for a population-based DNA biobank: the Marshfield clinic personalized medicine research project. Am J Med Genet A. 2008;146A(23):3026–3033. doi: 10.1002/ajmg.a.32559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty CA, Chisholm RL, Chute CG, Kullo IJ, Jarvik GP, Larson EB, Li R, Masys DR, Ritchie MD, Roden DM. The eMERGE Network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med Genomics. 2011;4:13. doi: 10.1186/1755-8794-4-13. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R. Wnt signaling in disease and in development. Cell Res. 2005;15(1):28–32. doi: 10.1038/sj.cr.7290260. [DOI] [PubMed] [Google Scholar]
- Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27(1):29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peissig PL, Rasmussen LV, Berg RL, Linneman JG, McCarty CA, Waudby C, Chen L, Denny JC, Wilke RA, Pathak J. Importance of multi-modal approaches to effectively identify cataract cases from electronic health records. J Am Med Inform Assoc. 2012;19(2):225–234. doi: 10.1136/amiajnl-2011-000456. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendergrass SA, Dudek SM, Crawford DC, Ritchie MD. Synthesis-view: visualization and interpretation of SNP association results for multi-cohort, multi-phenotype data and meta-analysis. BioData Min. 2010;3:10. doi: 10.1186/1756-0381-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendergrass SA, Frase A, Wallace J, Wolfe D, Katiyar N, Moore C, Ritchie MD. Genomic analyses with biofilter 2.0: knowledge driven filtering, annotation, and model development. BioData Min. 2013a;6(1):25. doi: 10.1186/1756-0381-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendergrass SA, Verma SS, Holzinger ER, Moore CB, Wallace J, Dudek SM, Huggins W, Kitchner T, Waudby C, Berg R. Next-generation analysis of cataracts: determining knowledge driven gene-gene interactions using Biofilter, and gene-environment interactions using the PhenX Toolkit. Pac Symp Biocomput. 2013b:147–158. others. [PMC free article] [PubMed] [Google Scholar]
- Perez-Moreno M, Jamora C, Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. Cell. 2003;112(4):535–548. doi: 10.1016/s0092-8674(03)00108-9. [DOI] [PubMed] [Google Scholar]
- Pontoriero GF, Smith AN, Miller LA, Radice GL, West-Mays JA, Lang RA. Co-operative roles for E-cadherin and N-cadherin during lens vesicle separation and lens epithelial cell survival. Dev Biol. 2009;326(2):403–417. doi: 10.1016/j.ydbio.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J. The Pfam protein families database. Nucleic Acids Res. 2012;40(Database issue):D290–D301. doi: 10.1093/nar/gkr1065. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen LV, Peissig PL, McCarty CA, Starren J. Development of an optical character recognition pipeline for handwritten form fields from an electronic health record. J Am Med Inform Assoc. 2012;19(e1):e90–e95. doi: 10.1136/amiajnl-2011-000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie MD. Using prior knowledge and genome-wide association to identify pathways involved in multiple sclerosis. Genome Med. 2009;1(6):65. doi: 10.1186/gm65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie MD. Using biological knowledge to uncover the mystery in the search for epistasis in genome-wide association studies. Ann Hum Genet. 2011;75(1):172–182. doi: 10.1111/j.1469-1809.2010.00630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie MD, Verma SS, Hall MA, Goodloe RJ, Berg RL, Carrell DS, Carlson CS, Chen L, Crosslin DR, Denny JC. Electronic medical records and genomics (eMERGE) network exploration in cataract: several new potential susceptibility loci. Mol Vis. 2014;20:1281–1295. others. [PMC free article] [PubMed] [Google Scholar]
