

Abstract
Objective
Activation of CYP2C29 releases superoxide during shear stress-induced dilation (SSID).
Methods
Mesenteric arteries isolated from female eNOS-KO and WT mice were cannulated and pressurized. Vasodilation and superoxide production in response to shear stress were assessed.
Results
SSID was significantly attenuated in vessels of eNOS-KO compared to WT mice, which was normalized by tempol and PEG-Catalase, in a PPOH (inhibitor of CYP2C29)-sensitive manner, but was unaffected by VAS2870 and allopurinol, inhibitors of NADPH oxidase and xanthine oxidase, respectively. NaNO2-induced dilation was comparable in both strains of mice. Confocal microscopy shows that SS-stimulated superoxide was increased particularly in the endothelium of eNOS-KO mice. HPLC analysis of 2-EOH indicated an increase in SS-stimulated superoxide in vessels of eNOS-KO mice, a response that was sensitive to PPOH. Inhibition of soluble epoxide hydrolase significantly enhanced SSID without affecting SS-stimulated superoxide production. CYP2C29 and catalase were upregulated, and exogenous H2O2 caused vasoconstriction in vessels of eNOS-KO mice.
Conclusion
CYP2C29 synthesizes EETs to mediate SSID, and simultaneously releases superoxide and sequential H2O2, which in turn, impair SSID.
Keywords: superoxide, shear stress, endothelium, CYP, catalase
Introduction
Cardiovascular protective effects of CYP/epoxygenase on catalyzing arachidonic acids to produce EETs have been well established [6,10]. The dark side of activation of CYP is that it continuously produces ROS via a consumption of NADPH by microsomal monooxygenases, implying that CYP enzyme(s) is the contributor(s) to the cellular production of ROS [6]. On the other hand, the ability of CYP enzymes to generate ROS is isoform-specific. Although the enzymes of CYP2E family have long been linked to oxidative stress [28,39], it is now believed that the CYP2C epoxygenases need to be taken seriously, as sources of ROS in the cardiovascular system. Non-specific CYP inhibitors in the prevention of ischemia/reperfusion-induced myocardial damage have been attributed to suppression of CYP-dependent ROS production [12], but direct administration of EETs protects against ischemia/reperfusion-, as well as hypoxia/reoxygenation-induced oxidative stress, endothelial injury and apoptosis [2,8,42]. CYP2C and 2J enzymes are the predominant arachidonic acid epoxygenase in the vasculature [3,4,10]. In human vascular endothelium, CYP2C9 has been demonstrated to be a major source of ROS [11], but CYP2J2 is not [30]. Overexpression of CYP2C8 in mouse heart worsens functional recovery and increases infarct size after ischemia/reperfusion, in an ROS-dependent manner [9], but transgenic mice with myocardial CYP2J2 expression improves functional recovery after ischemia [30]. Therefore, the enzymatic source of CYP may be a critical factor with respect to its net effect on cardiovascular system, in terms of divergent mechanisms that are beneficial for cardiovascular function of EETs and detrimental for simultaneous production of ROS.
CYP2C29 was the first mouse CYP2C member identified in 1994 [26] and has been mainly detected in liver, brain, kidney, heart, intestine, adrenals, aorta, seminal vesicles, testis and ovary [34,35]. We firstly reported the detection of CYP2C29 in vascular endothelium, as functions of estrogen and NO deficiency [33]. In vessels of female eNOS-KO mice, CYP2C29 enzyme releases EETs to mediate shear stress-induced dilation that otherwise, is mediated by NO and prostaglandins in their WT controls [15-18,40]. Notably, although shear stress-induced dilation was essentially preserved in NO deficient female arteries, via a CYP2C29/EET-mediated pathway, the pathological relevance of lacking NO was revealed by requiring a greater wall shear stress value than those of normal vessels to achieve a dilation of similar magnitude [15,18], implying an impaired endothelial sensitivity to shear stress. In this context, actions of CYP2C29 in the production of superoxide/hydrogen peroxide (H2O2), followed by consequential alterations in antioxidant signaling attracted considerable attention. We hypothesized therefore, that during the process of synthesis and release of EETs in response to shear stress, CYP2C29 simultaneously produces superoxide that accounts for the impaired shear stress-sensitive mechanism in NO deficient vessels. Consequently, the increased superoxide serves as an initiator to potentiate the metabolic pathway of O2-./SOD/H2O2/CAT/H2O+O2 by an adaptive regulation of SOD or catalase. To test these hypotheses, we measured superoxide production in shear stress-stimulated vessels isolated from female eNOS-KO mice, in which shear stress-induced vasodilation is solely mediated by CYP2C29-derived EETs [33]. Additionally, interactions between the activation of CYP2C29 and expression of ROS-metabolizing enzymes were also assessed. We believed that unlike the well-known effect of superoxide/H2O2 on NO-mediated responses, roles of superoxide/H2O2 in EET-mediated responses are yet, unknown.
Materials and Methods
Animals
12-14 weeks old female eNOS-KO (B6.129P2-Nos3tm1Unc/J; stock number: 002684) and wild-type (WT) mice (C57BL/6J) were purchased from Jackson Laboratories. One group of WT mice received L-NAME, an inhibitor of nitric oxide synthase, in their drinking water (50 mg/100ml) for three weeks. All protocols were approved by the Institutional Animal Care and Use Committee of New York Medical College and conform to the guidelines of the National Institutes of Health and the American Physiological Society for the use and care of laboratory animals.
Shear stress-induced vasodilation
As described previously [31], mice were sacrificed by inhalation of 100% CO2. After that, second-order mesenteric arteries (100-140 μm in active diameter) were isolated and cannulated in a vessel chamber and perfused with MOPS-buffered physiological salt solution (PSS) at 37°C, pH 7.4. Intravascular pressure was maintained constantly at 80 mmHg. After 1 h of stabilization, vessels developed spontaneous tone that reached to a basal tone of ∼65% their maximal diameter. Initial values of shear stresses (5, 10 and 20 dyne/cm2) were applied to the vessels, followed by recording changes in diameter. Wall shear stress was established by increasing perfusate flow via a syringe pump (Harvard Apparatus). The flow rate was calculated based on the diameter of each vessel. In separate experiments, shear stress-induced dilation was assessed in the presence of tempol (10-3M), a superoxide scavenger; VAS2870, (5×10-6M; Enzo Life Sciences), that has been used to inhibit vascular NADPH oxidase in cultured cells and isolated vessels, and has been demonstrated to have higher specificity in the inhibition of vascular NADPH oxidase than that of diphenylene iodonium or apocynin [20,37]; allopurinol (10-5M), an inhibitor of xanthine oxidase; PPOH, (5×10-5M), an inhibitor of CYP/epoxygenase; AUDA, (3×10-6M), an inhibitor of soluble epoxide hydrolase (sEH) that catalyzes EETs to DHETs, which in general, have less or lacking biological activity than EETs; or PEG-Catalase (100U/ml) respectively. Additionally, H2O2 (10-9-10-4M) was cumulatively administered to eNOS-KO vessels, followed by recording changes in diameter. Dilation to an NO donor (acidified NaNO2, 10-10-10-6M) was also assessed in vessels of both strains of mice. At the conclusion of experiments, vessels were incubated in a Ca2+-free solution with 1mM EGTA for 10 min and the passive diameter of vessels was then recorded.
Detection of Superoxide
Vascular superoxide production was assessed by a confocal microscopy of DHE staining method to identify the localization of superoxide in vessels [21], and also by a HPLC/fluorescence detector-based assay to quantitate 2-EOH (a superoxide-induced oxidative product of DHE) in isolated and perfused vessels [43], respectively. In order to obtain a sufficient amount of sample (internal area of vessels), first-order mesenteric arteries were used in the experiments.
Confocal Microscopy
Cannulated arteries were perfused with PSS at 80 mmHg. DHE (10 μM) was administered intra- and extraluminally for 30 min. After that, vessels were continuously stimulated with 10 dyne/cm2 shear stress for 10 min. Vessels were then cut longtitudinally and removed from the cannulae. Vessel segments were fixed in 4% paraformaldehyde for 10 min and adhered to glass slides with the endothelium facing up for fluorescent confocal microscopy (Bio-Rad MRC 1024ES/Olympus 1×70, UPlanFl 40× objective).
HPLC analysis
Isolated and pressured vessels were intraluminally administered with DHE (10 μM) for 30 min, followed by stimulation with 10 dyne/cm2 shear stress for 10 min. Vascular diameter and length were measured. Basal release of superoxide was also measured in DHE-treated vessels without shear stress stimulation. After that, vessels were washed and homogenized in a mixture of acetonitrile and water (1:1), and a 20 μl of the supernatant was injected into a HPLC with fluorescence detector (PU-2080 Plus/FP2020 Plus, Jasco). In separate experiments, shear stress-stimulated superoxide was assessed in the presence of VAS2870, AUDA or PPOH respectively. Standard curves of 2-EOH (0.3 - 5 pmol) were performed and concentrations of vascular superoxide were normalized to the internal surface area of vessels, expressed as pmol/mm2.
