

Abstract
Essential tremor (ET) is the most prevalent movement disorder affecting millions of people in the United States. Although a positive family history is one of the most important risk factors for ET, the genetic causes of ET remain unknown. In this study, whole exome sequencing and subsequent approaches were performed in a family with an autosomal dominant form of early-onset ET. Functional analyses including mutagenesis, cell culture, gene expression, enzyme-linked immunosorbent, and apoptosis assays were also performed. A disease-segregating mutation (p.Gly171Ala), absent in normal population, was identified in the SORT1 gene. The p.Gly171Ala mutation was shown not only to impair the expression of its encoding protein sortilin but also the mRNA levels of its binding partner p75 neurotrophin receptor that is known to be implicated in brain injury, neuronal apoptosis, and neurotransmission.
Keywords: SORT1 mutation, tremor, sortilin downregulation, p75NTR upregulation
Introduction
Essential tremor (ET) is one of the most common neurological disorders among adults whose incidence increases with age. Albeit its core motor symptom is an 8- to 12-Hz kinetic tremor of the arms, some patients with ET also develop other motor and nonmotor manifestations, including parkinsonism, myoclonus, dystonia, cerebellar dysfunction, sensory abnormalities, sleep disorders, and cognitive and psychiatric features (Louis, 2010). Despite many magnetic resonance imaging (MRI) studies have supported the hypothesis that the abnormalities of the cerebellothalamo-cortical motor pathway and the fronto-parietal circuit are involved in the functional pathological changes of ET (Bagepally et al., 2012), there is still a controversy as to whether there is an underlying neurodegenerative process of the cerebellum in ET (Buijink et al., 2013; Symanski et al., 2014). Consequently, there is growing evidence that ET may not be a single disease but rather a family of diseases (Benito-Leon, 2014).
Although familial aggregation has been long reported in ET, with 50% to 70% of patients having a familial form of ET, and a positive family history being one of the most important risk factor for ET (Louis et al., 2001), the genetic basis of ET remain elusive. This is partly due to misdiagnosis is a common feature in ET, with 37% to 50% of ET patients reported to be misdiagnosed (Schrag et al., 2000). Genetic variants within LINGO1 (MIM #609791), DRD3 (MIM #126451), and HS1-BP3 (MIM #609359) genes have been reported to have susceptibility for developing ET; however, these associations have not been consistently replicated (Zimprich, 2011; Kuhlenbaumer et al., 2014). Through genome-wide association and whole exome sequencing (WES) analyses, SLC1A2 (MIM #600300) and FUS (MIM #137070) genes have been reported to respectively carry susceptibility or causative alleles for ET (Merner et al., 2012; Thier et al., 2012). While the disease-associated mutation identified in the FUS gene was only present in 54% of individuals classified as “possible” ET, it fully segregated with disease in individuals with definite and probable ET (Merner et al., 2012). More recently, a disease-segregating mutation (p.G399S) in the HTRA2 gene has been reported in a large kindred featuring both ET and Parkinson’s disease (Unal Gulsuner et al., 2014). Taken together, these clinical barriers associated with ET make traditional gene discovery approaches not succeed in the identification of the causal gene defect. Therefore, in an attempt to identify a causative gene for ET, we performed WES and subsequent functional analyses in a family featuring an autosomal dominant form of early-onset ET.
Materials and Methods
Subjects
A Spanish ET family consisting of an affected father, an unaffected paternal aunt, a healthy mother as well as one affected and two unaffected siblings was clinically examined and subject to WES approaches (Figure 1(a)). Subjects were recruited from a descriptive study of familiar and sporadic ET cases and controls carried out in the Movement Disorders Unit at the Donostia University Hospital (San Sebastian, Spain). Genomic DNA samples from all family members in addition to samples from another 28 familial and 62 sporadic ET cases (n = 90) were available for study. A cohort composed of 188 control chromosomes of Spanish individuals without family history of any movement disorders was also available. The age at sample collection of the control cohort ranged from 60 to 93 years with an average of 69.1 years. The local ethics committee at the Donostia University Hospital approved this study and informed consent was obtained from all participants. DNA samples from all participating members were isolated from whole blood using standard procedures. All methods were carried out in accordance with the approved guidelines.
Figure 1.

Pedigree structure of a Spanish family with ET identified with a pathogenic SORT1 mutation. (a): Definitive affected cases are represented with black filled square (male) and circle (female) and a dot inside a square represents an individual male at risk. Wt/Mut indicates heterozygous carrier for the mutant allele while Wt/Wt indicates noncarrier. (b1): Sanger chromatograms of the human reference sequence (bottom) and heterozygous mutant sequence (top) with a red arrow highlighting the pathogenic mutation. (b2): The conservation of the p.G171A mutation among other orthologous is shown. HS: H. Sapiens; MM: M. Mulatta; CL: C. Lupus; BT: B. Taurus; Mm: M. musculus; RN: R. Norvegicus; DR: D. Rerio; XT: X. Tropicalis. 1(c): Diagram of sortilin structure. All protein domains are shown. TM: Transmembrane domain.
Clinical Examination
All participants in this study underwent a series of structured questionnaires and a comprehensive neurological and neuropsychological assessment undertaken at the Movement Disorders Unit by three experienced movement disorder specialist (JFM-M, JRM, and AB). The following standardized protocol was used: (a) demographic variables; (b) personal and family history and general medical health (Cumulative Illness Rating Scale score [range: 0–42], total number of prescription medications); (c) neurological assessment: a subjective motor complains questionnaire, the Activities-specific Balance Confidence scale, a motor examination including UPDRS part III and a general examination in order to detect dystonia, myoclonus, ataxia, and polyneuropathy as well as a specific tremor exploration that includes one test for postural tremor and five for kinetic tremor; (d) the use of medication (yes vs. no); (e) additional variables of interest (e.g., age of symptom onset); (f) the SCOPA-AUT assessment of autonomic dysfunction (Visser et al., 2004); and (g) the Pittsburgh sleep quality index (Buysse et al., 1989). The Montreal Cognitive Assessment (MoCA) that assesses different cognitive domains (http://www.mocatest.org/) was used to determine possible cognitive dysfunction and DSM-IV criteria were used for diagnosis of depression and anxiety disorders (American Psychiatric Association, 1994). In addition to the standard clinical exploration described above, evaluation of ET was carried out by recording the drawing of an “Archimedes spiral” (Elble et al., 1990) and using the Fahn–Tolosa–Marin tremor rating scale (TRS; Fahn et al 2003). Surface electromyography (EMG) was recorded from wrist extensor and flexor muscles using surface electrodes placed over the muscle bellies 3 cm apart. The filters were set with a bandpass of 10 Hz to 1 kHz. A triaxial accelerometer was placed over the first dorsal interossei muscle of the hand. In an effort to minimize diagnostic pitfalls, clinical criteria were comprehensively reviewed by the Consensus Statement of the Movement Disorder Society on Tremor (Deuschl et al., 1998) and the Washington Heights–Inwood Genetic Study of Essential Tremor criteria (Louis et al., 1997; Benito-Leon and Louis, 2006). Each patient received a diagnosis of ET from a movement disorders neurologist after the first evaluation which subsequently was confirmed by consensus with the rest of the team based on the review of the clinical data and electrophysiological records from the second evaluation using the formerly described diagnostic criteria. Criteria for definitive ET included abnormal bilateral postural or kinetic tremor of the hands in the absence of other neurological symptoms.
WES Approaches
Four DNA samples from two affected (Cases 1 and 3) and two unaffected cases (Cases 2 and 4) were subject to WES analyses (Figure 1(a)). The SureSelect Human All exon 50 Mb exon-capture kit was used for library enrichment (Agilent Technologies Inc., Santa Clara, CA) and captured libraries were sequenced on the HiSeq2000 according to the manufacturer’s instructions for paired-end 100-bp reads (Illumina Inc, San Diego, CA), using a single flow cell lane per sample. Sequencing data were put through a computational pipeline for WES data processing and analysis following the general workflow adopted by the 1000 genomes project (DePristo et al., 2011) and as described elsewhere (Marti-Masso et al., 2013). Each exome’s statistics were conducted in PICARD (http://picard.sourceforge.net/).
Filtering of Common Genetic Variation
Any potential mutation observed as common variation (frequency > 5%) in the latest dbSNP137 build, 1000 Genomes Project Phase 1, other public databases, such as the Exome Variant Server of the National Heart, Lung, and Blood Institute Exome Sequencing Project (http://evs.gs.washington.edu/EVS/; Exome Variant Server, 2015), or exomes generated in house (Marti-Masso et al., 2013) was removed from further analyses. Only genetic variants mapping to coding and splice site regions were considered causative.
Prediction of Mutation Pathogenicity
The pathogenicity of each novel disease-segregating mutation was predicted by four computational methods: MutPred (http://mutpred.mutdb.org/), SNPs&Go (http://snps-and-go.biocomp.unibo.it/snps-and-go/), Mutation taster (http://www.mutationtaster.org/), and SIFT (http://sift.jcvi.org/). The NCBI HomoloGene database was used to examine the conservation of novel SNVs identified in different species (http://www.ncbi.nlm.nih.gov/homologene). The professional Human Gene Mutation Database (https://portal.biobase-international.com/hgmd/pro/start.php) and the NCBI ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/) were used to determine if novel single nucleotide variations (SNVs) were already known to be associated with disease-related phenotypes.
Gene Screening Analyses
Genomic primers for Polymerase Chain Reaction (PCR) amplifications of the entire coding region and intron–exon boundaries of SORT1, LAPTM5 exon 5, and GRIN2D exon 13 were designed using a primer design public website (http://ihg.gsf.de/ihg/ExonPrimer.html; primer sequences available upon request). All purified PCR products were sequenced in both forward and reverse directions by Sanger sequencing using Applied Biosystems BigDye terminator v3.1 sequencing chemistry as per the manufacturer’s instructions and analyzed as described elsewhere (Marti-Masso et al., 2013).
Mutagenesis and Cell-Culture Assays
Human embryonic kidney cells HEK293 were cultured in Dulbecco’s modified eagle medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. The SORT1 p.Gly171Ala (p.G171A) mutation was introduced into the human pCMV6-XL5-SORT1 wild-type plasmid (Origene, Rockville, MD) by site-directed mutagenesis (QuikChangeII, Agilent Technologies, Santa Clara, CA). All generated constructs were verified in both directions by Sanger sequencing and using the human sequence GRCh37.p13 as a reference.
HEK293 cells were grown in 96-well and 12-well tissue culture plates. After reaching confluency (∼80%), cells were transiently transfected using FuGene 6 Transfection Reagent (Promega, Madison, WI) with 0.1 µg of either pCMV6-XL5-SORT1 wild-type or mutant human plasmids. After 24 hr, culture media was replaced and cells were treated independently with 10 ng/ml or 30 ng/ml of two different pro-neurotrophics factors: pro-nerve growth factor (proNGF) and pro-brain-derived neurotrophic factor (proBDNF), or with a combination of both of them (Novoprotein, Summit, NJ). Cells were then incubated at 37℃ for 72 hr except for apoptosis assays in which a 16 h incubation was used. Conditioned media was collected and cells were harvested.
Gene Expression Analyses
RNA extraction from HEK293 cells was performed using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) as per manufacturer’s instructions. Extracted RNA was transcribed into cDNA with SuperScript II reverse transcriptase (Life Technologies, Grand Island, NY). Gene expression was carried out by qPCR using an Eco Real-Time PCR System (Illumina, San Diego, CA), SYBRGreen PCR master mix (Applied Biosystems, Foster City, CA), and the following oligonucleotide sequences: SORT1-F 5′-GGGGACACATGGAGCATGG-3′ and SORT1-R 5′-GGAATAGACAATGCCTCGATCAT-3′ for SORT1 expression, B2MF1-F 5′-GGCCGAGATGTCTCGCTCCG-3′ and B2MR1 5′-TTGGAGTACGCTGGATAGCCTCC-3′ for B2M, and p75-F 5′-CCTACGGCTACTACCAGGATG-3′ and p75-R 5′-CACACGGTGTTCTGCTTGT-3′ for p75NTR. Samples were run in triplicates, analyzed using the standard curve method, and normalized to B2M housekeeping gene using the 2−ΔΔCT method (Livak and Schmittgen, 2001).
Immunoblot Analyses
Lysates from cells were prepared in radioactive immunoprecipitation assay buffer and treated with protease inhibitor cocktail. Equal amounts of proteins were added to Laemmli’s buffer (Bio-Rad, Hercules, CA), heated for 5 min at 95℃, and loaded into a 15-wells 4% to 12% Bis-Tris gel (Invitrogen, Carlsbad, CA). Proteins were then transferred to PVDF membranes (Bio-Rad, Hercules, CA), blocked with 5% nonfat dry milk in Phosphate Buffered Saline with Tween-20 (PBST), and incubated with primary antibody anti-SORT1 (1 µg/ml; (Abcam, Cambridge, MA; ab16640) and secondary antibody anti-Rabbit conjugated to horseradish peroxidase (1:5,000; Abcam; ab6721). Anti-GAPDH antibody (1:5000, Trevigen, Gaithersburg, MD) was used as loading control. Protein expression was detected by using Lumminata Forte (Millipore, Billerica, MA), gel images were obtained using a G:BOX Chemi image analyzer (Syngene, Frederick, MD), and densitometric analyses were performed through the ImageJ software (imagej.nih.gov).
Enzyme-Linked Immunosorbent Assay (ELISA)
Human progranulin (hPGRN) in conditioned medium from wild-type and mutant HEK-293 cells was detected using the DuoSet ELISA Development System kit (R&D Systems, Minneapolis, MN) following the manufacturer’s protocol. The hPGRN from five human samples (Cases 2–6; Figure 1(a)) was measured using the hPGRN ELISA kit from Adipogen (San Diego, CA). Absorbance values were measured using a M5 multimodal plate reader (Molecular Devices, Sunnyvale, CA) at wavelengths of 450 nm and 560 nm.
Apoptosis Assay by Flow Cytometry Analysis
Quantitative analysis of apoptotic cell death was done using the Alexa Fluor® 488 Annexin V/Dead Cell Apoptosis Kit according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA). HEK293 cells were treated with 30 ng/ml of proneurotrophins (proNTs) for 16 hr; cells were then harvested, stained with Alexa Fluor® 488-Annexin V and PI in annexin-binding buffer, and analyzed by flow cytometry using a BD LSR II flow cytometer and the BD FACSDiva software (BD Bioscience, San Jose, CA). Cells stained with only Annexin V were considered as being in early apoptosis, while cells stained with both Annexin V and PI were considered to be in late apoptosis or necrotic stage. HEK293 cells treated with 2 mM of hydrogen peroxide were used as positive control for apoptosis. Data analysis was performed using the FlowJo software version 9.3.2 (TreeStar, Ashland, OR).
Statistical Analyses
Statistical analyses were performed using the GraphPad Prism software version 6.00 (GraphPad, La Jolla, CA). Data on graphs are presented as mean ± SEM. Statistical differences between wild-type and mutant were calculated by using the Mann-Whitney nonparametric U test. For SORT1 mRNA expression using different proNT concentrations, statistical significance was determined using a linear regression model. Values of p ≤ .0001 were considered highly significant (****).
Results
Phenotypic Examination of a Spanish Family With ET
The reported ET family, who consisted of two “definitive” affected individuals as well as four unaffected members (Figure 1(a)), featured postural tremor of both hands with slow progression since childhood. Psychiatric features, such as depression and anxiety, were found in one unaffected member. Detailed clinical descriptions of all family members examined are described below.
Case 1
This is an affected male who was first seen in 2006 with postural and action tremor of both hands and without voice, head, or rest tremors. He reported to have tremor with slow progression since childhood. No other signs of motor impairment suggestive of parkinsonism, myoclonus, dystonia, ataxia, and polyneuropathy were identified, and he did not show any sign of autonomic involvement, sleep disorder (insomnia, drowsiness, REM sleep behavior disorder (RBD)), anxiety, or depression. He did not take any medication for tremor, but his tremor improved with alcohol intake. He reported hypercholesterolemia and had a myocardial infarction in 2000 and an atherothrombotic left pontine infarction in 2001. In 2008, he died of head injury with a left subdural hematoma and intracranial fronto-temporal hemorrhage at 73 years of age. He scored 36 in the TRS scale and had a MoCA value of 27/30 (normal value > 25). His drawing of right hand spiral is shown in Figure 2(a).
Figure 2.
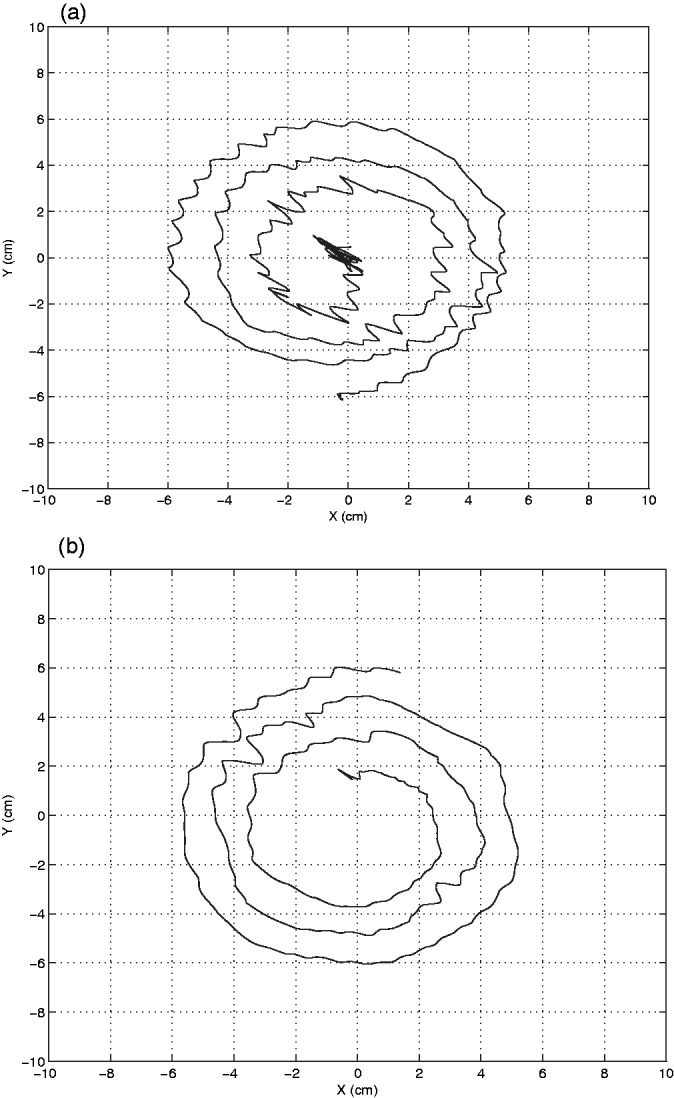
Archimedes spirals. Right hand spirals of affected Case 1 (a) and Case 3 (b) are shown. Archimedes spirals were drawn on a digitizing tablet.
Case 2
This is the wife of case 1 who did not show any sign of tremor or other neurological symptoms.
Case 3
This is a 48-year-old woman who suffers from tremor of both hands since childhood. During examination, she showed symmetric postural tremor involving hands without resting or action tremors and without affecting head, voice, and trunk. Her gait was normal and no clinical signs of parkinsonism, myoclonus, dystonia, ataxia, and polyneuropathy were found. She did not present any sign of autonomic involvement, sleep disorder (insomnia, drowsiness, RBD), anxiety, or depression. She scored 11 in the TRS scale and showed normal cognitive status with a MoCA value of 30/30 and a normal brain MRI. EMG recorded a postural tremor of 9.5 Hz of frequency and 46 mV of amplitude. Her total cholesterol was 244 mg/dl and low-density lipoprotein (LDL) cholesterol levels were slightly elevated (176 mg/dl). Her drawing of right hand spiral is shown in Figure 2(b).
Case 4
This is a 46-year-old woman who reported having mild tremor of both hands during her childhood when she was a regular tobacco smoker, but it greatly improved after smoking withdrawal. On a recent examination, she showed no tremor and a normal cognitive status (MoCA = 30/30). Her brain MRI, total cholesterol, and LDL levels were normal.
Case 5
This is a 43-year-old man who apparently has not reported tremor or an EMG-recorded tremor. His brain MRI was normal and showed a MoCA value of 30/30. He has high levels of total cholesterol and LDL and is under treatment with atorvastatine 20 mg/day.
Case 6
This is 67-year-old woman who has had anxiety and depression symptoms for 15 years and is being treated with 20 mg/day of paroxetine. On examination, she showed no tremor and her cognitive assessments (MoCA = 30/30) and LDL levels were normal.
None of the unaffected cases (Cases 4, 5, 6) showed any sign of motor involvement suggestive of parkinsonism, myoclonus, dystonia, ataxia, and polyneuropathy, nor nonmotor symptoms such as autonomic involvement, sleep disorders, anxiety, or depression.
WES and Subsequent Analyses
WES approaches performed in four family members captured between 91.33% (Case 1) and 88.30% (Case 2) of the target exome at 20-fold coverage or higher. Approximately 1,000 novel, coding SNVs, including only missense and nonsense genetic variants, were identified in each family member sequenced: 1062 SNVs were identified for Case 3, 1,024 for Case 1, 1,006 for Case 2, and 884 for Case 4. After adequate filtering and by keeping SNVs only shared by both affected individuals, we were left with six SNVs as potential disease-associated mutations. Two of these were not validated through Sanger sequencing and the other four were also present in the youngest family member (Case 5), who is apparently unaffected at his 43 years of age. Because the existence of a variable age at onset is common in families with tremor, every unaffected individual younger than 50 years should still be considered at risk (Merner et al., 2012). All four novel SNVs were then tested in 188 ethnicity-matched normal chromosomes, but only one was found present in the control population. A DNA sample from an additional unaffected member (Case 6) was then acquired and tested for these three novel SNVs; she was found to be a noncarrier for any of them. By using four different computational programs, only two SNVs were predicted to be deleterious by all programs. Although one of this (p.Gly171Ala) was reported with very low frequency (5.769E-05) in the Exome Aggregation Consortium (ExAC) that contains exome data for a variety of rare diseases including rare neuromuscular diseases (ExAC, 2015), none of them were reported in the European population. These two SNVs were also highly conserved across different species while the other one reported in European population (ExAC data) and situated in the GRIN2D gene, which encodes a class of ionotropic glutamate receptor and has been associated with schizophrenia susceptibility in the Japanese population (Makino et al., 2005), was not. The two pathogenic-predicted, highly conserved SNVs were located within LAPTM5 and SORT1 genes (Table 1; Figure 1(b) and (c)). The LAPTM5 gene encodes for a lysosomal-associated multispanning membrane protein that is known to be associated with spontaneous regression of neuroblastomas, pigmented villonodular synovitis, and lung cancer (Finis et al., 2006; Cortese et al., 2008; Inoue et al., 2009). The SORT1 gene encodes for sortilin, a member of a family of cellular vacuolar protein sorting 10 domain receptors that is expressed in neurons of the central nervous system, including cortical, hippocampal, and cerebellar neurons, and peripheral nervous system. SORT1 is known to regulate both neuronal viability and function through its regulation of both protein transport and signal transduction (Willnow et al., 2008) and is known to bind proNTs to control neuronal survival and cessation (Nykjaer and Willnow, 2012). Sortilin, which has multifunctional roles in intracellular trafficking of polipeptide from the golgy apparatus to secretory and endocytic pathways (Nielsen et al., 2001; Chen et al., 2005), has already been implicated in Alzheimer’s disease (AD) and fronto-temporal lobar degeneration. In AD, sortilin has been shown to represent a major endocytic pathway for the clearance of apoE/Aβ complexes (Carlo et al., 2013) and to control APP processing to Aβ (Gustafsen et al., 2013), while in fronto-temporal lobar degeneration it has been shown to regulate extracellular levels of progranulin (PGRN; Hu et al., 2010). Sortilin has also been postulated as a risk factor for hypercholesterolemia and myocardial infarction (Musunuru et al., 2010). Interestingly, all of our SORT1 mutation carriers were shown later to carry elevated levels of LDL cholesterol, suggesting a possible role of cholesterol homeostasis in the pathogenesis of tremor.
Table 1.
Heterozygous Disease-Segregating Mutations Identified Through WES. All Variants Were Absent in Large Number of Control Individuals.
| Ch | Position | Gene | Nucleotide change | Protein change | Protein domain | Conservationa | Pathogenicity predictionb | ExAC data (EU fqcy) | Human brain expressionc | Associated mechanisms/diseases |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 31,211,818 | LAPTM5 | c.479C > T | p.T160I | Mtp (28-261) | High | Deleterious | Not present | Medium | Lysosomal destabilization/ neuroblastomas, PVNS, autoimmune diseases, and lung cancer |
| 1 | 109,898,020 | SORT1 | c.512G > C | p.G171A | Vps10p (133-745aa) | High | Deleterious | Not present | High | NPs ligand, lysosomal sorting, endocytosis/ FTD, heart diseases |
| 19 | 48,946,504 | GRIN2D | c.3321T > A | p.D1107E | None | Medium | Neutral | 2/15,812 | Medium | Schizophrenia |
Note. EU fqcy refers to the frequency of the variants in the European population assessed in the ExAC; Mtp refers to Golgi 4-transmembrane spanning transporter.
WES = whole exome sequencing. VPS10p = vacuolar sorting protein 10 p; NPs = neuropeptides; PVNS = pigmented villonodular synovitis; FTD = fronto-temporal dementia; ExAC = Exome Aggregation Consortium.
Conservation outcomes across different species were taken from the homologene NCBI database (http://www.ncbi.nlm.nih.gov/homologene).
Prediction of pathogenicity was determined by the following computational programs (See methods): MutPred, SNPs&Go, Mutation Taster, and SIFT.
Human Brain Expression data were acquired from the Human protein atlas (http://www.proteinatlas.org/).
The entire coding region of SORT1 was then examined in 61 Spanish ET cases (13 familial and 48 sporadic), but no pathogenic mutation was identified. Likewise, the exon 4 containing the SORT1 mutation (p.Gly171Ala) was tested in additional 90 ET cases (28 familial ET and 62 sporadic), but no additional mutation carrier was identified.
Functional Assays
The SORT1 p.G171A mutation downregulates its mRNA and protein expression levels
To further test the pathogenecity of the SORT1 p.G171A mutation, HEK293 cells were transfected with either wild-type or mutant (p.G171A) plasmids and quantitative PCR (qPCR) on cDNA extracted from wild-type and mutant cells was carried out. Sortilin mRNA levels cells were found nearly three-fold lower in p.G171A mutation carriers than in noncarriers (p < .0001; Figure 3(a)), and a similar decrease of two-fold between wild-type and mutant cells was observed at the protein level (p < .05; Figure 3(b)). Because sortilin binds to the prodomains of both proNGF and proBDNF to regulate sorting of mature NGF and BDNF to appropriate endocytic and secretory pathways (Chen et al., 2005), transfected cells were also treated with proNGF, proBDNF, and with a combination of proNGF/proBNDF. Sortilin downregulation was consistent in both treated and untreated cells, suggesting that the treatment with proNTs has no effect on its mutant-related differential expression (Figure 3(a)–(c)).
Figure 3.

SORT1 and sortilin expression in untreated and treated cells with proNTs factors. Gray columns represent untrasfected HEK293 cells, blue columns HEK293 cells transfected with SORT1 wild-type plasmid, and orange columns HEK293 cells transfected with SORT1 G171A mutant plasmid. Real time PCR analyses for SORT1 mRNA expression of HEK293 cells untreated and treated with proNTs are shown in (a) (10 ng/ml) and (c) (x-axis represents different concentrations of proNGF, proBDNF, and proNGF/proBDNF (5, 10, 30, or 60 ng/ml). The housekeeping gene β2 microglobulin (B2M) was used as control gene. Values represent the mean ± SEM of two different experiments with nine replicates each. (b) Western blot analysis of sortilin and GAPDH proteins in cell lysates obtained from untransfected and transfected HEK293 cells with wild-type and mutant sortilin cultured for 4 days after transfection in the absence and presence of 10 ng/ml of proNGF, proBDNF, and a combination of proNGF/proBDNF. The full-length of sortilin (90 KDa) and GAPDH protein (37 KDa) that was used as a loading control are shown. Results are representative of three separate experiments. Values represent the mean ± SEM. ****p < .0001, while **p < .05.
The p75NTR mRNA expression is increased in SORT1 p.G171A mutant cells
Given the role of sortilin in proNGF- and proBDNF-induced neuronal apoptosis via its interaction with p75NTR (Nykjaer et al., 2004; Teng et al., 2005) and the effects of the SORT1 p.G171A mutation on mRNA and protein expression levels, we sought to determine whether this mutation may also alter the p75NTR mRNA levels and the levels of apoptosis. The expression of p75NTR in untransfected cells was first tested by direct qPCR: HEK293 cells expressed p75NTR, and then examined in both wild-type and mutant SORT1 transfected cells: p75NTR mRNA levels were significantly increased in mutant cells when compared with their wild-type counterparts (Figure 4(a); p < .05, p < .01). This increase was more significant in cells treated with proBDNF and proNGF/proBDNF (p < .01). Although a slightly increase in apoptotic levels was found in p.G171A mutant cells treated with proNGF and proNGF/proBDNF together, these differences were not statistically significant (Figure 4(b)).
Figure 4.
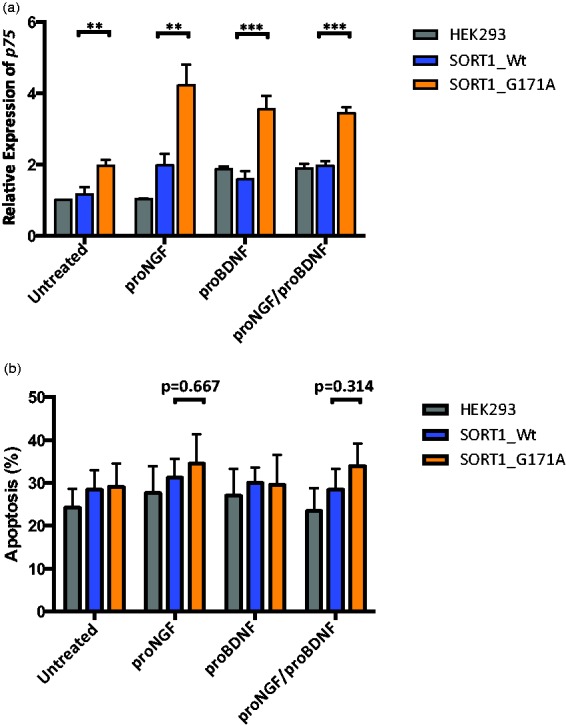
P75NTR mRNA expression and apoptotic cell death levels in wild-type and mutant SORT1 cells. Gray columns represent untrasfected HEK293 cells, blue columns HEK293 cells transfected with SORT1 wild-type plasmid, and orange columns HEK293 cells transfected with SORT1 mutant plasmid. (a) Relative quantification of p75NTR mRNA expression in untransfected and transfected HEK293 cells with either wild-type or mutant SORT1 alleles. The y-axis shows the relative expression of the target gene (P75NTR) relative to the internal control gene (B2M) and the x-axis shows treated and untreated cells with different proNTs (10 ng/ml). (b) Percentage of apoptosis in untransfected and transfected wild-type and mutant SORT1 cells. The y-axis shows the percentage of cells in early apoptosis and the x-axis represents different proNT treatments. ***p < .01, **p < .05.
PGRN levels are not altered in SORT1 mutant cells or human mutation carriers
An exciting area of research is the role of sortilin function in the metabolism of PGRN beacuse sortilin has been shown to regulate extracellular levels of PGRN in vivo (Hu et al., 2010) and in vitro (Lee et al., 2013). Thus, to determine whether the SORT1 p.G171A mutation, which downregulates SORT1 mRNA and protein levels and upregulates p75NTR mRNA levels, may also be associated with increased PGRN levels in culture and human serum samples, PGRN levels were measured by ELISA. No differences in PGRN levels between wild-type and mutant cells (Figure 5(a)) and SORT1 p.G171A mutation carriers versus noncarriers (p = .65; Figure 5(b)) were found.
Figure 5.
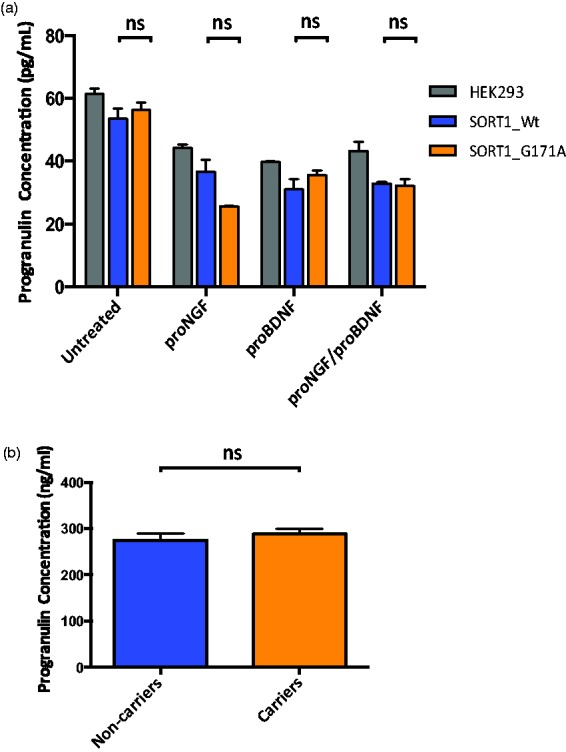
Progranulin expression in HEK293 cells and human serum samples. (a) PGRN levels in cells. Gray columns represent untransfected HEK293 cells, blue columns HEK293 cells transfected with SORT1 wild-type plasmid, and orange columns HEK293 cells transfected with SORT1 mutant plasmid. Values on the graph represent the mean ± SEM of three independent experiments. (b) PGRN levels in human serum. The blue column represents PGRN levels for SORT1 p.G171A noncarriers while the orange column is shown PGRN levels for p.G171A carriers. Values on the graph represent the mean ± SEM of duplicate samples from each individual. Statistical analysis was done with GraphPad software. Non-parametric Mann-Whitney test were performed. ns = non significant.
Discussion
Despite significant efforts made to identify the genetic basis of ET, the results are limited with linkage, genome wide association, and candidate gene studies failing to detect reliable risk and causative alleles for ET. Here, we examined a family presenting with a mild form of ET characterized by slow progression and beneficial response to alcohol consumption. A disease-segregating mutation resulting in p.Gly171Ala was identified in the SORT1 gene through WES analyses. Because the expression of sortilin is altered in aging of the nervous system and under pathological conditions (Jansen et al., 2007), we first investigated whether the SORT1 p.G171A mutation may also alter sortilin expression levels. A highly significant reduction of mRNA and sortilin expression levels was observed in mutant cells when compared with their wild-type counterparts (Figure 3(a) and (b)), suggesting that the SORT1 p.G171A mutation identified in our ET patients is implicated in sortilin dysregulation. Since the expression of p75NTR, which binds to sortilin to mediate neuronal cell death (Nykjaer et al., 2004) and modulates cholinergic transmission (Yang et al., 2002), was also found significantly elevated in mutant cells when compared with wild-type (Figure 4(a)), one may commend that under pathological conditions dysregulation of sortilin may upregulate p75NTR expression levels. Considering that p75NTR is not only upregulated in pathological conditions (Nykjaer et al., 2005) but also in impaired GABAergic transmission (Riffault et al., 2014), the sortilin deficiency caused by the p.G171A mutation might also be responsible for defects in neurotransmission, resulting in both the development of tremor and high p75NTR expression.
Given the role of sortilin as a principal neuronal binding site for PGRN and that a dramatic consequence of such binding is the rapid endocytosis of PGRN by sortilin (Hu et al., 2010), extracellular PGRN levels were also examined. Although PGRN levels were not disrupted in mutant cells versus wild-type (Figure 5(a)) or in mutation carriers versus noncarriers (Figure 5(b)), it has previously been reported that SORT1 mutants known to disrupt endocytosis do not have altered PGRN levels (Nielsen et al., 2001; Hu et al., 2010), thus leaving the possibility that our SORT1 mutation could still affect sortilin-mediated endocytosis, sorting, and trafficking, regardless of its effect on PGRN levels.
Lastly, noncoding SORT1 variation has recently been associated with high cholesterol levels and heart diseases (Musunuru et al., 2010). There are mounting evidence that aberrations in cholesterol homeostasis can lead to severe neurological diseases and impairments in synaptic plasticity (Lane-Donovan et al., 2014); along these lines, high levels of serum total cholesterol have been associated with an increased risk for Parkinson’s disease in Finnish population (Hu et al., 2008). The importance of cholesterol homeostasis for brain function was first highlighted by Niemann-Pick type C disease, in which pathogenic mutations result in impaired cholesterol trafficking and progressive neurodegeneration (Carstea et al., 1997; Naureckiene et al., 2000). The association of a specific apolipoprotein E allele (ɛ4) with the risk for both sporadic and familial AD also raised the possibility that dysfunction in the lipid transport system may cause defects in the brain lipid homeostasis of AD patients (Poirier et al., 2014). Nowadays, it is known that lipoprotein receptors, which participate in synapse development, cargo trafficking, and signal transduction, are key players in AD pathogenesis and neurodegeneration; indeed, the association between impaired lipid metabolism and brain diseases including but not limited to AD, PD and Huntington’s disease is well reported and documented (Lane-Donovan et al., 2014).
In conclusion, we here identified a disease-segregating SORT1 mutation in a small family with early-onset ET (Figure 1(a)). The pathogenicity of the SORT1 p.G171A mutation is supported by its absence in neurologically normal population, its prediction as pathogenic by different computational programs, its high conservation across other orthologs (Figure 1(b)), its location in a relevant functional domain (Figure 1(c)), and its effects on both sortilin downregulation (Figure 3(a)–(c)) and p75NTR upregulation (Figure 4(a)). While further research is still required, given the role of sortilin downregulation and p75NTR upregulation in central nervous system impairment and neurodegeneration (Harrington et al., 2004; Jansen et al., 2007; Willnow et al., 2008) and taking into account that changes in the expression level of p75NTR are correlated with functional changes in the GABAergic inhibitory neurotransmission (Blaesse et al., 2009), we hypothesize that the sortilin deficiency caused by the SORT1 p.G171A mutation may lead to defects in neurotransmission, giving rise to the development of tremor.
Acknowledgments
The authors thank the patients, relatives, and other participants for their cooperation in this study.
Footnotes
*These authors contributed equally to this work.
Author Contributions
E. S., J. F. M.-M., and C. P. R. participated in the conception and design of the research project. E. S., C. E. K., and C. P. R. performed experiments. E. S., V. M., A. C., and C. P. R. analyzed experimental data. A. B., J. R. M., A. L. M., and J. F. M.-M. conducted clinical assessments. A. G. contributed with reagents and materials and performed P. G. R. N. assays in human serum samples. C. P. R. supervised experiments. J. F. M.-M. supervised clinical assessments. C. P. R. wrote the manuscript with help of E. S., A. B., and J. F. M.-M.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the “Instituto de Salud Carlos III” (FIS PI10/02714; JFM-M) and the Institute of Neurological Disorders and Stroke of the National Institute of Health (R21NS082881, R01NS079388; CPR).
References
- American Psychiatric Association (1994) Diagnostic and statistical manual of mental disorders: DSM-IV, Washington, DC: Author. [Google Scholar]
- Bagepally B. S., Bhatt M. D., Chandran V., Saini J., Bharath R. D., Vasudev M. K., Pal P. K. (2012) Decrease in cerebral and cerebellar gray matter in essential tremor: A voxel-based morphometric analysis under 3T MRI. Journal of Neuroimaging 22: 275–278. [DOI] [PubMed] [Google Scholar]
- Benito-Leon J. (2014) Essential tremor: A neurodegenerative disease? Tremor and Other Hyperkinetic Movements 4(252): 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito-Leon J., Louis E. D. (2006) Essential tremor: Emerging views of a common disorder. Nature Clinical Practice Neurology 2: 666–678. quiz 662p following 691. [DOI] [PubMed] [Google Scholar]
- Blaesse P., Airaksinen M. S., Rivera C., Kaila K. (2009) Cation-chloride cotransporters and neuronal function. Neuron 61: 820–838. [DOI] [PubMed] [Google Scholar]
- Buijink A. W., Caan M. W., Tijssen M. A., Hoogduin J. M., Maurits N. M., van Rootselaar A. F. (2013) Decreased cerebellar fiber density in cortical myoclonic tremor but not in essential tremor. Cerebellum 12: 199–204. [DOI] [PubMed] [Google Scholar]
- Buysse D. J., Reynolds C. F., 3rd, Monk T. H., Berman S. R., Kupfer D. J. (1989) The Pittsburgh Sleep Quality Index: A new instrument for psychiatric practice and research. Psychiatry Research 28: 193–213. [DOI] [PubMed] [Google Scholar]
- Carlo A. S., Gustafsen C., Mastrobuoni G., Nielsen M. S., Burgert T., Hartl D., Willnow T. E. (2013) The pro-neurotrophin receptor sortilin is a major neuronal apolipoprotein E receptor for catabolism of amyloid-beta peptide in the brain. Journal of Neuroscience 33: 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstea E. D., Morris J. A., Coleman K. G., Loftus S. K., Zhang D., Cummings C., Tagle D. A. (1997) Niemann-Pick C1 disease gene: Homology to mediators of cholesterol homeostasis. Science 277: 228–231. [DOI] [PubMed] [Google Scholar]
- Chen Z. Y., Ieraci A., Teng H., Dall H., Meng C. X., Herrera D. G., Lee F. S. (2005) Sortilin controls intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. Journal of Neuroscience 25: 6156–6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese R., Hartmann O., Berlin K., Eckhardt F. (2008) Correlative gene expression and DNA methylation profiling in lung development nominate new biomarkers in lung cancer. International Journal of Biochemistry & Cell Biology 40: 1494–1508. [DOI] [PubMed] [Google Scholar]
- DePristo M. A., Banks E., Poplin R., Garimella K. V., Maguire J. R., Hartl C., Daly M. J. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuschl G., Bain P., Brin M. (1998) Consensus statement of the Movement Disorder Society on Tremor. Ad Hoc Scientific Committee. Movement Disorders: Official Journal of the Movement Disorder Society 13(Suppl 3): 2–23. [DOI] [PubMed] [Google Scholar]
- Elble R. J., Sinha R., Higgins C. (1990) Quantification of tremor with a digitizing tablet. Journal of Neuroscience Methods 32: 193–198. [DOI] [PubMed] [Google Scholar]
- Exome Aggregation Consortium (ExAC). (2015). Cambridge, MA. Retrieved from http://exac.broadinstitute.org.
- Exome Variant Server (2015) NHLBI Exome Sequencing Project (ESP), Seattle, WA: Author. [Google Scholar]
- Fahn S., Tolosa E., Marin C. (2003) Clinical rating scale for tremor, Baltimore, MD: Williams and Wilkins. [Google Scholar]
- Finis K., Sultmann H., Ruschhaupt M., Buness A., Helmchen B., Kuner R., Berger I. (2006) Analysis of pigmented villonodular synovitis with genome-wide complementary DNA microarray and tissue array technology reveals insight into potential novel therapeutic approaches. Arthritis & Rheumatology 54: 1009–1019. [DOI] [PubMed] [Google Scholar]
- Gustafsen C., Glerup S., Pallesen L. T., Olsen D., Andersen O. M., Nykjaer A., Petersen C. M. (2013) Sortilin and SorLA display distinct roles in processing and trafficking of amyloid precursor protein. Journal of Neuroscience 33: 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington A. W., Leiner B., Blechschmitt C., Arevalo J. C., Lee R., Morl K., Giehl K. M. (2004) Secreted proNGF is a pathophysiological death-inducing ligand after adult CNS injury. Proceedings of the National Academy of Sciences of the United States of America 101: 6226–6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F., Padukkavidana T., Vaegter C. B., Brady O. A., Zheng Y., Mackenzie I. R., Strittmatter S. M. (2010) Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68: 654–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G., Antikainen R., Jousilahti P., Kivipelto M., Tuomilehto J. (2008) Total cholesterol and the risk of Parkinson disease. Neurology 70: 1972–1979. [DOI] [PubMed] [Google Scholar]
- Inoue J., Misawa A., Tanaka Y., Ichinose S., Sugino Y., Hosoi H., Inazawa J. (2009) Lysosomal-associated protein multispanning transmembrane 5 gene (LAPTM5) is associated with spontaneous regression of neuroblastomas. PLoS One 4: e7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen P., Giehl K., Nyengaard J. R., Teng K., Lioubinski O., Sjoegaard S. S., Nykjaer A. (2007) Roles for the pro-neurotrophin receptor sortilin in neuronal development, aging and brain injury. Nature Neuroscience 10: 1449–1457. [DOI] [PubMed] [Google Scholar]
- Kuhlenbaumer G., Hopfner F., Deuschl G. (2014) Genetics of essential tremor: Meta-analysis and review. Neurology 82: 1000–1007. [DOI] [PubMed] [Google Scholar]
- Lane-Donovan C., Philips G. T., Herz J. (2014) More than cholesterol transporters: Lipoprotein receptors in CNS function and neurodegeneration. Neuron 83: 771–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. C., Almeida S., Prudencio M., Caulfield T. R., Zhang Y. J., Tay W. M., Petrucelli L. (2013) Targeted manipulation of the sortilin-progranulin axis rescues progranulin haploinsufficiency. Human Molecular Genetics 23: 1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Louis E. D. (2010) Essential tremor: Evolving clinicopathological concepts in an era of intensive post-mortem enquiry. The Lancet Neurology 9: 613–622. [DOI] [PubMed] [Google Scholar]
- Louis E. D., Ford B., Frucht S., Barnes L. F., X-Tang M., Ottman R. (2001) Risk of tremor and impairment from tremor in relatives of patients with essential tremor: A community-based family study. Annals of Neurology 49: 761–769. [DOI] [PubMed] [Google Scholar]
- Louis E. D., Ottman R., Ford B., Pullman S., Martinez M., Fahn S., Hauser W. A. (1997) The Washington Heights-Inwood Genetic Study of Essential Tremor: Methodologic issues in essential-tremor research. Neuroepidemiology 16: 124–133. [DOI] [PubMed] [Google Scholar]
- Makino C., Shibata H., Ninomiya H., Tashiro N., Fukumaki Y. (2005) Identification of single-nucleotide polymorphisms in the human N-methyl-D-aspartate receptor subunit NR2D gene, GRIN2D, and association study with schizophrenia. Psychiatric Genetics 15: 215–221. [DOI] [PubMed] [Google Scholar]
- Marti-Masso J. F., Bergareche A., Makarov V., Ruiz-Martinez J., Gorostidi A., de Munain A. L., Paisan-Ruiz C. (2013) The ACMSD gene, involved in tryptophan metabolism, is mutated in a family with cortical myoclonus, epilepsy, and parkinsonism. Journal of Molecular Medicine (Berlin) 91: 1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merner N. D., Girard S. L., Catoire H., Bourassa C. V., Belzil V. V., Riviere J. B., Rouleau G. A. (2012) Exome sequencing identifies FUS mutations as a cause of essential tremor. American Journal of Human Genetics 91: 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K., Strong A., Frank-Kamenetsky M., Lee N. E., Ahfeldt T., Sachs K. V., Rader D. J. (2010) From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 466: 714–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naureckiene S., Sleat D. E., Lackland H., Fensom A., Vanier M. T., Wattiaux R., Lobel P. (2000) Identification of HE1 as the second gene of Niemann-Pick C disease. Science 290: 2298–2301. [DOI] [PubMed] [Google Scholar]
- Nielsen M. S., Madsen P., Christensen E. I., Nykjaer A., Gliemann J., Kasper D., Petersen C. M. (2001) The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. The EMBO Journal 20: 2180–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nykjaer A., Lee R., Teng K. K., Jansen P., Madsen P., Nielsen M. S., Petersen C. M. (2004) Sortilin is essential for proNGF-induced neuronal cell death. Nature 427: 843–848. [DOI] [PubMed] [Google Scholar]
- Nykjaer A., Willnow T. E. (2012) Sortilin: A receptor to regulate neuronal viability and function. Trends in Neurosciences 35: 261–270. [DOI] [PubMed] [Google Scholar]
- Nykjaer A., Willnow T. E., Petersen C. M. (2005) p75NTR—live or let die. Current Opinion in Neurobiology 15: 49–57. [DOI] [PubMed] [Google Scholar]
- Poirier J., Miron J., Picard C., Gormley P., Theroux L., Breitner J., Dea D. (2014) Apolipoprotein E and lipid homeostasis in the etiology and treatment of sporadic Alzheimer’s disease. Neurobiology of Aging 35(Suppl 2): S3–S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riffault B., Medina I., Dumon C., Thalman C., Ferrand N., Friedel P., Porcher C. (2014) Pro-brain-derived neurotrophic factor inhibits GABAergic neurotransmission by activating endocytosis and repression of GABAA receptors. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience 34: 13516–13534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrag A., Munchau A., Bhatia K. P., Quinn N. P., Marsden C. D. (2000) Essential tremor: An overdiagnosed condition? Journal of Neurology 247: 955–959. [DOI] [PubMed] [Google Scholar]
- Symanski C., Shill H. A., Dugger B., Hentz J. G., Adler C. H., Jacobson S. A., Beach T. G. (2014) Essential tremor is not associated with cerebellar Purkinje cell loss. Movement Disorders: Official Journal of the Movement Disorder Society 29: 496–500. [DOI] [PubMed] [Google Scholar]
- Teng H. K., Teng K. K., Lee R., Wright S., Tevar S., Almeida R. D., Hempstead B. L. (2005) ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience 25: 5455–5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thier S., Lorenz D., Nothnagel M., Poremba C., Papengut F., Appenzeller S., Kuhlenbaumer G. (2012) Polymorphisms in the glial glutamate transporter SLC1A2 are associated with essential tremor. Neurology 79: 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal Gulsuner H., Gulsuner S., Mercan F. N., Onat O. E., Walsh T., Shahin H., Tekinay A. B. (2014) Mitochondrial serine protease HTRA2 p.G399S in a kindred with essential tremor and Parkinson disease. Proceedings of the National Academy of Sciences of the United States of America 111: 18285–18290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser M., Marinus J., Stiggelbout A. M., Van Hilten J. J. (2004) Assessment of autonomic dysfunction in Parkinson’s disease: The SCOPA-AUT. Movement Disorders: Official Journal of the Movement Disorder Society 19: 1306–1312. [DOI] [PubMed] [Google Scholar]
- Willnow T. E., Petersen C. M., Nykjaer A. (2008) VPS10P-domain receptors – Regulators of neuronal viability and function. Nature Reviews Neuroscience 9: 899–909. [DOI] [PubMed] [Google Scholar]
- Yang B., Slonimsky J. D., Birren S. J. (2002) A rapid switch in sympathetic neurotransmitter release properties mediated by the p75 receptor. Nature Neuroscience 5: 539–545. [DOI] [PubMed] [Google Scholar]
- Zimprich A. (2011) Genetics of Parkinson’s disease and essential tremor. Current Opinion in Neurobiology 24: 318–323. [DOI] [PubMed] [Google Scholar]