

Abstract
We have previously observed that in vivo exposure to growing melanoma tumors fundamentally alters activated T cell homeostasis by suppressing the ability of naïve T cells to undergo antigen-driven proliferative expansion. We hypothesized that exposure of T cells in later stages of differentiation to melanoma would have similar suppressive consequences. C57BL/6 mice were inoculated with media or syngeneic B16F10 melanoma tumors 8 or 60 days after infection with lymphocytic choriomeningitis virus (LCMV), and splenic populations of LCMV-specific T cells were quantified using flow cytometry 18 days after tumor inoculation. Inoculation with melanoma on post-infection day 8 potentiated the contraction of previously activated T cells. This enhanced contraction was associated with increased apoptotic susceptibility among T cells from tumor-bearing mice. In contrast, inoculation with melanoma on post-infection day 60 did not affect the ability of previously established memory T cells to maintain themselves in stable numbers. In addition, the ability of previously established memory T cells to respond to LCMV challenge was unaffected by melanoma. Following adoptive transfer into melanoma-bearing mice, tumor-specific memory T cells were significantly more effective at controlling melanoma growth than equivalent numbers of tumor-specific effector T cells. These observations suggest that memory T cells are uniquely resistant to suppressive influences exerted by melanoma on activated T cell homeostasis; these findings may have implications for T cell–based cancer immunotherapy.
Keywords: Cancer, T cell, Melanoma, LCMV, Memory, Immunotherapy
Introduction
Following antigenic stimulation, naïve resting CD8+ T cells undergo profound activation-driven proliferative expansion. After clonal expansion, activated T cells enter a phase of rapid contraction, during which the vast majority of short-lived effector T cells undergo apoptotic cell death. A small subset of memory precursor effector T cells (MPEC) survives contraction and eventually differentiates into memory T cells, which are capable of prolonged homeostatic proliferation and rapid expansion in response to repeat exposure to their cognate antigen [1–6]. The ability to identify and expand large populations of activated CD8+ T cells specific for cancer antigens has motivated great interest in the use of adoptive cell transfer as a novel means of treating cancer [7–14]. Following adoptive transfer, cancer-specific T cells must overcome an array of immunosuppressive obstacles in order for them to exert their therapeutic effect. In addition to being capable of evading immunological detection, growing tumors eventually elaborate a number of immunosuppressive mechanisms that can neutralize the efficacy of cancer-specific T cells [15–17].
Our laboratory has also demonstrated that in vivo interaction with cancer impairs the ability of naïve T cells to undergo maximal antigen-driven proliferative expansion. This effect appears to be mediated by increased susceptibility of activated T cells to apoptotic cell death [18]. The magnitude of this suppression is proportional to the extent of tumor burden; moreover, this influence is exerted in a globally non-specific manner that affects tumor antigen-specific T cells to the same degree as non-tumor antigen-specific T cells [19]. These observations indicate that the presence of cancer fundamentally alters the homeostasis of newly activated T cells; indeed, we have found that the ability of mice to generate new MPEC is markedly impaired in the presence of melanoma tumors [18]. Another implication of these findings is that previous exposure to tumor may negatively impact the ability of expand sufficient numbers of tumor-specific T cells that would be needed for successful adoptive immunotherapy.
In this report, we tested the influence that in vivo exposure to cancer has on T cells in other states of differentiation. We previously observed that exposure of naïve T cells to melanoma suppresses their ability to undergo antigen-driven proliferative expansion due to upregulated apoptosis; we therefore hypothesized that exposure of acutely activated effector T cells to melanoma would potentiate apoptotic contraction. We further hypothesized that in vivo exposure of previously established memory T cells to melanoma would impair their ability to undergo both homeostatic proliferation in the absence of antigen stimulation and proliferative expansion in response to antigen challenge.
Materials and methods
Mice
Seven- to eight-week-old female C57BL/6 mice were purchased from Taconic (Hudson, NY) and maintained in pathogen-free conditions. All animal work was performed in strict accordance with the guidelines of the University of Wisconsin and William S. Middleton Memorial VA Hospital Animal Care and Use Committees.
Tumor cell lines and virus
B16F10, a poorly immunogenic melanoma cell line derived from C57BL/6 mice, was maintained in RPMI-1640 medium (Mediatech, Herndon, VA) supplemented with 100U/mL penicillin, 100 μg/mL streptomycin, and 2 mM l-glutamine (Life Technologies, Inc., Grand Island, NY) and 10 % fetal bovine serum (HyClone Laboratories, South Logan, UT). A single inoculum of 106 B16F10 cells suspended in serum-free RPMI1640 media was injected subcutaneously into C57BL/6 mice and tumors were measured using electronic calipers every 3 days. Mice were infected with 2x105 PFU Armstrong strain lymphocytic choriomeningitis virus (LCMV) by intraperitoneal injection. To test LCMV-specific memory T cell responses, LCMV-immune mice were infected with 106 PFU clone 13 strain LCMV by intravenous injection. Mice were also inoculated with 106 B16GP33 cells, previously generated in our laboratory by stable transfecting B16F10 cells to express low levels of the LCMV epitope peptide GP33 [19].
Apoptosis assay
Isolated splenocytes (106 cells/well) were incubated in flat-bottom 96-well plates at 37 °C in RPMI with 10 % FBS for 5 h. Cell surface staining with anti-CD8-PerCP, anti-CD44-PE, and APC conjugated MHC class I tetramers loaded with LCMV epitope peptides (NP396, GP33, or GP276) was followed by permeabilization with Cytofix/Cytoperm solution (BD Biosciences) and intracellular staining with anti-caspase 3-FITC. All antibodies were purchased from BD Biosciences Pharmingen (San Diego, CA).
Flow cytometry
MHC class I tetramers loaded with various LCMV antigen peptides were prepared as previously described [1]. Single-cell suspensions of splenocytes were stained with fluorophore-labeled MHC class I(Db) tetramers loaded with class I-restricted LCMV epitope peptides (NP396, GP33, or GP276), as well as anti-CD8, anti-CD62L, anti-CD44, anti-CD127, or anti-KLRG-1 antibodies. Alternatively, freshly harvested splenocytes (106 cells/well) were stimulated with or without various LCMV epitope peptides (NP396, GP33, GP34, or GP276) at a concentration of 0.1 μg/mL in the presence of brefeldin A and human recombinant IL-2 (10 U/well) at 37 °C for 5 h in flat-bottomed 96-well plates. Cells were stained with anti-CD8 and anti-CD62L antibodies, and then permeabilized and stained for intracellular cytokines using anti-IFNγ, anti-TNFα, and anti-IL-2 antibodies using the Cytofix/Cytoperm kit purchased from BD Biosciences Pharmingen. Stained cells were acquired on a FACSCalibur flow cytometer (BD Biosciences) and resulting data were analyzed using FlowJo software (Tree Star, Inc., Ashland, OR). LCMV antigen-specific T cell populations were identified either by their expression of T cell receptors specific for LCMV antigen peptides or by their ability to elaborate inflammatory cytokines in response to LCMV antigen peptide stimulation. All reagents and antibodies were purchased from BD Biosciences Pharmingen (San Diego, CA) with the exception of anti-CD127 and anti-granzyme B antibodies, which were purchased from eBioscience, Inc. (San Diego, CA), and Invitrogen, Inc. (Carlsbad, CA), respectively.
Adoptive transfer
Mice bearing B16F10 or B16GP33 melanoma tumors underwent intravenous injection of serum-free media or 104 CD8+ GP33-specific T cells of effector or memory phenotype. Effector T cells were isolated from splenocytes harvested from C57BL/6 mice 8 days after LCMV infection, and memory T cells were isolated from splenocytes harvested from C57BL/6 mice 50 days after LCMV infection. Splenocytes were enriched for CD8 expression using magnetic bead separation columns from Miltenyi (Auburn, CA) and flow cytometry was used to quantify CD8+ GP33-specific T cell populations.
Statistical analysis
Experimental data were analyzed using IBM SPSS statistical software version 19 (Armonk, NY). Groups were compared using an analysis of variance (ANOVA), and pair-wise comparisons were made using Fisher’s protected least significant difference tests and Tukey’s adjustment. All data were log transformed prior to analysis in order to better meet the assumptions of ANOVA. All p values reported are two sided, and significance was defined as p < 0.05. All error bars in graphical representations of data indicate standard errors of the mean.
Results
In vivo exposure to melanoma potentiates apoptotic contraction of acutely activated T cells
CD8+ T cell responses to acute LCMV infection occur in three distinct phases: an expansion phase (occurring on days 0–8 post-infection), a contraction phase (days 8–30 post-infection), and a memory phase (>30 days post-infection). To assess the influence of melanoma on contraction of LCMV-specific effector CD8+ T cells, mice were inoculated with media or B16F10 melanoma 8 days after LCMV infection, which corresponds to the onset of T cell contraction. In this way, equivalent numbers of LCMV-specific T cells were present in all mice at the moment of peak T cell expansion and melanoma inoculation. Splenocyte analysis was performed 18 days after tumor inoculation, which corresponds to the same duration of tumor growth tested in our previous analysis of naïve T cells [18]. LCMV-specific T cells were identified and quantified based on staining with MHC tetramers loaded with LCMV epitope peptides. At 18 days after tumor inoculation, significantly smaller numbers of LCMV-specific T cells as detected by CD8 expression and staining positivity for MHC tetramers loaded with LCMV epitope peptides were observed in tumor-bearing mice compared with controls (Fig. 1a, b). Comparison of effector activation status as measured by CD44high and CD62Llow expression identified no differences among LCMV-specific T cell populations between controls and tumor-bearing mice (data not shown). To determine if the reduced number of LCMV-specific CD8+ T cells might be due to enhanced apoptosis, expression of activated caspase-3 was measured after 5 h of in vitro culture. The proportions of caspase-3-expressing cells were greater among LCMV-specific CD8+ T cells from tumor-bearing mice as compared with controls (Fig. 2). Although slightly smaller percentages of LCMV-specific T cells bore memory precursor phenotype (CD127high/KLRGlow) in tumor-bearing mice compared with controls, the predominant difference was in the absolute numbers of LCMV-specific T cells present following contraction (Fig. 3).
Fig. 1.

Contraction of activated T cells is potentiated in the presence of melanoma. C57BL/6 mice were infected with LCMV then inoculated with media (n = 4) or B16F10 melanoma (n = 4) on post-infection day 8, at which time expansion of LCMV-specific CD8+ T cells reaches its peak and LCMV is cleared. Splenocytes were harvested 18 days later on post-infection day 26, at which time contraction of LCMV-specific T cells has occured. LCMV-specific CD8+ T cells were identified by staining cells with fluorophore-labeled antibodies against CD8 and fluorophore-labeled MHC tetramers loaded with MHC class I-restricted LCMV epitope peptides, and then quantified by flow cytometric analysis. a Representative dot plots demonstrate smaller percentages of CD8+ T cells specific for the immunodominant LCMV epitope peptide NP396 (gated on lymphocytes) among splenocytes harvested from melanoma-bearing mice compared with control mice. b Histograms of absolute numbers of LCMV-specific CD8+ T cells per spleen (calculated as the percentage of cells by flow cytometry multiplied by the total number of splenocytes) demonstrate smaller populations of LCMV-specific CD8+ T cells for all LCMV epitope peptides tested. This experiment was performed four times. (*p < 0.05)
Fig. 2.

Potentiated contraction in the presence of melanoma is associated with increased susceptibility of activated T cells to apoptotic cell death. C57BL/6 mice were infected with LCMV, and then inoculated with media (n = 4) or B16F10 melanoma (n = 4) on post-infection day 8. Splenocytes were harvested 18 days later and cultured in full media in the absence of stimulation for 5 h to promote apoptotic cell death. Cells were stained with fluorophore-labeled antibodies against CD8 and activated caspase-3 and fluorophore-labeled MHC tetramers loaded with NP396, and then quantified by flow cytometric analysis. Representative zebra plots (a) and histograms of mean data (b) demonstrate higher relative expression of activated caspase-3high expression (gated on CD8+ T cells specific for NP396) among splenocytes harvested from melanoma-bearing mice compared with controls. This experiment was performed three times. (*p < 0.05)
Fig. 3.

Potentiated contraction in the presence of melanoma results in marked diminution in the size of memory T cell populations. C57BL/6 mice were infected with LCMV, and then inoculated with media (n = 4) or B16F10 melanoma (n = 4) on post-infection day 8. Splenocytes were harvested 18 days later and splenocytes were stained with fluorophore-labeled antibodies against CD8, KLRG1, CD127, and fluorophore-labeled MHC tetramers loaded with MHC class I-restricted LCMV epitope peptides, and then quantified by flow cytometric analysis. a Representative zebra plots demonstrate smaller percentages of KLRG1low and CD127high memory phenotype (gated on CD8+ T cells specific for NP396) among splenocytes harvested from melanoma-bearing mice compared with control mice (gated on lymphocytes). b Histograms of absolute numbers of LCMV-specific CD8+ memory T cells per spleen demonstrate smaller populations of LCMV-specific CD8+ memory T cells for all LCMV epitope peptides tested. This experiment was performed three times. (*p < 0.05)
In vivo exposure to melanoma does not impair the persistence of previously established memory T cells
To assess the effect of melanoma on preexisting CD8+ memory T cells, mice were inoculated with media or B16F10 melanoma 60 days after LCMV infection. Splenocyte analysis was again performed 18 days after tumor inoculation, and LCMV-specific T cells were identified and quantified based on staining with MHC tetramers loaded with LCMV epitope peptides. No significant differences were observed in the numbers of LCMV-specific T cells present in tumor-bearing mice compared with controls (Fig. 4). In order to verify that LCMV-specific T cells exhibit a functional phenotype consistent with memory T cells at this time point, explanted splenocytes were stimulated for 4 h in vitro in the presence of LCMV epitope peptides, after which intracellular levels of IL-2 were measured. Memory T cells are unique in their ability to elaborate relatively high levels of IL-2 in response to antigen stimulation; although LCMV-specific T cells harvested from tumor-bearing and control mice were both capable of relatively high levels of IL-2 production in response to antigenic stimulation, levels of IL-2 expression were not significantly different between groups. Similarly, no differences in expression of activated caspase-3 were observed after 5 h in vitro culture among LCMV-specific T cell populations harvested from tumor-bearing mice compared with controls (Fig. 5).
Fig. 4.
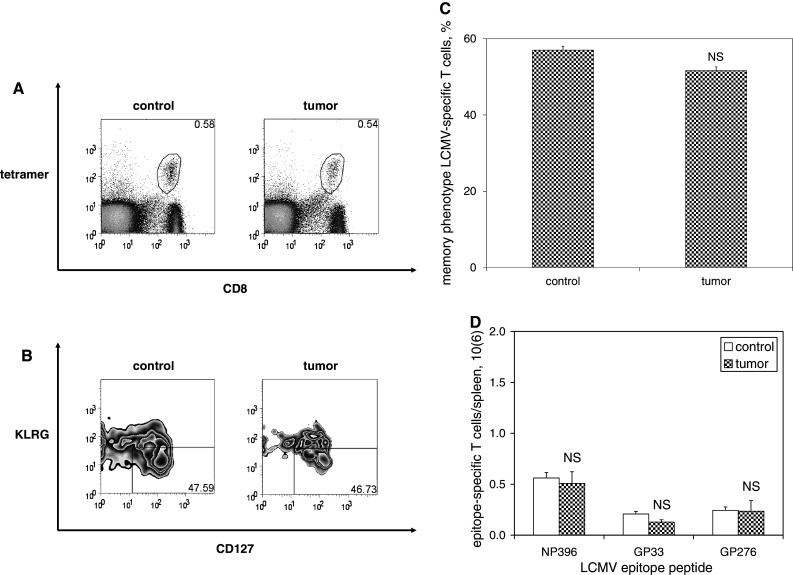
Maintenance of memory T cells is unaffected by the presence of melanoma. C57BL6/mice were infected with LCMV, and then inoculated with media (n = 4) or B16F10 melanoma (n = 4) on post-infection day 60. Splenocytes were harvested 18 days later and splenocytes were stained with fluorophore-labeled antibodies against CD8, KLRG1, CD127, and fluorophore-labeled MHC tetramers loaded with MHC class I-restricted LCMV epitope peptides, and then quantified by flow cytometric analysis. a Representative dot plots demonstrate similar percentages of NP396-specific CD8+ T cells among melanoma-bearing and control mice. b Representative zebra plots demonstrate similar percentages of KLRG1low and CD127high memory phenotype (gated on CD8+ T cells specific for NP396) among splenocytes harvested from control and melanoma-bearing mice (gated on lymphocytes). c Histograms of absolute numbers of LCMV-specific CD8+ memory T cells per spleen demonstrate similar populations of LCMV-specific CD8+ memory T cells for all LCMV epitope peptides tested. This experiment was performed three times. (NS p ≥ 0.05)
Fig. 5.

Functional capacity and apoptotic susceptibility of memory T cells are unaffected by the presence of melanoma. Single-cell suspensions were prepared from spleens harvested from mice as described in Fig. 4. a Splenocytes were stimulated with or without the immunodominant LCMV epitope peptide NP396 (0.1 μg/mL) in the presence of brefeldin A and IL-2 at 37 °C for 5 h, and then analyzed by flow cytometry. Representative zebra plots demonstrate similar levels of IL-2 expression (gated on CD8+ NP396-specific IFNγ-producing T cells) in control and melanoma-bearing mice. b Alternatively, splenocytes were cultured in full media in the absence of stimulation for 5 h to promote apoptotic cell death. Cells were stained with fluorophore-labeled antibodies against CD8 and activated caspase-3 and fluorophore-labeled MHC tetramers loaded with NP396, and then quantified by flow cytometric analysis. Histograms of mean data demonstrate similar relative expression levels of activated caspase-3high expression (gated on CD8+ T cells specific for NP396) among splenocytes harvested from control and melanoma-bearing mice. This experiment was performed three times. (NS p ≥ 0.05)
In vivo exposure to melanoma does not impair the ability of previously established memory T cells to undergo antigen challenge-driven proliferative re-expansion
Mice were inoculated with media or B16F10 melanoma 60 days after LCMV infection, and then re-infected with clone 13 strain LCMV 10 days after tumor inoculation. Splenocyte analysis was performed 5 days after challenge infection, and LCMV-specific T cells were identified and quantified based on their ability to elaborate IFNγ in response to 4 h in vitro stimulation with LCMV epitope peptides. No significant differences were observed in the numbers of LCMV-specific T cells expanded in response to LCMV challenge in tumor-bearing mice compared with controls (Fig. 6). To address the functional competence of activated T cells expanded in response to antigenic challenge, intracellular levels of granzyme B expression were measured; no differences in granzyme B expression were observed among activated LCMV-specific T cells in tumor-bearing mice compared with controls (Fig. 7).
Fig. 6.

The ability of memory T cells to expand in response to antigen challenge is unaffected by the presence of melanoma. C57BL/6 mice were infected with LCMV, and then inoculated with media (n = 4) or B16F10 (n = 4) on post-infection day 60. Mice were infected with clone 13 LCMV 10 days later, and splenocytes were harvested 5 days after LCMV re-infection (at the time of peak re-expansion of LCMV-specific T cells). Splenocytes were stimulated with or without various LCMV epitope peptides in the presence of brefeldin A and IL-2 at 37 °C for 5 h, and then analyzed by flow cytometry. a Representative dot plots demonstrate similar percentages of CD8+ T cells expressing IFNγ in response to stimulation with the immunodominant LCMV epitope peptide NP396 (gated on lymphocytes) among splenocytes harvested from control and melanoma-bearing mice. b Histograms of absolute numbers of LCMV-specific CD8+ T cells per spleen (calculated as the percentage of cells by flow cytometry multiplied by the total number of splenocytes) demonstrate similar populations of CD8+ T cells expressing IFNγ in response to stimulation with all LCMV epitope peptides tested. This experiment was performed two times. (NS p ≥ 0.05)
Fig. 7.

Granzyme B expression levels among effector T cells expanded by LCMV-immune mice in response to LCMV challenge are unaffected by the presence of melanoma. Splenocytes harvested from mice described in Fig. 6 were stained with fluorophore-labeled antibodies against CD8 and granzyme B (or isotype controls) and fluorophore-labeled MHC tetramers loaded with the immunodominant LCMV epitope peptide NP396 and analyzed by flow cytometry. Representative histograms (a) and histograms of mean data (b) demonstrate comparable levels of upregulated granzyme B expression among expanded CD8+ T cells from control and melanoma-bearing LCMV-immune mice. (*p < 0.05)
Memory T cells are more effective than effector T cells in an experimental model of melanoma adoptive immunotherapy
Splenocytes were harvested from mice either 8 or 50 days after LCMV infection, at which time LCMV-specific CD8+ T cells were exclusively of effector or memory phenotype, respectively. Harvested splenocytes were enriched for CD8 expression using magnetic bead separation, and representative aliquots were analyzed for specificity against GP33 by flow cytometric analysis of staining with MHC tetramers loaded with GP33 peptide. Expression levels of CD127 and KLRG-1 were used to verify effector (CD127low/KLRGhigh) and memory (CD127low/KLRGhigh) phenotype (data not shown). One day after inoculation with either B16F10 or B16GP33 melanoma, a separate group of mice received intravenous injections of serum-free media (control) or 104 CD8+ GP33-specific effector or memory T cells, and B16GP33 melanoma tumor growth was assessed by serial tumor measurements. B16F10 melanoma tumor growth was comparable in all three groups (data not shown). B16GP33 melanoma growth was significantly impaired in mice treated with adoptive transfer of GP33-specific T cells; comparison of tumor growth curves in mice treated with adoptive T cell transfer identified significantly more potent suppression of B16GP33 melanoma growth among mice treated with memory T cells (Fig. 8).
Fig. 8.

Melanoma-specific memory T cells are more effective than effector T cells at controlling melanoma tumor growth following adoptive transfer. 104 CD8+ GP33-specific T cells harvested from mice 8 or 50 days after LCMV infection were adoptively transferred into mice 1 day after inoculation with B16GP33 melanoma. Control mice received intravenous injections of serum-free media alone in place of adoptive transfer immunotherapy. a Tumor growth curves demonstrate significant impairment of B16GP33 melanoma tumor growth in mice receiving effector and memory T cell immunotherapy as compared with controls. Comparison of B16GP33 melanoma tumor growth curves following immunotherapy demonstrates significantly greater tumor control with the use of melanoma-specific memory T cells as compared with equal numbers of melanoma-specific effector T cells. (*p < 0.05 compared with controls; † p < 0.05 compared with effector T cells)
Discussion
Ample evidence indicates that tumor antigen-specific T cells must overcome a number of physiological barriers in order for them to be able to exert any potential therapeutic benefit. Downregulated tumoral expression of MHC class I molecules and antigenic variation resulting from high frequency somatic mutations allow tumors to elude T cell recognition, and active immunosuppressive mechanisms elaborated by growing tumors further thwart the ability of T cells to initiate a meaningful immune response [15–17]. Adoptive immunotherapy has been proposed as a means of overcoming these barriers by repopulating the host T cell repertoire with sufficiently large populations of tumor-specific T cells harvested from tumor-infiltrating lymphocytes (TILs) [7–14].
In response to antigen encounter, resting CD8+ T cells are stimulated to undergo rapid proliferative expansion. Following expansion, acutely activated T cell populations enter a phase of programmed contraction, during which the vast majority of antigen-specific short-lived effector T cells undergo apoptotic cell death. A small subset of memory precursor effector T cells survive contraction and eventually differentiate into memory T cells capable of long-term survival, even in the absence of antigenic stimulation [1–6]. We have previously found that a critical window of in vivo interaction exists between resting T cells and growing tumors, after which resting T cells are no longer capable of maximal proliferative expansion in response to antigenic stimulation. Importantly, this suppression is observed even when the in vivo interaction is halted, either by resecting tumor or by taking resting T cells out of the tumor environment and transferring them into a tumor-free environment [18]. An implication of this suppression is that tumor-specific T cells harvested from TILs may be ill-equipped to undergo the extent of proliferative expansion needed to therapeutically engage and eliminate tumor antigen. In the present analysis, we sought to expand on our previous observations by examining the in vivo influence exerted by growing tumors on T cells in later phases of differentiation: acutely activated T cells and memory T cells.
We hypothesized that in vivo exposure of previously activated T cells to melanoma would potentiate the contraction phase of T cell homeostasis. To test this hypothesis, mice were inoculated with melanoma tumors 8 days after LCMV infection (at which point activated LCMV-specific T cells have undergone maximal expansion and LCMV has been cleared). Indeed, in vivo exposure of activated T cells to melanoma ultimately resulted in significantly smaller populations of LCMV-specific T cells after contraction had taken place (26 days after LCMV infection). As we had previously observed among activated T cells undergoing proliferative expansion [18], the effect of melanoma appeared to impact the size of T cell pools following contraction and not the activation status of individual T cells. LCMV-specific T cells harvested from melanoma-bearing mice were much more susceptible to apoptotic cell death than comparable T cells harvested from control mice. We previously observed accelerated apoptosis among activated LCMV-specific T cells in melanoma-bearing mice during proliferative expansion [18], suggesting that in vivo exposure to melanoma broadly and non-specifically upregulates the apoptotic susceptibility of acutely activated T cells.
By inhibiting expansion and augmenting contraction of T cells following antigen encounter, the presence of melanoma can be expected to have long-term consequences on later phases of activated T cell homeostasis. The size of a memory T cell population is a function of the clonal burst size of T cells present at the moment of maximal proliferative expansion and the rate of apoptotic contraction that takes place thereafter; by negatively impacting both of these variables, the suppressive influence of melanoma would be expected to markedly impair the generation of new T cell memory [3–6]. We previously observed significantly smaller numbers of LCMV-specific MPECs 10 days after viral infection; in the present analysis, we confirmed that there are markedly smaller numbers of LCMV-specific memory T cells present 26 days after viral infection [18]. Although these findings are most applicable to the generalized ability of cancer patients to establish new T cell memory to novel antigens, another possible implication of this observation is that the ability of adoptive immunotherapy to promote durable tumor immunity through the induction of tumor-specific memory T cells may be undercut by the presence of tumor.
We hypothesized that melanoma would also exert a non-specific suppressive influence on the maintenance of previously established memory T cells. In contrast to acute effector T cells, memory T cell populations are capable of prolonged persistence in the absence of antigenic stimulation through a process of homeostatic proliferation that is largely driven by in vivo exposure to IL-7 and IL-15 [20–22]. When melanoma tumors were inoculated into mice 60 days after LCMV infection, at which time remaining LCMV-specific T cells are of memory phenotype, populations of LCMV-specific memory T cells after 18 days of tumor growth (the same interval used in our previous experiments examining resting and acutely activated T cells) were no different than in tumor-free control mice. No differences in apoptotic susceptibility were seen between LCMV-specific memory T cells from control versus tumor-bearing mice. Moreover, the ability of mice to re-expand acutely activated populations of LCMV-specific T cells in response to challenge infection was unaffected by melanoma. These observations suggest that, once established, memory T cells may be uniquely resistant to the suppressive influences exerted by melanoma tumors.
We explored a potential clinical implication of these findings: the possibility that tumor-specific T cells of memory phenotype may represent an ideal subset for use in clinical adoptive immunotherapy. Recent analyses of resected melanoma, renal cell, and colon carcinomas have found the presence of memory T cells within TILs to be associated with favorable prognostic outcome measures [23–25], highlighting a potential oncological advantage for tumor-specific memory T cells. Indeed, it has been speculated that adoptive transfer of memory T cells may be more effective than acute effector T cells in promoting tumor clearance [26]. A recent investigation using a murine model of adoptive cell transfer immunotherapy identified no obvious advantage to the use of memory T cells over effector T cells [27]. Importantly, this model employed aggressive in vitro stimulation of effector and memory T cell populations prior to adoptive transfer; consequently, it is possible that this preparative regimen effectively enriched homogenous populations of acutely activated effector T cells in both experimental groups, diluting any potential therapeutic benefit that memory T cells may inherently offer. We previously described the selection of B16F10 melanoma cell line clones stably transfected to express very low and poorly immunogenic levels of the LCMV epitope peptide GP33 (B16GP33) [19]. By rendering GP33 a novel tumor antigen and harvesting effector or memory phenotype GP33-specific CD8+ T cells from mice 8 or >30 days after LCMV infection, respectively, our model enables us to precisely control the phenotype of adoptively transferred tumor antigen-specific T cells. When equivalent numbers of GP33-specific CD8+ T cells were adoptively transferred, memory T cells were far more effective than effector T cells at controlling B16GP33 tumor growth. Interestingly, the kinetics of B16GP33 tumor growth were essentially superimposable for the first 8 days after adoptive transfer of acute and memory T cells, after which tumors progressed in mice treated with effector T cells but regressed in mice treated with memory T cells. These preliminary data verify a therapeutic advantage for memory T cell–based immunotherapy. This advantage may reflect the enhanced durability of memory T cells as compared with short-lived effector T cells in tumor-bearing mice; this possibility will require closer analysis.
In summary, our observations indicate that in vivo exposure to melanoma alters activated T cell homeostasis by blunting the transition of resting to acutely activated T cells and weakening the transition of acutely activated T cells to memory T cells, but appears to exert no effect on the maintenance and activation of previously established memory T cells. Preliminary exploration of the preclinical implications of this finding suggests that this physiological advantage of memory T cells may make them optimal for use in adoptive immunotherapy. Ongoing investigations in our laboratory will examine how these observations may be applied to the design and implementation of novel adoptive immunotherapeutic strategies for cancer.
Acknowledgments
This work was supported by grant support from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Science Research and Development Service, Career Development Award (CDA-2), American College of Surgeons Faculty Research Fellowship, and Central Surgical Association Foundation Grant to CSC, and by support from NIH Grant AI48785 to MS.
Conflicts of interest
The authors declare they have no conflict of interest; the contents of this work do not represent the views of the Department of Veterans Affairs or the United States Government.
References
- 1.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, Ahmed R. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/S1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 2.Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2:251–262. doi: 10.1038/nri778. [DOI] [PubMed] [Google Scholar]
- 3.Wherry EJ, Teichgräber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–234. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 4.Sarkar S, Teichgräber V, Kalia V, Polley A, Masopust D, Harrington LE, Ahmed R, Wherry EJ. Strength of stimulus and clonal competition impact the rate of memory CD8 T cell differentiation. J Immunol. 2007;179:6704–6714. doi: 10.4049/jimmunol.179.10.6704. [DOI] [PubMed] [Google Scholar]
- 5.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh A, Jatzek A, Plisch EH, Srinivasan R, Svaren J, Suresh M. Regulation of memory CD8 T-cell differentiation by cyclin-dependent kinase inhibitor p27Kip1. Mol Cell Biol. 2010;30:5145–5159. doi: 10.1128/MCB.01045-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg SA, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol. 2009;21:233–240. doi: 10.1016/j.coi.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudley ME, Gross CA, Langhan MM, Garcia MR, Sherry RM, Yang JC, Phan GQ, Kammula US, Hughes MS, Citrin DE, Restifo NP, Wunderlich JR, Prieto PA, Hong JJ, Langan RC, Zlott DA, Morton KE, White DE, Laurencot CM, Rosenberg SA. CD8+ enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin Cancer Res. 2010;16:6122–6131. doi: 10.1158/1078-0432.CCR-10-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garbe C, Eigentler TK, Keilholz U, Hauschild A, Kirkwood JM. Systematic review of medical treatment in melanoma: current status and future prospects. Oncologist. 2011;16:5–24. doi: 10.1634/theoncologist.2010-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dudley ME, Rosenberg SA. Adoptive cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3:666–675. doi: 10.1038/nrc1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred cells. Proc Natl Acad Sci USA. 2002;99:16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 16.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and progression. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 17.Quezada SA, Peggs KS, Simpson TR, Allison JR. Shifting the equilibrium in cancer immunoediting: from tumor tolerance to eradication. Immunol Rev. 2011;241:104–118. doi: 10.1111/j.1600-065X.2011.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russ AJ, Wentworth L, Xu K, Rakhmilevich A, Seroogy CM, Sondel PM, Suresh M, Cho CS. Suppression of T-cell expansion by melanoma is exerted on resting cells. Ann Surg Oncol. 2011;18:3848–3857. doi: 10.1245/s10434-011-1667-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russ AJ, Xu K, Wentworth L, Alam S, Meyers JV, Macklin MD, Rakhmilevich AL, Suresh M, Cho CS. Melanoma-induced suppression of tumor antigen-specific T cell expansion is comparable to suppression of global T cell expansion. Cell Immunol. 2011;271:104–109. doi: 10.1016/j.cellimm.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan JT, Ernst B, Kieper WC, LeRoy E, Sprent J, Surh CD. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J Exp Med. 2002;195:1523–1532. doi: 10.1084/jem.20020066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Surh CD, Bowman O, Purton JF, Sprent J. Homeostasis of memory T cells. Immunol Rev. 2006;211:154–163. doi: 10.1111/j.0105-2896.2006.00401.x. [DOI] [PubMed] [Google Scholar]
- 22.Nanjappa SG, Walent JH, Morre M, Suresh M. Effects of IL-7 on memory CD8 T cell homeostasis are influenced by the timing of therapy in mice. J Clin Invest. 2008;118:1027–1039. doi: 10.1172/JCI32020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou J, Nagarkatti P, Zhong Y, Nagarkatti M. Characterization of T-cell memory phenotype after in vitro expansion of tumor-infiltrating lymphocytes from melanoma patients. Anticancer Res. 2011;31:4099–4109. [PMC free article] [PubMed] [Google Scholar]
- 24.Hotta K, Sho M, Fujimoto K, Shimada K, Yamato I, Anai S, Konishi N, Hirao Y, Nonomura K, Nakajima Y. Prognostic significance of CD45RO+ memory T cells in renal cell carcinoma. Br J Cancer. 2011;105:1191–1196. doi: 10.1038/bjc.2011.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peng RQ, Wu XJ, Ding Y, Li CY, Yu XJ, Zhang X, Pan ZZ, Wan DS, Zheng LM, Zeng YX, Zhang XS. Co-expression of nuclear and cytoplasmic HMGB1 is inversely associated with infiltration of CD45RO+ T cells and prognosis in patients with stage IIIB colon cancer. BMC Cancer. 2010;10:496. doi: 10.1186/1471-2407-10-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perret R, Ronchese F. Memory T cells in cancer immunotherapy: which CD8 T-cell population provides the best protection against tumours? Tissue Antigens. 2008;72:187–194. doi: 10.1111/j.1399-0039.2008.01088.x. [DOI] [PubMed] [Google Scholar]
- 27.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, Waldhamm TA, Restifo NP. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci USA. 2005;102:9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]