- Roden DM, Pulley JM, Basford MA, Bernard GR, Clayton EW, Balser JR, Masys DR. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther. 2008;84(3):362–369. doi: 10.1038/clpt.2008.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan S, Conley KW, Le TT, Donner AL, Maas RL, Brown NL. Notch signaling regulates growth and differentiation in the mammalian lens. Dev Biol. 2008;321(1):111–122. doi: 10.1016/j.ydbio.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito R, Smoot ME, Ono K, Ruscheinski J, Wang PL, Lotia S, Pico AR, Bader GD, Ideker T. A travel guide to Cytoscape plugins. Nat Methods. 2012;9(11):1069–1076. doi: 10.1038/nmeth.2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Breitkreutz BJ, Chatr-Aryamontri A, Boucher L, Oughtred R, Livstone MS, Nixon J, Van Auken K, Wang X, Shi X. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011;39(Database issue):D698–D704. doi: 10.1093/nar/gkq1116. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Lu Q, Mukheerjee S, Crane PK, Elston R, Ritchie MD. Analysis pipeline for the epistasis search—statistical versus biological filtering. Front Genet. 2014;5:106. doi: 10.3389/fgene.2014.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner SD, Berg RL, Linneman JG, Peissig PL, Crawford DC, Denny JC, Roden DM, McCarty CA, Ritchie MD, Wilke RA. Knowledge-driven multi-locus analysis reveals gene-gene interactions influencing HDL cholesterol level in two independent EMR-linked biobanks. PLoS ONE. 2014a;6(5):e19586. doi: 10.1371/journal.pone.0019586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner S, Armstrong LL, Bradford Y, Carlson CS, Crawford DC, Crenshaw AT, de Andrade M, Doheny KF, Haines JL, Hayes G. Quality control procedures for genome-wide association studies. Curr Protoc Hum Genet Chapter. 2011b;1(Unit1)):19. doi: 10.1002/0471142905.hg0119s68. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma SSdAM, Tromp G, Kuivaniemi H, Pugh E, Namjou B, Mukherjee S, Jarvik GP, Kottyan LC, Burt A. Imputation and quality control steps for combining multiple genome-wide datasets. Front Genet. 2014;5(370) doi: 10.3389/fgene.2014.00370. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Xu W, San Lucas FA, Liu Y. Incorporating prior knowledge into Gene Network Study. Bioinformatics. 2013;29(20):2633–2640. doi: 10.1093/bioinformatics/btt443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CJ, Xu X, Lo CW. Connexins and cell signaling in development and disease. Annu Rev Cell Dev Biol. 2004;20:811–838. doi: 10.1146/annurev.cellbio.19.111301.144309. [DOI] [PubMed] [Google Scholar]
- White TW, Paul DL. Genetic diseases and gene knockouts reveal diverse connexin functions. Annu Rev Physiol. 1999;61:283–310. doi: 10.1146/annurev.physiol.61.1.283. [DOI] [PubMed] [Google Scholar]
- White TW, Goodenough DA, Paul DL. Targeted ablation of connexin50 in mice results in microphthalmia and zonular pulverulent cataracts. J Cell Biol. 1998;143(3):815–825. doi: 10.1083/jcb.143.3.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao K, Ye PP, Tan J, Tang XJ, Shen Tu XC. Involvement of PI3K/Akt pathway in TGF-beta2-mediated epithelial mesenchymal transition in human lens epithelial cells. Ophthalmic Res. 2008;40(2):69–76. doi: 10.1159/000113884. [DOI] [PubMed] [Google Scholar]
- Yu X, Lin J, Zack DJ, Qian J. Computational analysis of tissue-specific combinatorial gene regulation: predicting interaction between transcription factors in human tissues. Nucleic Acids Res. 2006;34(17):4925–4936. doi: 10.1093/nar/gkl595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: genetic interactions create phantom heritability. Proc Natl Acad Sci USA. 2012;109(4):1193–1198. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuvich RL, Armstrong LL, Bielinski SJ, Bradford Y, Carlson CS, Crawford DC, Crenshaw AT, de Andrade M, Doheny KF, Haines JL. Pitfalls of merging GWAS data: lessons learned in the eMERGE network and quality control procedures to maintain high data quality. Genet Epidemiol. 2011;35(8):887–898. doi: 10.1002/gepi.20639. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix