

Abstract
X-linked adrenoleukodystrophy is a neurometabolic disorder caused by inactivation of the peroxisomal ABCD1 transporter of very long-chain fatty acids. In mice, ABCD1 loss causes late onset axonal degeneration in the spinal cord in association with locomotor disability resembling the most common phenotype in patients, adrenomyeloneuropathy. Increasing evidence indicates that oxidative stress and bioenergetic failure play major roles in the pathogenesis of X-linked adrenoleukodystrophy. In this study, we aimed to evaluate whether mitochondrial biogenesis is affected in X-linked adrenoleukodystrophy. We demonstrated that Abcd1 null mice show reduced mitochondrial DNA concomitant with downregulation of mitochondrial biogenesis pathway driven by PGC-1α/PPARγ and reduced expression of mitochondrial proteins cytochrome c, NDUFB8 and VDAC. Moreover, we show that the oral administration of pioglitazone, an agonist of PPARγ, restored mitochondrial content and expression of master regulators of biogenesis, neutralized oxidative damage to proteins and DNA, and reversed bioenergetic failure in terms of ATP levels, NAD+/NADH ratios, pyruvate kinase and glutathione reductase activities. Most importantly, the treatment halted locomotor disability and axonal damage in X-linked adrenoleukodystrophy mice. These results lend support to the use of pioglitazone in clinical trials with patients with adrenomyeloneuropathy and reveal novel molecular mechanisms of action of pioglitazone in neurodegeneration. Future studies should address the effects of this anti-diabetic drug on other axonopathies in which oxidative stress and mitochondrial dysfunction are contributing factors.
Keywords: mitochondrial biogenesis, oxidative stress, axonal degeneration, pioglitazone, X-linked adrenoleukodystrophy
Introduction
Impaired bioenergetics in combination with oxidative stress commonly underlies age-related neurodegenerative diseases such as Parkinson’s disease, Huntington’s disease, Alzheimer’s disease and amyotrophic lateral sclerosis (Lin and Beal, 2006; Martinez et al., 2010). A common theme among these diseases, as well as in the prototypic demyelinating disease multiple sclerosis, is axonal degeneration (Mandelkow and Mandelkow, 1998; Braak et al., 1999; Li et al., 2001; McSharry, 2010). In addition, it has often been noted that mitochondrial dysfunction promotes axonal loss or damage in multiple sclerosis (Lassmann, 2011) and amyotrophic lateral sclerosis (Shi et al., 2010). Axons are highly vulnerable because their high metabolic demands and unusual length render them susceptible to oxidative damage, ischaemia and metabolic defects.
X-linked adrenoleukodystrophy (McKusick no. 300100) is a rare, fatal disease characterized by central inflammatory demyelination in the brain or slowly progressive spastic paraparesis resulting from axonal degeneration in the spinal cord (Powers et al., 2000; Moser et al., 2001; Ferrer et al., 2010). X-linked adrenoleukodystrophy is the most frequently inherited leukodystrophy, with a minimal incidence of 1 in 17 000 males. All patients with X-linked adrenoleukodystrophy have mutations in the ABCD1 gene that encodes the ATP-binding cassette transporter, which is an integral peroxisomal membrane protein involved in the importation of very long-chain fatty acids (C≥22:0) and very long-chain fatty acids-CoA esters into the peroxisome for degradation (Hettema et al., 1996; van Roermund et al., 2008). The defective function of the ABCD1 transporter leads to accumulation of very long-chain fatty acids due to impaired import into the peroxisome and subsequent lack of substrate for the peroxisomal beta-oxidation enzymes (Singh et al., 1984; Wanders et al., 1987; Fourcade et al., 2009) in most patients’ organs, fibroblasts and plasma, particularly affecting hexacosanoic acid or C26:0, which serves as a pathognomonic biomarker for the biochemical diagnosis of the disease.
Inactivation of the ABCD1 transporter in the mouse leads to late-onset neurodegeneration with axonopathy in the spinal cord but without inflammatory demyelination in the brain; this condition resembles the most frequent X-linked adrenoleukodystrophy phenotype, adult adrenomyeloneuropathy (Pujol et al., 2002, 2004). The mouse model of X-linked adrenoleukodystrophy presents the first signs of lipoxidative modifications to proteins as early as 3.5 months of age, which is >1 year before the neuropathological and neurological signs of the disease appear (at 15 and 20 months of age, respectively) (Fourcade et al., 2008). Signs of oxidative damage have also been found in post-mortem brain and blood samples from patients with X-linked adrenoleukodystrophy (Vargas et al., 2004; Powers et al., 2005; Fourcade et al., 2010). We recently showed that early oxidative damage and bioenergetic dysfunction with decreased ATP and NADH levels underlie the late onset degeneration of axons observed in the mouse model of the disease (Fourcade et al., 2008; Singh and Pujol, 2010; Galino et al., 2011; Lopez-Erauskin et al., 2011; Galea et al., 2012; Lopez-Erauskin et al., 2012).
We report here that (i) X-linked adrenoleukodystrophy mice exhibit mitochondrial depletion at 12 months of age, concomitant with a downregulation of the peroxisome proliferator activated receptor-γ (PPARγ) coactivator 1-alpha (PGC-1α) pathway leading to a mitochondrial biogenesis defect; and (ii) that treatment with pioglitazone, an activator of PPARγ/PGC-1α-dependent pathways (Hondares et al., 2006), halts axonal degeneration and disability by preventing mitochondrial depletion, metabolic failure and oxidative stress in a X-linked adrenoleukodystrophy mouse model. These results lend support to the use of pioglitazone in neurodegenerative diseases in which mitochondrial dysfunction contributes to axonal damage.
Materials and methods
Human brain samples
Brain tissues from patients with cerebral adrenomyeloneuropathy were obtained from the Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD, USA as described (Lopez-Erauskin et al., 2012). Informed written consent was obtained from all patients or their legal representatives and the local ethics committee approved the studies.
Mouse strain
The generation and genotyping of Abcd1− and Abcd1−/−/Abcd2−/− mice have been previously described (Lu et al., 1997; Pujol et al., 2002, 2004). The mice used for the study were of a pure C57BL/6J background. The animals were sacrificed and the tissues were recovered and stored at −80°C. All of the methods employed in this study were in accordance with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publications No. 85-23, revised 1996) and the guidelines of the ethical committees of IDIBELL and the Generalitat de Catalunya, Spain.
Treatment of mice
Two mouse models were used. The first model was Abcd1−/− mice at 12 months of age. These mice already show biochemical signs of pathology, including oxidative stress (Fourcade et al., 2008) and alterations in energy homeostasis (Galino et al., 2011), although the first clinical signs of adrenomyeloneuropathy (i.e. axonopathy and locomotor impairment) appear at 20 months (Pujol et al., 2002, 2004). We characterized the biochemical signs of adult X-linked adrenoleukodystrophy in these mice. The second model was mice having a double knockout of both the ABCD1 and ABCD2 transporters (Abcd1−/−/Abcd2−/−). The double knockout mice, compared with the Abcd1−/− mice, display greater accumulation of very long-chain fatty acids in the spinal cord (Pujol et al., 2004), higher levels of oxidative damage to proteins (Fourcade et al., 2010), and a more severe adrenomyeloneuropathy-like pathology with earlier onset (at 12 months) (Pujol et al., 2002, 2004; Ferrer et al., 2005; Lopez-Erauskin et al., 2011). We assessed the clinical signs of adrenomyeloneuropathy in these mice.
Pioglitazone (Actos) was mixed into AIN-76A chow (Dyets) at 0.012% w/w, which corresponds to a dose of 9 mg/kg/day per mouse (Yan et al., 2003). Male mice were randomly assigned to one of the following groups: wild-type mice (n = 12) that received normal AIN-76A chow, Abcd1−/− mice (n = 12) that received normal AIN-76A chow and Abcd1−/− mice (n = 12) that received AIN-76A chow containing pioglitazone. We treated the animals for 2 months starting at 10.5 months of age to assess its effect on the progression of the biochemical signs of X-linked adrenoleukodystrophy. The double knockout mice and their control littermates were randomly assigned to one of the following groups: wild-type mice (n = 25) that received normal AIN-76A chow; double knockout mice (n = 17) that received normal AIN-76A chow; and double knockout mice (n = 17) that received AIN-76A chow containing pioglitazone. We treated the animals for 4 months starting at 13 months of age to coincide with the onset of clinical signs. Pioglitazone had no effect on weight or food intake under either treatment protocol.
Western blotting
Tissues were removed from euthanized mice and flash-frozen in liquid nitrogen. Frozen tissues were homogenized in RIPA buffer, boiled for 5 min and centrifuged as previously described (Galino et al., 2011). The following antibodies were used for western blots: mouse anti-NADH-ubiquinol oxidoreductase [NDUFB8 (complex I)]: dilution 1/2000 [459210 (Molecular Probes)]; mouse anti-Porin 31HL (voltage dependant anion channel, VDAC): dilution 1/1000 [529536 (Calbiochem)]; rabbit anti-pyruvate kynase: dilution 1/500 [ab38237 (Abcam)]; mouse anti-MnSOD (SOD2): dilution: 1/1000 [611581 (BD Pharmigen)]; rabbit anti-GPX1: dilution: 1/200 [LF-PA0019 (Labfrontier)]; rabbit anti-glutathione reductase (GR): dilution 1/1000 [ab16801 (Abcam)]; mouse anti-γ-tubulin dilution 1/5000 [T6557, clone GTU-88 (Sigma)] were used as primary antibodies. Goat anti-rabbit IgG linked to horseradish peroxidase, dilution: 1/15 000 [P0448 (Dako)] and goat anti-mouse IgG linked to horseradish peroxidase, dilution: 1/15 000 [G21040 (Invitrogen)] were used as secondary antibodies.
RNA and DNA extraction
Total RNA was extracted using RNeasy® Kit (Qiagen) according to the manufacturer’s instructions. Total DNA was extracted using Gentra® Puragene® Tissue Kit (Qiagen) according to the manufacturer’s instructions.
Quantitative real-time polymerase chain reaction
One microgram of RNA was transcribed into complementary DNA using SuperScript® II reverse transcription reagents in a final volume of 25 µl (Invitrogen). Complementary DNA (0.1–0.2 µl) and 100 ng DNA were used to measure gene expression and mitochondrial DNA levels, respectively. TaqMan® real-time PCR was performed in the ABI PRISM 7300HT sequence detection system using the TaqMan® Universal PCR master mix and the standardized primers for mouse PGC-1α (Mm00447183), PGC-1β (Mm01258518), ERRα (Mm00433143), NRF-1 (Mm00447996), TFAM (Mm00447485), PPARα (Mm00440939), PPARβ (Mm00803184), PPARγ (Mm01184322), iNOS (Mm00440502) and COX2 (Mm00478374). To quantify mouse and human mitochondrial DNA content, two probes [mouse cytochrome b (cytb) and human cytochrome c oxidoreductase subunit II (COXII)] were designed (Custom TaqMan® Gene Expression Assays; Applied Biosystems). The sequences for mouse cytb were: ATGACCCCAATACGCAAAATTA (forward) and GGAGGACATAGCCTATGAAGG (reverse) and the FAM-labelled probe was TTGCAACTATAGCAACAG. The sequences for human COXII were: CAAACCACTTTCACCGCTACAC (forward) and GGACGATGGGCATGAAACTGT (reverse), and the FAM-labelled probe was AAATCTGTGGAGCAAACC. Quantification of mitochondrial DNA was referred to nuclear DNA as determined by the amplification of the intronless nuclear gene C/EBPα (mouse and human CEBPα are Mm00514283 and Hs00269972, respectively) (Cote et al., 2011). Expression of the genes of interest was normalized to that of the reference control mouse RPL0 (Mm01974474). Each sample was run in duplicate, and the mean value of the duplicate was used to calculate the mitochondrial DNA amount or messenger RNA expression using the comparative (2−ΔCt) method, according to the manufacturer’s instructions.
ATP levels, NAD+/NADH determination and pyruvate kinase activity
ATP levels, NAD+/NADH determination and pyruvate kinase activity were quantified as previously described (Galino et al., 2011).
Glutathione reductase activity
Glutathione reductase activity was determined with a spectrophotometrical method (NWLSSTM Glutathione Reductase Activity microplate assay, NWK-GR01, Northwest). Ninety micrograms of tissue in the same buffer as for pyruvate kinase activity was added to 100 µl of a reaction buffer containing GSSG and NADPH. NADPH was spectophotometrically recorded at 340 nm in a microplate spectrophotometer (PowerWave Microplate Spectrophotometer, BioTek). All assays were performed in triplicate at room temperature. Results were expressed as U/mg tissue.
Measurement of glutamic semialdehyde, aminoadipic semialdehyde, Nε-(carboxymethyl)-lysine and Nε-malondialdehyde-lysine
GSA (glutamic semialdehyde), AASA (aminoadipic semialdehyde), CML [Nε-(carboxymethyl)-lysine] and MDAL (Nε-malondialdehyde-lysine) concentrations in total proteins from spinal cord homogenates were measured by gas chromatography/mass spectrometry (GC/MS) (Pamplona et al., 2005; Fourcade et al., 2008, 2010; Lopez-Erauskin et al., 2011). The product levels were expressed as the ratio of micromoles of GSA, AASA, CML or MDAL to 1 mole of lysine.
Measurements of very long-chain fatty acids
Content of very long-chain fatty acids in total lipids from spinal cord was analysed as methyl ester derivative by gas chromatography (Pamplona et al., 2005). Separation was performed by a DBWAX capillary column (30 m × 0.25 mm × 0.20 μm) in a GC System 7890A with a Series Injector 7683B and a FID detector (Agilent Technologies). The injection port was maintained at 220°C, and the detector at 250°C; the temperature program was 5 min at 145°C, then 2°C/min to 240°C with a hold of 10 min, then 0.5°C/min to 250°C, and finally hold at 250°C for 5 min. Identification of fatty acid methyl esters was made by comparison with authentic standards (Sigma and Larodan Fine Chemicals). Results are expressed as mol%.
Immunohistochemistry
Spinal cords were harvested from 12-month-old wild-type and Abcd1−/− or 18-month-old wild-type, Abcd1−/−/Abcd2−/− and Abcd1−/−/Abcd2−/− mice fed with pioglitazone for 4 months, after perfusion with 4% paraformaldehyde, as described (Pujol et al., 2004; Ferrer et al., 2005; Lopez-Erauskin et al., 2011). Spinal cords were embedded in paraffin and serial sections (5-µm thick) were cut in a transversal or longitudinal (1-cm long) plane. The number of abnormal specific profiles was counted at every 10 sections for each stain. At least three sections of the spinal cord were analysed per animal and per stain. The sections were stained with haematoxylin and eosin and Sudan black, or processed for immunohistochemistry to glial fibrillary acidic protein (GFAP) (Dako, rabbit polyclonal, 1:500), amyloid precursor protein (Boehringer, 1:10), synaptophysin (Dako, monoclonal, 1:500), with lectin Lycopericon esculentum (Sigma, L-0651, 1:200) used as a marker of microglial cells, 8-oxodG (Abcam, 1:1000), SMI-32 (Sternberger Monoclonals, mouse, 1:3000) and cytochrome c (BD Pharmingern, mouse 1:500). The results were expressed as the mean ± standard deviation.
Behavioural testing
Treadmill test
Treadmill apparatus consisted of a variable speed belt varying in terms of speed and slope. An electrified grid was located to the rear of the belt on which footshocks (0.2 mA) were administered whenever the mice fell off the belt. The treadmill apparatus (Panlab) consisted of a belt (50 cm long and 20 cm wide) varying in terms of speed (5 to 150 cm/s) and slope (0–25°) enclosed in a plexiglass chamber (Martinez de Lágran et al., 2004). The latency to falling off the belt (time of shocks in seconds) and the number of received shocks were measured. The mice were placed on the top of the already moving belt facing away from the electrified grid and in the direction opposite to the movement of the belt. Thus, to avoid the footshocks, the mice had to move forward.
The mice were evaluated in five trials in a single day session. In the first trial, the belt speed was set at 20 cm/s and the inclination at 5°. In the second and third trials, the belt speed was 10 cm/s and the slope was increased to 10° and 20°, respectively. For the fourth and the fifth trials, the inclination was maintained at 20° and the belt speed was increased to 20 and 30 cm/s, respectively. For the three first trials, mice ran for 1 min. The fourth and fifth trials were 3 and 7 min, respectively. The time between each test was 1, 1, 5 and 20 min, respectively. When the mice were subjected to consecutive trials at increasing speeds up to 20 cm/s and a 20° slope, no differences were detected from one session to another between the wild-type and Abcd1−/−/Abcd2−/− mice. However, when the belt speed was increased up to 30 cm/s and the slope was 20°, differences were detected between the Abcd1−/−/Abcd2−/− mice and the control mice because this task requires greater coordination. These conditions were therefore chosen to assess the effects of pioglitazone.
The training session performance was normal for all groups, indicating that correct acquisition of the skill had occurred (data not shown). The ratio between the time of shocks and the number of shocks was used as a locomotor deficit index.
Horizontal bar cross test
The bar cross test was carried out using a wooden bar of 100 cm in length and 2 cm in width (diameter). This bar is just wide enough for mice to stand on with their hind feet hanging over the edge such that any slight lateral misstep will result in a slip. The bar was elevated 50 cm from the bench surface, so that animals did not jump off, yet were not injured upon falling from the bar. The mice are put on one end of the bar and expected to cross to the other end. To eliminate the novelty of the task as a source of slips, all animals were given four trials on the bar the day before and at the beginning of the testing session. In an experimental session, the number of hind limb lateral slips and falls from the bar was counted on four consecutive trials. If an animal fell, it was placed back on the bar at the point at which it fell and was allowed to complete the task. The bar was cleaned with ethanol after each animal.
Statistical analysis
Data are presented as the mean ± standard deviation (SD). Significant differences were determined by Student’s t-test or one-way ANOVA followed by Tukey HSD post hoc (*P < 0.05, **P < 0.01, ***P < 0.001) after verifying normality. Statistical analyses were performed using the software program SPSS 12.0.
Results
Mitochondrial depletion in Abcd1−/− mouse spinal cords
We recently reported a transcriptomic analysis of 12-month-old Abcd1−/− spinal cords, in which a significant reduction of messenger RNA expression in ∼40% of the genes encoding mitochondrial oxidative phosphorylation proteins was revealed (Schluter et al., 2012). To investigate whether this decrease was associated with a reduced number of mitochondria, we quantified (i) the level of respiratory chain complex I (NDUFB8) protein; (ii) the protein content of the voltage-dependent anion channel, a mitochondrial porin whose expression is correlated with mitochondrial abundance (Mahad et al., 2009); (iii) the mitochondrial DNA/nuclear DNA ratio; and (iv) the level of mitochondrial cytochrome c protein by immunohistological analysis. We observed reductions in these four parameters in Abcd1−/− mouse spinal cords compared with control mice (Fig. 1A–C), confirming the depletion in mitochondria suggested by transcriptomics studies (Schluter et al., 2012). The mitochondrial depletion appears to be specific to the spinal cord, as we did not observe a similar depletion in the cerebral cortex or in the liver of Abcd1−/− mice at the same age (Supplementary Fig. 1).
Figure 1.
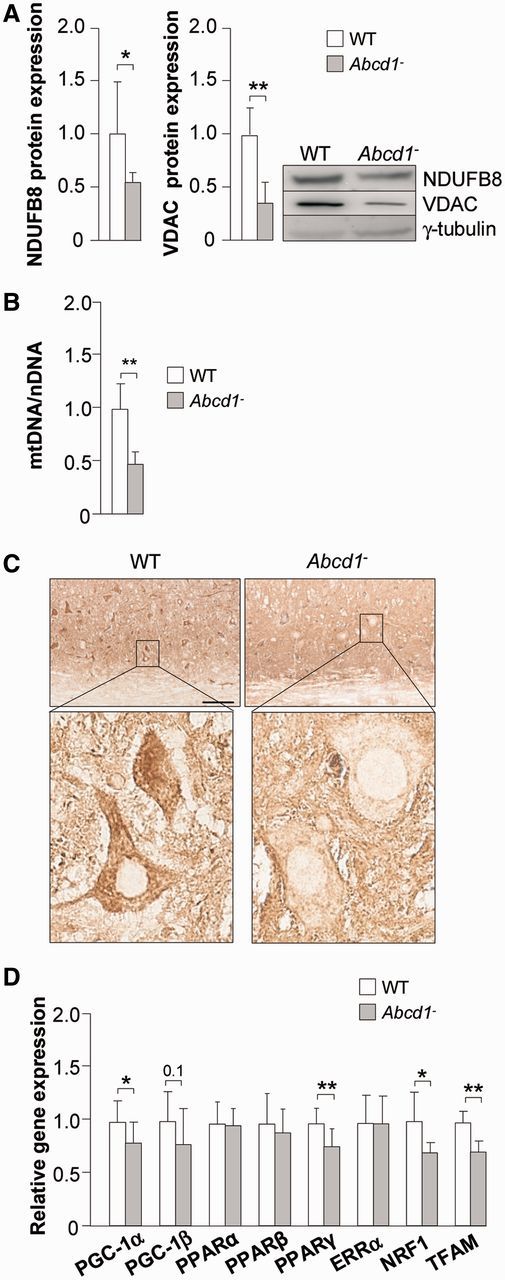
Mitochondrial DNA and mitochondrial protein levels are reduced in spinal cords of Abcd1−/− mice at 12 months of age. Wild-type (n = 8) and Abcd1−/− (n = 8) mice at 12 months of age. (A) NDUFB8 and VDAC protein levels, (B) mitochondrial DNA (mtDNA) levels, (C) cytochrome c quantification by immunostaining in mice spinal cord histological slides and (D) relative gene expression of PGC-1α, PGC-1β, PPARα, PPARβ, PPARγ, ERRα, NRF1 and TFAM. Scale bar = 100 µm (low magnification) / 20 µm (high magnification). Data represent mean ± SD (*P ≤ 0.05, **P ≤ 0.01). WT = wild-type.
Pathways controlled by PGC-1α are altered in X-linked adrenoleukodystrophy
We next investigated the mechanisms governing the decrease in mitochondrial number in the spinal cords of X-linked adrenoleukodystrophy mice. The primary and best-characterized master regulators of mitochondrial biogenesis are PGC-1α/PGC-1β. These factors regulate the contents and/or activity of several transcriptional regulators of mitochondrial components, including (i) nuclear hormone receptors such as PPARα/β/γ and ERRα (oestrogen-related receptor-α); (ii) nuclear-encoded NRF1 (nuclear respiratory factor 1), which activates the transcription of nuclear genes encoding for respiratory and detoxifying proteins; and (iii) the mitochondrial transcription factor TFAM. This transcription factor controls the replication and transcription of mitochondrial DNA, which in turn encodes certain subunits of the electron transport chain complexes (Wu et al., 1999). NRF1 also induces the expression of these mitochondrial transcription factors (Wu et al., 1999), thereby coordinating the nuclear and mitochondrial events that lead to the complete synthesis of mitochondrial components. We found that, at the messenger RNA level, PGC-1α was reduced in the spinal cords of Abcd1−/− mice (Fig. 1D). Consistent with these findings, we found that the messenger RNA levels of the transcriptional targets of PGC-1α, the transcription factors NRF1, TFAM and PPARγ were reduced in Abcd1−/− spinal cords (Fig. 1D), indicating that this PGC-1α signalling pathway is impaired in X-linked adrenoleukodystrophy.
Importantly, reductions in the levels of mitochondrial DNA (Fig. 2A) and NDUFB8 and VDAC (Fig. 2B) proteins were also observed in the affected brain white matter of patients with X-linked adrenoleukodystrophy. These findings support the hypothesis that mitochondrial depletion may play a central role in the pathogenesis of X-linked adrenoleukodystrophy.
Figure 2.
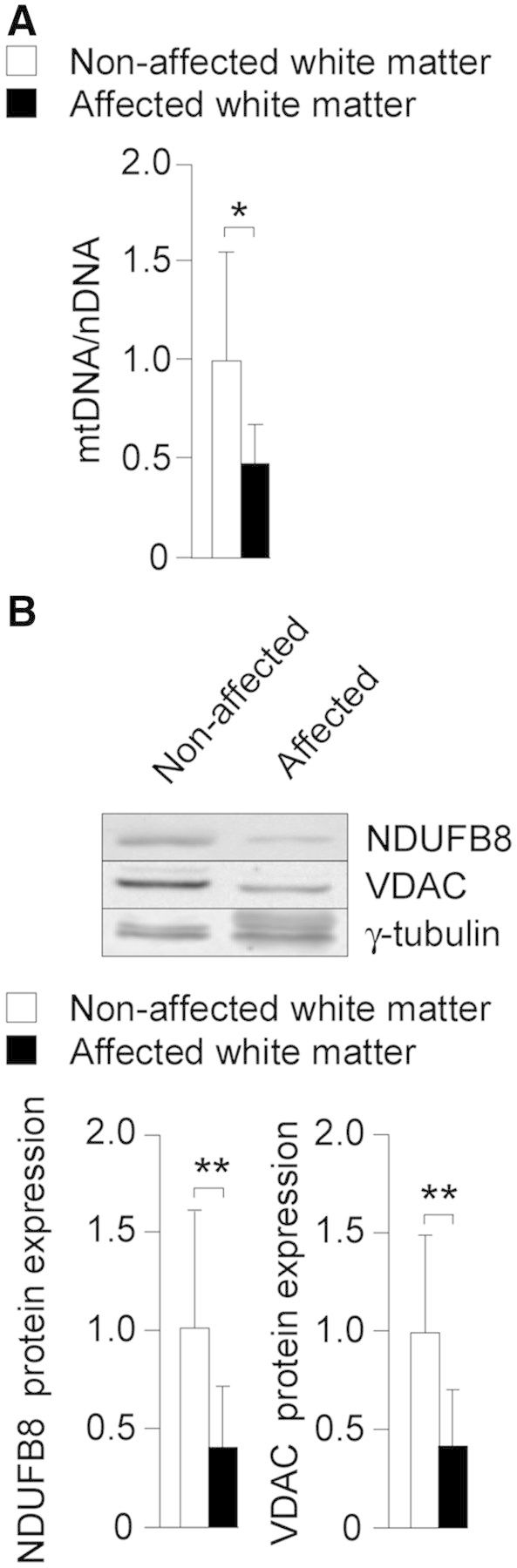
Mitochondrial DNA and mitochondrial protein levels are reduced in the affected white matter of patients with X-linked adrenoleukodystrophy. Affected (n = 4) and normal-appearing white matter (n = 4) of patients with X-linked adrenoleukodystrophy. (A) Mitochondrial DNA (mtDNA) levels and (B) NDUFB8 and VDAC protein levels. Data represent mean ± SD (*P ≤ 0.05, **P ≤ 0.01).
Pioglitazone prevents mitochondrial depletion in Abcd1−/− mice
The abovementioned results demonstrate that X-linked adrenoleukodystrophy mice show mitochondria depletion associated with impairment of the PGC-1α pathway. It follows that the stimulation of PGC-1α-dependent pathways could restore mitochondrial biogenesis and function by coordinating the actions of transcriptional networks. Pioglitazone is a PPARγ agonist that has been shown to be able to regulate PGC-1α-dependent pathways in vitro in both human neuroblastoma SHY5Y cells (Miglio et al., 2009) and neuron-like NT2 cells (Ghosh et al., 2007) and in vivo in human subcutaneous adipose tissue (Bogacka et al., 2005).
Because pioglitazone has been successfully tested in animal models of Alzheimer’s disease (Heneka et al., 2005), amyotrophic lateral sclerosis (Schutz et al., 2005) and multiple sclerosis (Feinstein et al., 2002), we examined the possible therapeutic benefits of this drug in the X-linked adrenoleukodystrophy models. In order to demonstrate that the dietary administration of pioglitazone was effective, we measured expression of two classical PPARγ targets, iNOS and COX2 (Schutz et al., 2005). Interestingly, pioglitazone negatively modulated iNOS and COX2 levels in spinal cord (Fig. 3A).
Figure 3.
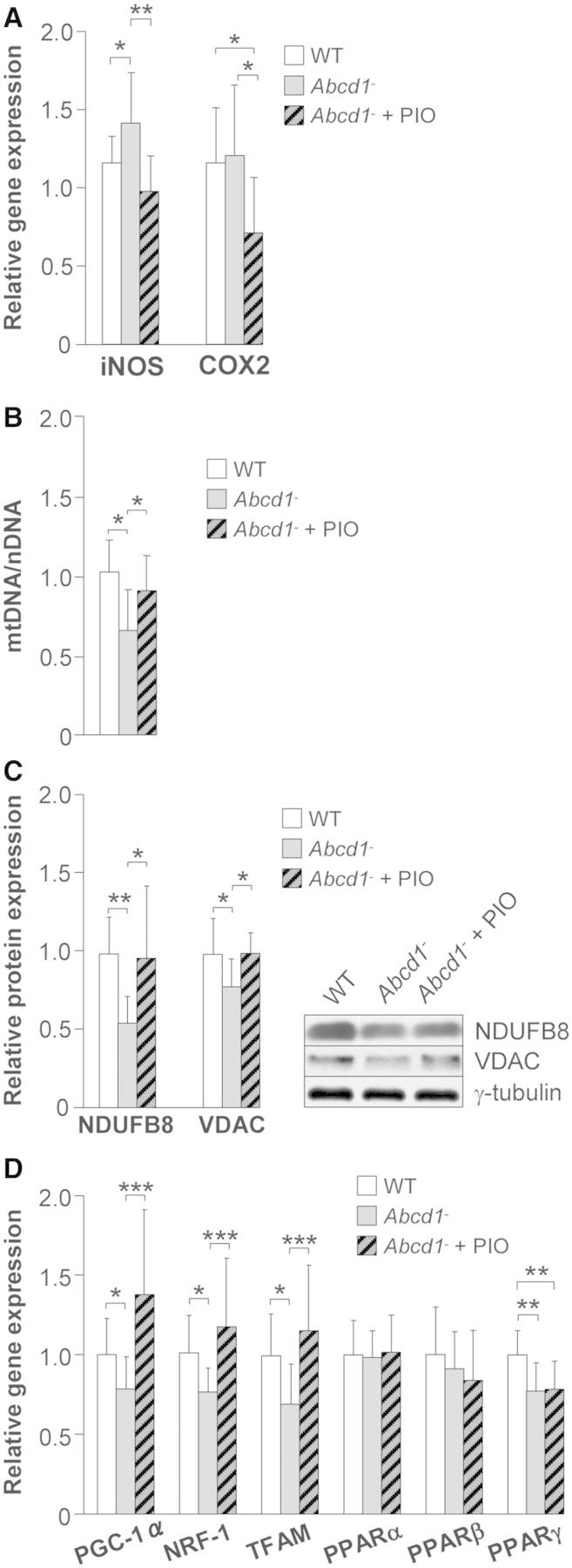
Pioglitazone normalizes mitochondrial DNA and mitochondrial protein levels in spinal cords of Abcd1−/− mice. Wild-type (n = 8), Abcd1−/− (n = 8) and Abcd1−/− mice fed for 2 months with pioglitazone (Abcd1− + PIO) (n = 8) at 12 months of age. (A) Relative gene expression of iNOS and COX2, (B) mitochondrial DNA (mtDNA) level, (C) NDUFB8 and VDAC protein levels and (D) relative gene expression of PGC-1α, NRF1, TFAM and PPARα, PPARβ and PPARγ. Data represent mean ± SD (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). WT = wild-type.
Next, we examined whether pioglitazone was able to activate the mitochondrial biogenesis program in X-linked adrenoleukodystrophy mice. We found pioglitazone normalized the mitochondrial DNA/nuclear DNA ratio (Fig. 3B) and increased the levels of the mitochondrial proteins NDUFB8 and VDAC (Fig. 3C) and the expression of PGC-1α-dependent factors such as NRF1, TFAM as well as PGC-1α itself (Fig. 3D).
Pioglitazone reverses metabolic failure in Abcd1−/− mice
The NAD+/NADH ratio is a sensitive indicator of energy metabolism and the redox state of the cell because it links the citric acid cycle with oxidative phosphorylation within mitochondria. A condition of oxidative stress and energy metabolism impairment is reflected by reduced levels of NADH and ATP. We previously reported decreased levels of both of these metabolites in the spinal cords of Abcd1−/− mice (Galino et al., 2011), suggesting that a deficit in energy homeostasis is a key feature in X-linked adrenoleukodystrophy pathology. The levels and activity of pyruvate kinase, a key enzyme in glycolysis, were also found to be reduced in Abcd1−/− mice (Galino et al., 2011). In the present study, we examined the possibility that pioglitazone might prevent bioenergetic failure in Abcd1−/− mice by measuring ATP, NADH and pyruvate kinase activity. Pioglitazone was found to normalize the levels of ATP and NADH in accordance with the correction of mitochondrial levels (Fig. 4A and B), as well as the expression and activity of pyruvate kinase (Fig. 4C and D).
Figure 4.

Pioglitazone normalizes ATP, NADH levels and pyruvate kinase in spinal cords of Abcd1 null mice. Wild-type (n = 8), Abcd1−/− (n = 8) and Abcd1−/− mice fed for 2 months with pioglitazone (Abcd1− + PIO) (n = 8) at 12 months of age. (A) ATP, (B) NAD+ and NADH levels, (C) pyruvate kinase activity and (D) pyruvate kinase protein levels. Data represent mean ± SD (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). WT = wild-type.
Pioglitazone normalizes the levels of oxidative stress biomarkers in Abcd1−/− mice
Because pioglitazone has been shown to exert antioxidant effects in vitro and in vivo (Collino et al., 2006; Rahimian et al., 2009), we quantified these effects using gas chromatography/mass spectrometry markers of oxidative lesions to proteins, which are abnormally elevated in Abcd1−/− mice (Fourcade et al., 2008; Lopez-Erauskin et al., 2011). We found that the contents of GSA, AASA, CML (glycoxidative) and MDAL (lipoxidative) were normalized by pioglitazone (Fig. 5A). Pioglitazone also normalized the content of the antioxidant enzyme GPX1 but not SOD2, whose levels are altered in Abcd1−/− mice (Fourcade et al., 2008) (Fig. 5B). Moreover, we measured the activity of glutathione reductase, the enzyme that generates reduced glutathione, the main antioxidant protector of the cell. Interestingly, the activity of this enzyme was slowed in X-linked adrenoleukodystrophy mice and normalized with pioglitazone treatment (Fig. 5C). Reduction of oxidative stress biomarkers appears most likely independent of C26:0 levels, since the accumulation of this toxic metabolite is only very slightly decreased by pioglitazone treatment (Supplementary Fig. 2A and Supplementary Table 1). This is concomitant with a diminished expression of the enzyme responsible for elongation of very long-chain fatty acids Elovl3 (Kihara, 2012) (Supplementary Fig 2B).
Figure 5.
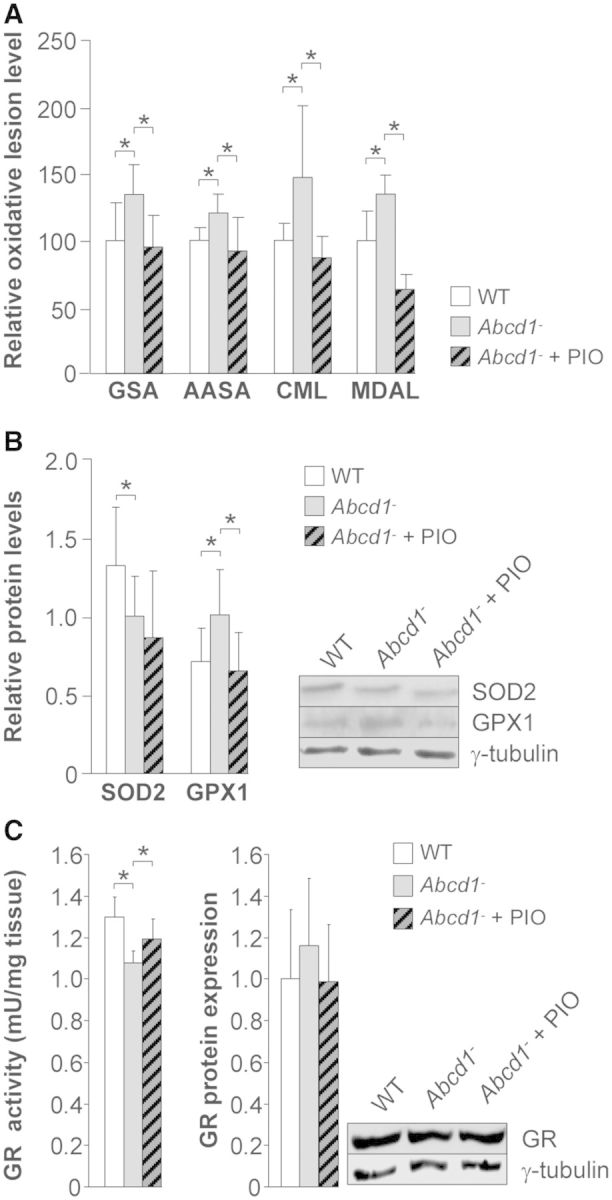
Pioglitazone reverses oxidative lesions in spinal cords of Abcd1−/− mice. Wild-type (n = 8), Abcd1−/− (n = 8) and Abcd1−/− mice fed for 2 months with pioglitazone (Abcd1− + PIO) (n = 8) at 12 months of age. (A) GSA, AASA, CML and MDAL levels. (B) SOD2 and GPX1 protein levels. (C) Glutathione reductase (GR) activity and protein levels. Data represent mean ± SD (*P ≤ 0.05).
Pioglitazone prevents axonal degeneration and increases mitochondria levels in Abcd1−/−/Abcd2−/− mice
Abcd1−/−/Abcd2−/− (double knockout) mice present an overt neuropathological phenotype at 16 months of age, characterized by (i) axonal damage, as suggested by the accumulation of amyloid precursor protein (APP) and synaptophysin in axonal swellings; (ii) scattered myelin debris, as revealed by Sudan black; (iii) astrocytosis and microgliosis, as revealed by GFAP and lectin staining, respectively; (iv) increased labelling of 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG), a marker of oxidative DNA damage, in motor neurons (Pujol et al., 2004; Lopez-Erauskin et al., 2011); (v) decreased levels of cytochrome c, a marker of mitochondrial content; and (vi) unhealthy motor neurons, denoted by reduced staining for SMI-32, an antibody that recognizes a non-phosphorylated epitope in neurofilament H. The most affected areas for both the axonal and accompanying reactive glial changes are the pyramidal tracts and the dorsal fascicles (Pujol et al., 2004; Lopez-Erauskin et al., 2011). This model is used as a bona fide surrogate for the Abcd1 null mouse, for instance in gene therapy and pharmacological preclinical tests (Mastroeni et al., 2009; Lopez-Erauskin et al., 2011). The accumulation of markers of axonal damage and DNA oxidation and the numbers of reactive astrocytes and reactive microglia were strikingly reduced to control levels with pioglitazone treatment. Likewise, pioglitazone was able to recover mitochondria levels and improve motor neurons health in spinal cord from X-linked adrenoleukodystrophy mice (Fig. 6A–Y and Table 1).
Figure 6.
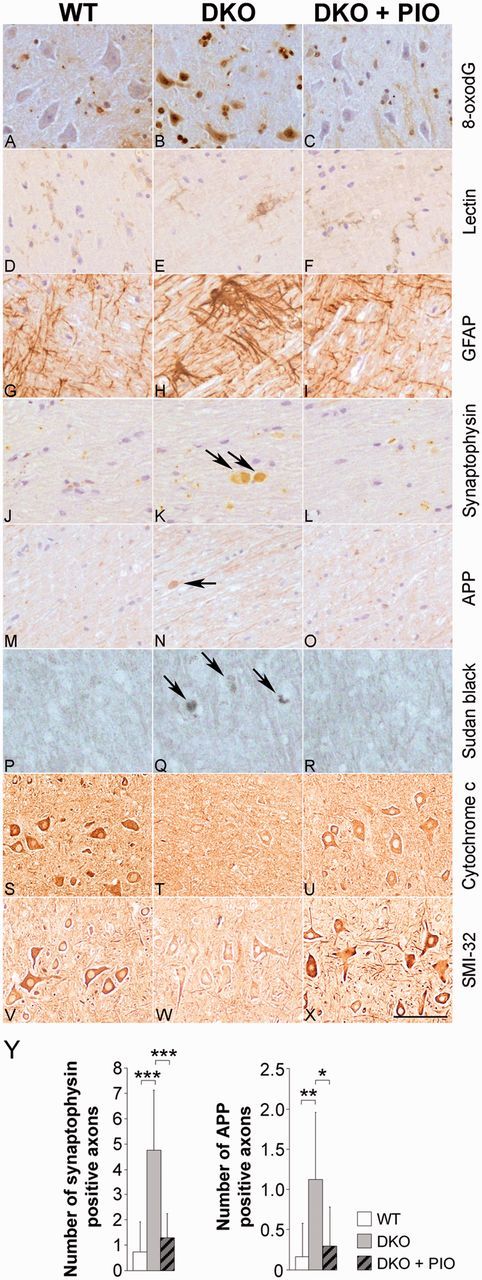
Oxidative stress, myelin and axonal pathologies and mitochondrial depletion are prevented by pioglitazone in spinal cords of 18-month-old Abcd1−/−/Abcd2−/− mice. Longitudinal and transversal sections of the spinal cord in wild-type (A, D, G, J, M, P, S, V), Abcd1−/−/Abcd2−/− (DKO) (B, E, H, K, N, Q, T, W) and Abcd1−/−/Abcd2−/− + PIO (DKO + PIO) (C, F, I, L, O, R, U, X) mice processed for 8-oxodG (A–C), lectin Lycopericon esculentum (D–F), GFAP (G–I), synaptophysin (J–L), APP (M–O), Sudan black (P–R), cytochrome c (S–U), and SMI-32 (V–X). Scale bar = 100 µm. (Y) Quantification of amyloid precursor protein (APP) and synaptophysin accumulation in axonal swellings in wild-type (WT), double knockout (DKO) and double knockout + PIO (DKO + PIO) mice. Significant differences were determined as described in the ‘Materials and methods’ section (n = 5–6 mice per genotype and condition; *P < 0.05, **P < 0.01, ***P < 0.001). DKO = double knockout.
Table 1.
Summary of the major histopathological findings in mice spinal cord at 18 months of age
| Wild-type | DKO | DKO + PIO | |
|---|---|---|---|
| 8-oxodG | Normal | Increased | Normal |
| Lectin | Normal | Increased | Normal |
| GFAP | Normal | Increased | Normal |
| Sudan black | Normal | Increased | Normal |
| Cytochrome c | Normal | Reduced | Normal |
| SMI-32 | Normal | Reduced | Normal |
DKO = double knockout = Abcd1−/−/Abcd2−/−; double knockout + PIO = Abcd1−/−/Abcd2−/− + PIO; (n = 5–6 mice per genotype and condition).
Pioglitazone stops the progression of locomotor deficits in Abcd1−/−/Abcd2−/− mice
Locomotor deficits in Abcd1−/−/Abcd2−/− (double knockout) mice were evaluated by treadmill and bar cross tests before treatment (at 13 months of age) and again after 2 months (at 15 months of age) and 4 months (at 17 months of age) of treatment. In the treadmill test, the double knockout mice at 13 months of age showed an increase in the ratio between the time length of the shocks and the number of shocks, reflecting a locomotor disability. After 2 months of treatment, this ratio was partially reduced in double knockout animals treated with pioglitazone. Interestingly, after 4 months of treatment, this ratio was totally normalized, indicating that pioglitazone treatment had stopped the progression of locomotor deficits in X-linked adrenoleukodystrophy mice (Fig. 7A).
Figure 7.
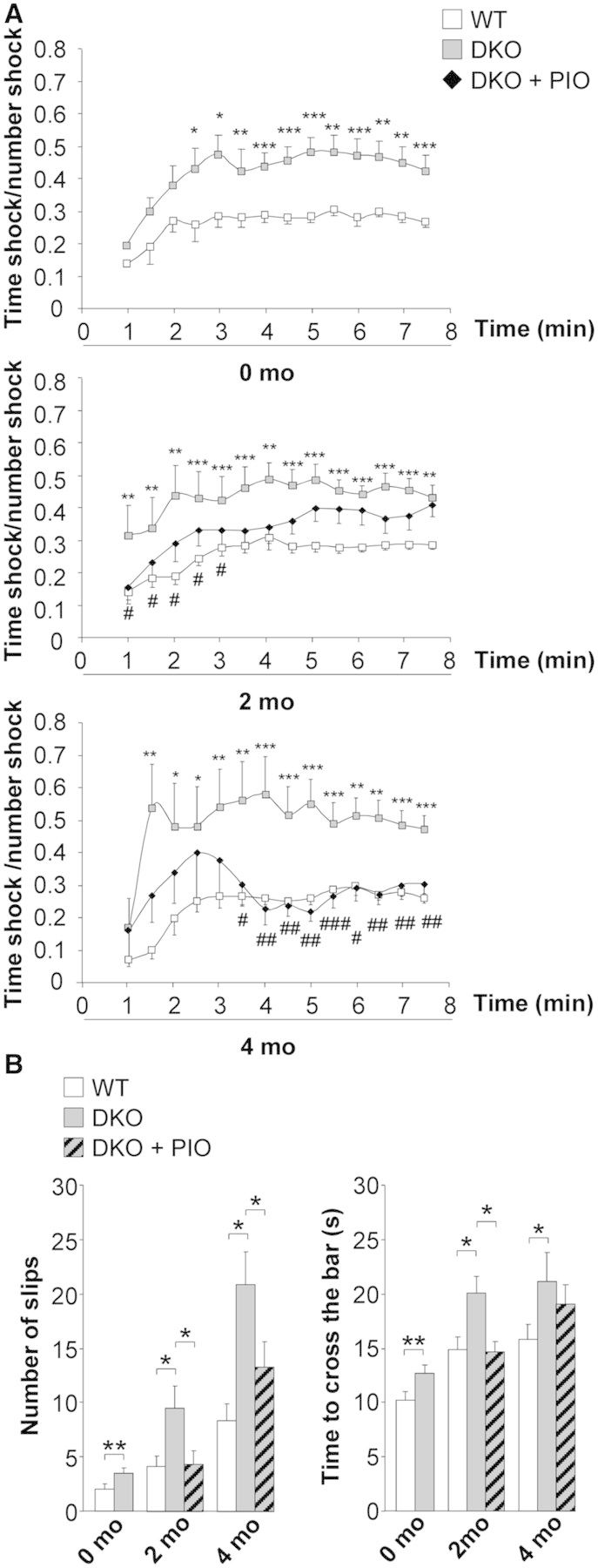
Pioglitazone improves locomotor disability in an X-linked adrenoleukodystrophy mouse model. (A) Treadmill and (B) bar-cross test in wild-type (WT) and Abcd1−/−/Abcd2−/− mice (DKO) mice at 13 months of age (before treatment) and after 2 and 4 months (mo) of pioglitazone treatment (DKO + PIO). Data represent mean ± SD. Statistical analysis was carried out with Student’s t-test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001) or ANOVA followed by Tukey HSD post hoc (wild-type versus DKO = *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001; DKO versus DKO + PIO = #P ≤ 0.05, ##P ≤ 0.01). DKO = double knockout.
In the bar cross experiments, double-knockout mutants on vehicle often failed to maintain their balance, and they displayed a greater tendency to slip off the bar and longer time latencies in reaching the platform at the opposite end of the bar. Some of the mice displayed ventral recumbence by wrapping the hind and front limbs laterally around the bar, as previously described (Ferrer et al., 2005; Lopez-Erauskin et al., 2011). Following pioglitazone treatment, the time needed to cross the bar and the number of slips reached full normalization after 2 months (Fig. 7B).
Discussion
In this work, we have observed mitochondrial depletion shown by decreased levels of mitochondrial DNA and mitochondrial proteins in the brains of patients with X-linked adrenoleukodystrophy and spinal cords of the corresponding mouse model, together with a decrease in cytochrome c immunoreactivity in spinal cord motor neurons. These results reveal similarities between this prototypical peroxisomal disease and classical mitochondrial DNA depletion syndromes, which are a group of early-lethal, autosomal-recessive diseases characterized by a low mitochondrial DNA copy number in specific tissues and that often lead to hepatocerebral disorders (Spinazzola et al., 2009). The defective genes encode proteins that are involved in the de novo biosynthesis of deoxynucleotides, mitochondrial DNA replication or mitochondrial enzymatic activity (Spinazzola et al., 2009). Our findings support the hypothesis that the dysfunction of a non-mitochondrial protein such as the ABCD1 transporter, which is not directly involved in mitochondrial metabolism or mitochondrial DNA maintenance, may affect the biogenesis of mitochondria. Moreover, it has been reported that excess of C26:0 reduces OXPHOS protein levels (complex III subunit core 2 and complex IV subunit II) in the human neuroblastoma cell line SK-NB-E (Zarrouk et al., 2012). Along similar lines, we expand these results by describing a joint downregulation of several transcription factors that control the expression of structural or functional components of mitochondria in the Abcd1−/− mouse model. This indicates that impairment in PGC-1α-dependent pathways underlies the decrease in mitochondrial DNA and mitochondrial protein levels. This may suggest that the excess of C26:0 could negatively regulate the expression of PGC-1α, perhaps in a similar manner than a high fat diet or saturated fatty acid treatment does (Mootha et al., 2003; Richardson et al., 2005 Crunkhorn et al., 2007). This phenomenon is not limited to X-linked adrenoleukodystrophy because decreased PGC-1α activity has also been reported in several neurodegenerative disorders including Huntington’s disease (Cui et al., 2006), Parkinson’s disease (Shin et al., 2011), amyotrophic lateral sclerosis (Zhao et al., 2011), Friedreich ataxia (Coppola et al., 2009) and Alzheimer’s disease (Qin et al., 2009).
In addition, we demonstrated that treatment of X-linked adrenoleukodystrophy mice with pioglitazone, an activator of PPARγ that increases the expression of PGC-1α (Bogacka et al., 2005; Ghosh et al., 2007; Miglio et al., 2009), prevented mitochondrial depletion, normalized the signs of oxidative damage and oxidative lesions to proteins and DNA, preserved energetic homeostasis (NADH and ATP levels), restored enzymatic activities of glutathione reductase and pyruvate kinase expression and activity, and, more importantly, halted the progression of axonal degeneration and locomotor disabilities. We propose that a direct mechanism to mediate the protective effect of pioglitazone in Abcd1 null mice could be the increase of PGC-1α expression, most likely through a positive regulatory loop between PPARγ and its co-activator, as described (Hondares et al., 2006). Notwithstanding, to establish a direct causality link, to order the events chronologically in the cascade, or even to highlight a major insult over the others, and therefore underscore one precise beneficial effect of pioglitazone over the rest, may be too premature at this stage. Rather, we tend to think that the simultaneous restoration of energetic and redox homeostasis exerted by pioglitazone is most likely a key factor for arresting disease progression.
We believe that the amelioration of clinical outcome of the disease was independent on the levels of C26:0 and very long-chain fatty acids, which remained close to the levels of untreated Abcd1 null mice, although showing a slight decrease. Because the expression levels of the partially overlapping Abcd2 gene (Pujol et al., 2004; Fourcade et al., 2009) were decreased (Supplementary Fig 2B), this could be attributed to a repressive effect of pioglitazone on the expression of the fatty acid elongase Elovl3, which is responsible for elongation of very long-chain fatty acids (Kihara, 2012). Along the same lines, the treatment with the PPAR pan-agonist bezafibrate exerted an inhibitory effect on ELOVL1 activity in X-linked adrenoleukodystrophy fibroblasts, leading to a reduction of C26:0 (Engelen et al., 2012).
Moreover, potent antioxidant effects of pioglitazone have been previously reported in Alzheimer’s disease-related cerebrovascular alterations (Nicolakakis et al., 2008) and in models of ischaemia/reperfusion in rats (Collino et al., 2006; Rahimian et al., 2009). The latter studies also suggested direct reactive oygen species-scavenging capacity by pioglitazone, although the molecular effects of pioglitazone on mitochondrial biogenesis, or energetic metabolism on parameters such as ATP or NADH levels, or enzymatic measurements of pyruvate kinase or glutathione reductase were not addressed in previous reports. Thus, our study broadens the current knowledge on the molecular mechanisms driving pioglitazone’s pleiotropic effects.
Besides fostering of PGC-1α pathways, we should not minimize the impact of pioglitazone on reducing inflammation, which would synergistically add to the beneficial effects of the drug. The anti-inflammatory effect of pioglitazone has been described in a model of Alzheimer’s disease in which activated microglial cells were found in close contact with degenerating motor neurons. Treatment with pioglitazone reduced the number of fully activated microglia and thereby protected neurons (Schutz et al., 2005). Similar results were observed by Dehmer et al. (2004) in a model of Parkinson’s disease. These authors suggested that pioglitazone may act sequentially through PPARγ activation, IκBα induction, and inhibition of NFκB activation (Dehmer et al., 2004; Schutz et al., 2005). These results are consistent with our observation of the reduction of both microglial activation, astrocytosis, and some inflammatory PPARγ target genes (iNOS and COX2) upon pioglitazone treatment (Fig. 3A and Fig. 6D–F). Finally, a new target of pioglitazone, the outer mitochondrial membrane protein MitoNEET, could constitute an additional mediator of the positive effects of the drug in this model. This is an iron-containing protein participating in redox signalling and oxidative capacity regulation (Paddock et al., 2007; Wiley et al., 2007). This appealing possibility will be examined in future studies.
Taken together, our findings strongly suggest that an early and carefully tailored interventional treatment using pioglitazone is an attractive therapeutic option for patients with X-adrenomyeloneuropathy and deserves to be translated into clinical trials. The biological effects could be easily monitored by quantitative measurement of the biomarkers of oxidative damage in the peripheral blood mononuclear cells or plasma of patients with X-adrenomyeloneuropathy, as previously described (Fourcade et al., 2010). The therapeutic implications of this work may be extrapolated to other diseases that exhibit both axonal degeneration as a significant component of clinical progression and oxidative stress/mitochondrial depletion or failure as major early contributing pathogenic factors.
Funding
This work was supported by grants from the European Commission [FP7-241622], the European Leukodystrophy Association [ELA2009-036C5, ELA2008-040C4], the Spanish Institute for Health Carlos III [FIS PI11/01043], the Autonomous Government of Catalonia [2009SGR85 to A.P.], the Spanish Institute for Health Carlos III [Miguel Servet program CP11/00080 to S.F.], the COST action [BM0604 to A.P.]. S.F. was a fellow of the European Leukodystrophy Association [ELA 2010-020F1]. L.M. is a fellow of the Spanish Ministry of Education [FPU program: AP2008-03728]. J.G. was a fellow of IDIBELL. The studies conducted at the Experimental Medicine Department were supported in part by R+D grants from the Spanish Ministry of Science and Innovation [BFU2009-11879/BFI], the Spanish Ministry of Health [PI081843], the Autonomous Government of Catalonia [2009SGR735], the ‘La Caixa' Foundation and COST B35 Action of the European Union. The CIBER on Rare Diseases (CIBERER) and the CIBER on Neurodegenerative Diseases (CIBERNED) are initiatives of the ISCIII.
Supplementary material
Supplementary material is available at Brain online.
References
- Bogacka I, Xie H, Bray GA, Smith SR. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes. 2005;54:1392–9. doi: 10.2337/diabetes.54.5.1392. [DOI] [PubMed] [Google Scholar]
- Braak H, Sandmann-Keil D, Gai W, Braak E. Extensive axonal Lewy neurites in Parkinson's disease: a novel pathological feature revealed by alpha-synuclein immunocytochemistry. Neurosci Lett. 1999;265:67–9. doi: 10.1016/s0304-3940(99)00208-6. [DOI] [PubMed] [Google Scholar]
- Collino M, Aragno M, Mastrocola R, Gallicchio M, Rosa AC, Dianzani C, et al. Modulation of the oxidative stress and inflammatory response by PPAR-gamma agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur J Pharmacol. 2006;530:70–80. doi: 10.1016/j.ejphar.2005.11.049. [DOI] [PubMed] [Google Scholar]
- Coppola G, Marmolino D, Lu D, Wang Q, Cnop M, Rai M, et al. Functional genomic analysis of frataxin deficiency reveals tissue-specific alterations and identifies the PPARgamma pathway as a therapeutic target in Friedreich's ataxia. Hum Mol Genet. 2009;18:2452–61. doi: 10.1093/hmg/ddp183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote HC, Gerschenson M, Walker UA, Miro O, Garrabou G, Hammond E, et al. Quality assessment of human mitochondrial DNA quantification: MITONAUTS, an international multicentre survey. Mitochondrion. 2011;11:520–7. doi: 10.1016/j.mito.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunkhorn S, Dearie F, Mantzoros C, Gami H, da Silva WS, Espinoza D, et al. Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J Biol Chem. 2007;282:15439–50. doi: 10.1074/jbc.M611214200. [DOI] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Dehmer T, Heneka MT, Sastre M, Dichgans J, Schulz JB. Protection by pioglitazone in the MPTP model of Parkinson's disease correlates with I kappa B alpha induction and block of NF kappa B and iNOS activation. J Neurochem. 2004;88:494–501. doi: 10.1046/j.1471-4159.2003.02210.x. [DOI] [PubMed] [Google Scholar]
- Engelen M, Schackmann MJ, Ofman R, Sanders RJ, Dijkstra IM, Houten SM, et al. Bezafibrate lowers very long-chain fatty acids in X-linked adrenoleukodystrophy fibroblasts by inhibiting fatty acid elongation. J Inherit Metab Dis. 2012;35:1137–45. doi: 10.1007/s10545-012-9471-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein DL, Galea E, Gavrilyuk V, Brosnan CF, Whitacre CC, Dumitrescu-Ozimek L, et al. Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann Neurol. 2002;51:694–702. doi: 10.1002/ana.10206. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Aubourg P, Pujol A. General aspects and neuropathology of X-linked adrenoleukodystrophy. Brain Pathol. 2010;20:817–30. doi: 10.1111/j.1750-3639.2010.00390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Kapfhammer JP, Hindelang C, Kemp S, Troffer-Charlier N, Broccoli V, et al. Inactivation of the peroxisomal ABCD2 transporter in the mouse leads to late-onset ataxia involving mitochondria, Golgi and endoplasmic reticulum damage. Hum Mol Genet. 2005;14:3565–77. doi: 10.1093/hmg/ddi384. [DOI] [PubMed] [Google Scholar]
- Fourcade S, Lopez-Erauskin J, Galino J, Duval C, Naudi A, Jove M, et al. Early oxidative damage underlying neurodegeneration in X-adrenoleukodystrophy. Hum Mol Genet. 2008;17:1762–73. doi: 10.1093/hmg/ddn085. [DOI] [PubMed] [Google Scholar]
- Fourcade S, Ruiz M, Camps C, Schluter A, Houten SM, Mooyer PA, et al. A key role for the peroxisomal ABCD2 transporter in fatty acid homeostasis. Am J Physiol Endocrinol Metab. 2009;296:E211–21. doi: 10.1152/ajpendo.90736.2008. [DOI] [PubMed] [Google Scholar]
- Fourcade S, Ruiz M, Guilera C, Hahnen E, Brichta L, Naudi A, et al. Valproic acid induces antioxidant effects in X-linked adrenoleukodystrophy. Hum Mol Genet. 2010;19:2005–14. doi: 10.1093/hmg/ddq082. [DOI] [PubMed] [Google Scholar]
- Galea E, Launay N, Portero-Otin M, Ruiz M, Pamplona R, Aubourg P, et al. Oxidative stress underlying axonal degeneration in adrenoleukodystrophy: a paradigm for multifactorial neurodegenerative diseases? Biochim Biophys Acta. 2012;1822:1475–88. doi: 10.1016/j.bbadis.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Galino J, Ruiz M, Fourcade S, Schluter A, Lopez-Erauskin J, Guilera C, et al. Oxidative damage compromises energy metabolism in the axonal degeneration mouse model of x-adrenoleukodystrophy. Antioxid Redox Signal. 2011;15:2095–107. doi: 10.1089/ars.2010.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Patel N, Rahn D, McAllister J, Sadeghi S, Horwitz G, et al. The thiazolidinedione pioglitazone alters mitochondrial function in human neuron-like cells. Mol Pharmacol. 2007;71:1695–702. doi: 10.1124/mol.106.033845. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, et al. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–53. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- Hettema EH, van Roermund CW, Distel B, van den Berg M, Vilela C, Rodrigues-Pousada C, et al. The ABC transporter proteins Pat1 and Pat2 are required for import of long-chain fatty acids into peroxisomes of Saccharomyces cerevisiae. EMBO J. 1996;15:3813–22. [PMC free article] [PubMed] [Google Scholar]
- Hondares E, Mora O, Yubero P, Rodriguez de la Concepcion M, Iglesias R, Giralt M, et al. Thiazolidinediones and rexinoids induce peroxisome proliferator-activated receptor-coactivator (PGC)-1alpha gene transcription: an autoregulatory loop controls PGC-1alpha expression in adipocytes via peroxisome proliferator-activated receptor-gamma coactivation. Endocrinology. 2006;147:2829–38. doi: 10.1210/en.2006-0070. [DOI] [PubMed] [Google Scholar]
- Kihara A. Very long-chain fatty acids: elongation, physiology and related disorders. J Biochem. 2012;152:387–95. doi: 10.1093/jb/mvs105. [DOI] [PubMed] [Google Scholar]
- Lassmann H. Mechanisms of neurodegeneration shared between multiple sclerosis and Alzheimer's disease. J Neural Transm. 2011;118:747–52. doi: 10.1007/s00702-011-0607-8. [DOI] [PubMed] [Google Scholar]
- Li H, Li SH, Yu ZX, Shelbourne P, Li XJ. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington's disease mice. J Neurosci. 2001;21:8473–81. doi: 10.1523/JNEUROSCI.21-21-08473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–95. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Lopez-Erauskin J, Fourcade S, Galino J, Ruiz M, Schluter A, Naudi A, et al. Antioxidants halt axonal degeneration in a mouse model of X-adrenoleukodystrophy. Ann Neurol. 2011;70:84–92. doi: 10.1002/ana.22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Erauskin J, Galino J, Bianchi P, Fourcade S, Andreu AL, Ferrer I, et al. Oxidative stress modulates mitochondrial failure and cyclophilin D function in X-linked adrenoleukodystrophy. Brain. 2012;135:3584–98. doi: 10.1093/brain/aws292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JF, Lawler AM, Watkins PA, Powers JM, Moser AB, Moser HW, et al. A mouse model for X-linked adrenoleukodystrophy. Proc Natl Acad Sci USA. 1997;94:9366–71. doi: 10.1073/pnas.94.17.9366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahad DJ, Ziabreva I, Campbell G, Lax N, White K, Hanson PS, et al. Mitochondrial changes within axons in multiple sclerosis. Brain. 2009;132:1161–74. doi: 10.1093/brain/awp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow EM, Mandelkow E. Tau in Alzheimer's disease. Trends Cell Biol. 1998;8:425–7. doi: 10.1016/s0962-8924(98)01368-3. [DOI] [PubMed] [Google Scholar]
- Martinez A, Portero-Otin M, Pamplona R, Ferrer I. Protein targets of oxidative damage in human neurodegenerative diseases with abnormal protein aggregates. Brain Pathol. 2010;20:281–97. doi: 10.1111/j.1750-3639.2009.00326.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez de Lagrán M, Altafaj X, Gallego X, Marti E, Estivill X, Sahun I, et al. Motor phenotypic alterations in TgDyrk1a transgenic mice implicate DYRK1A in Down syndrome motor dysfunction. Neurobiol Dis. 2004;15:132–42. doi: 10.1016/j.nbd.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Mastroeni R, Bensadoun JC, Charvin D, Aebischer P, Pujol A, Raoul C. Insulin-like growth factor-1 and neurotrophin-3 gene therapy prevents motor decline in an X-linked adrenoleukodystrophy mouse model. Ann Neurol. 2009;66:117–22. doi: 10.1002/ana.21677. [DOI] [PubMed] [Google Scholar]
- McSharry C. Multiple sclerosis: axonal loss linked to MS disability. Nat Rev Neurol. 2010;6:300. doi: 10.1038/nrneurol.2010.63. [DOI] [PubMed] [Google Scholar]
- Miglio G, Rosa AC, Rattazzi L, Collino M, Lombardi G, Fantozzi R. PPARgamma stimulation promotes mitochondrial biogenesis and prevents glucose deprivation-induced neuronal cell loss. Neurochem Int. 2009;55:496–504. doi: 10.1016/j.neuint.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Moser H, Smith KD, Watkins PA, Powers J, Moser AB. X-linked adrenoleukodystrophy. In: Scriver C, editor. The metabolic and molecular bases of inherited disease. Vol. II. New-York: McGraw-Hill; 2001. pp. 3257–301. [Google Scholar]
- Nicolakakis N, Aboulkassim T, Ongali B, Lecrux C, Fernandes P, Rosa-Neto P, et al. Complete rescue of cerebrovascular function in aged Alzheimer's disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J Neurosci. 2008;28:9287–96. doi: 10.1523/JNEUROSCI.3348-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddock ML, Wiley SE, Axelrod HL, Cohen AE, Roy M, Abresch EC, et al. MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc Natl Acad Sci USA. 2007;104:14342–7. doi: 10.1073/pnas.0707189104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamplona R, Dalfo E, Ayala V, Bellmunt MJ, Prat J, Ferrer I, et al. Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation. Effects of Alzheimer disease and identification of lipoxidation targets. J Biol Chem. 2005;280:21522–30. doi: 10.1074/jbc.M502255200. [DOI] [PubMed] [Google Scholar]
- Powers JM, DeCiero DP, Ito M, Moser AB, Moser HW. Adrenomyeloneuropathy: a neuropathologic review featuring its noninflammatory myelopathy. J Neuropathol Exp Neurol. 2000;59:89–102. doi: 10.1093/jnen/59.2.89. [DOI] [PubMed] [Google Scholar]
- Powers JM, Pei Z, Heinzer AK, Deering R, Moser AB, Moser HW, et al. Adreno-leukodystrophy: oxidative stress of mice and men. J Neuropathol Exp Neurol. 2005;64:1067–79. doi: 10.1097/01.jnen.0000190064.28559.a4. [DOI] [PubMed] [Google Scholar]
- Pujol A, Ferrer I, Camps C, Metzger E, Hindelang C, Callizot N, et al. Functional overlap between ABCD1 (ALD) and ABCD2 (ALDR) transporters: a therapeutic target for X-adrenoleukodystrophy. Hum Mol Genet. 2004;13:2997–3006. doi: 10.1093/hmg/ddh323. [DOI] [PubMed] [Google Scholar]
- Pujol A, Hindelang C, Callizot N, Bartsch U, Schachner M, Mandel JL. Late onset neurological phenotype of the X-ALD gene inactivation in mice: a mouse model for adrenomyeloneuropathy. Hum Mol Genet. 2002;11:499–505. doi: 10.1093/hmg/11.5.499. [DOI] [PubMed] [Google Scholar]
- Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, et al. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol. 2009;66:352–61. doi: 10.1001/archneurol.2008.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimian R, Fakhfouri G, Rasouli MR, Nouri M, Nezami BG, Paydar MJ, et al. Effect of pioglitazone on sciatic nerve ischemia/reperfusion injury in rats. Pediatr Neurosurg. 2009;45:126–31. doi: 10.1159/000209287. [DOI] [PubMed] [Google Scholar]
- Richardson DK, Kashyap S, Bajaj M, Cusi K, Mandarino SJ, Finlayson J, et al. Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J Biol Chem. 2005;280:10290–7. doi: 10.1074/jbc.M408985200. [DOI] [PubMed] [Google Scholar]
- Schluter A, Espinosa L, Fourcade S, Galino J, Lopez E, Ilieva E, et al. Functional genomic analysis unravels a metabolic-inflammatory interplay in adrenoleukodystrophy. Hum Mol Genet. 2012;21:1062–77. doi: 10.1093/hmg/ddr536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutz B, Reimann J, Dumitrescu-Ozimek L, Kappes-Horn K, Landreth GE, Schurmann B, et al. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J Neurosci. 2005;25:7805–12. doi: 10.1523/JNEUROSCI.2038-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P, Gal J, Kwinter DM, Liu X, Zhu H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim Biophys Acta. 2010;1802:45–51. doi: 10.1016/j.bbadis.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, et al. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh I, Moser AE, Moser HW, Kishimoto Y. Adrenoleukodystrophy: impaired oxidation of very long chain fatty acids in white blood cells, cultured skin fibroblasts, and amniocytes. Pediatr Res. 1984;18:286–90. doi: 10.1203/00006450-198403000-00016. [DOI] [PubMed] [Google Scholar]
- Singh I, Pujol A. Pathomechanisms underlying X-adrenoleukodystrophy: a three-hit hypothesis. Brain Pathol. 2010;20:838–44. doi: 10.1111/j.1750-3639.2010.00392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinazzola A, Invernizzi F, Carrara F, Lamantea E, Donati A, Dirocco M, et al. Clinical and molecular features of mitochondrial DNA depletion syndromes. J Inherit Metab Dis. 2009;32:143–58. doi: 10.1007/s10545-008-1038-z. [DOI] [PubMed] [Google Scholar]
- van Roermund CW, Visser WF, Ijlst L, van Cruchten A, Boek M, Kulik W, et al. The human peroxisomal ABC half transporter ALDP functions as a homodimer and accepts acyl-CoA esters. FASEB J. 2008;22:4201–8. doi: 10.1096/fj.08-110866. [DOI] [PubMed] [Google Scholar]
- Vargas CR, Wajner M, Sirtori LR, Goulart L, Chiochetta M, Coelho D, et al. Evidence that oxidative stress is increased in patients with X-linked adrenoleukodystrophy. Biochim Biophys Acta. 2004;1688:26–32. doi: 10.1016/j.bbadis.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Wanders RJ, van Roermund CW, van Wijland MJ, Nijenhuis AA, Tromp A, Schutgens RB, et al. X-linked adrenoleukodystrophy: defective peroxisomal oxidation of very long chain fatty acids but not of very long chain fatty acyl-CoA esters. Clin Chim Acta. 1987;165:321–9. doi: 10.1016/0009-8981(87)90177-x. [DOI] [PubMed] [Google Scholar]
- Wiley SE, Murphy AN, Ross SA, van der Geer P, Dixon JE. MitoNEET is an iron-containing outer mitochondrial membrane protein that regulates oxidative capacity. Proc Natl Acad Sci USA. 2007;104:5318–23. doi: 10.1073/pnas.0701078104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–24. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, et al. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci. 2003;23:7504–9. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrouk A, Vejux A, Nury T, El Hajj HI, Haddad M, Cherkaoui-Malki M, et al. Induction of mitochondrial changes associated with oxidative stress on very long chain fatty acids (C22:0, C24:0, or C26:0)-treated human neuronal cells (SK-NB-E) Oxid Med Cell Longev. 2012;2012:623257. doi: 10.1155/2012/623257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Varghese M, Yemul S, Pan Y, Cheng A, Marano P, et al. Peroxisome proliferator activator receptor gamma coactivator-1alpha (PGC-1alpha) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Mol Neurodegener. 2011;6:51. doi: 10.1186/1750-1326-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]