Western blots
Isolated mesenteric arteries were incubated in Laemmli buffer at 4° C for one hour, and then subjected to a one-minute sonication with a 10-minute interval in ice for two times, followed by 5-min boiling. Samples were loaded on a 10% SDS-PAGE gel. Proteins were transferred to a PVDF membrane, and probed with antibodies to CuZn-SOD, Mn-SOD (Calbiochem Novabiochem Co. CA), gp91phox (Santa Cruz biotechnology, Inc. CA), human CYP2C9 (Biodesign International, Saco, ME), catalase or xanthine oxidase (Abcam Inc. MA) respectively, and secondary antibodies. Highly homologous and comparable product profiles between human CYP2C9 and mouse CYP2C29 have already been proven [29,33]. Specific bands were normalized to GAPDH.
Real time RT-PCR (LightCyclerTM Roche Diagnostics)
Oligonucleotide primers for CYP2C29 were purchased from Qiagen (QIAGEN, #QT01076880). Primers for catalase (sigma Genosys) were designed as follows: 5′-GTGCTAATGAAGACAATGTCACTC-3′ (sense) and 5′-TCAGCGTTGTACTTGTCCAGAAG-3′ (antisense). Gene expressions were normalized to GAPDH. A relative quantitation method (ΔΔCt) was used to evaluate the relative expression of each gene in two groups of mice. All primer products were verified on a 1.5% Agarose gel.
Calculation and Statistical analysis
Changes in diameter in response to increases in shear stress in each vessel were normalized to its passive diameter. Intraluminal flow (Q) was calculated based on the equation of τ=4ηQ/πr3, where τ is the level of shear stress, η is the viscosity of the perfusate solution (0.007 poise at 37°C), and r is the radius of vessels [15,18]. Data are expressed as means ± SEM. N/n refers to the number of mice/vessels. Statistical analysis was performed using repeated-measures of ANOVA followed by the Tukey-Kramer post hoc test and Student's t-test. Statistical significance was accepted at a level of p<0.05.
Results
A total of 26 female eNOS-KO and 23 female WT mice were used. The average active (125 ± 7.4 vs 120 ± 6.5 μm) and passive (211 ±10 μm vs 210 ± 11 μm) diameter of mesenteric arteries, at 80 mmHg of intravascular pressure, were comparable in two groups of mice.
Reduced endothelium-dependent shear stress-induced dilation in vessels of eNOS-KO mice
To evaluate shear stress sensitive mechanism of vessels, shear stress-induced dilator responses were assessed. Figure 1 shows that 5, 10 and 20 dyne/cm2 shear stress dose-dependently initiated vasodilation in both strains of mice. However, at each value of shear stress, vessels of eNOS-KO mice exhibited significantly attenuated dilator responses compared to WT mice, indicating a reduced endothelial sensitivity to shear stress. This result is consistent with our previous findings that vessels lacking NO require a greater value of shear stress than normal vessels to achieve a dilation of similar magnitude [15,18]. However, the endothelium-independent vasodilation to acidified NaNO2 was comparable in both strains of mice, suggesting normal smooth muscle responsiveness. Since CYP2C29-derived EETs are the only mediators responsible for the shear stress-induced dilation in vessels of female eNOS-KO mice [33], we speculate that activated CYP2C29 simultaneously releases superoxide that in turn, impairs endothelial sensitivity to shear stress. To test this hypothesis, shear stress-induced vasodilation was performed in the presence of tempol. As shown in Figure 2 that tempol normalized dilator responses in vessels of eNOS-KO mice, but did not affect responses of WT mice. Moreover, neither VAS2870 nor allopurinol affected, but PPOH abolished the tempol-induced improvement of vasodilation in eNOS-KO mice, confirming a CYP2C29-dependent response. In line with the result, protein expression of CYP2C29 was significantly increased in vessels of eNOS-KO compared to WT mice, whereas expressions of NADPH oxidase subunit gp91 and xanthine oxidase in both strains of mice were comparable (Figure 3).
Figure 1.

Shear stress (a) (5, 10 and 20 dyme/cm2)- and NaNO2 (b)-induced dilations in mesenteric arteries of female WT (N/n=15/19) and eNOS-KO (N/n=14/20) mice. *Significant difference from WT mice.
Figure 2.

Shear stress-induced dilation in mesenteric arteries of female WT (N/n=6/7) and eNOS-KO (N/n=6/8) mice, in controls and in the presence of tempol, and tempol plus PPOH (a), or in the presence of VAS2870 or allopurinol (b) respectively (N/n=5/6 in each group). *Significant difference from controls of eNOS-KO mice.
Figure 3.
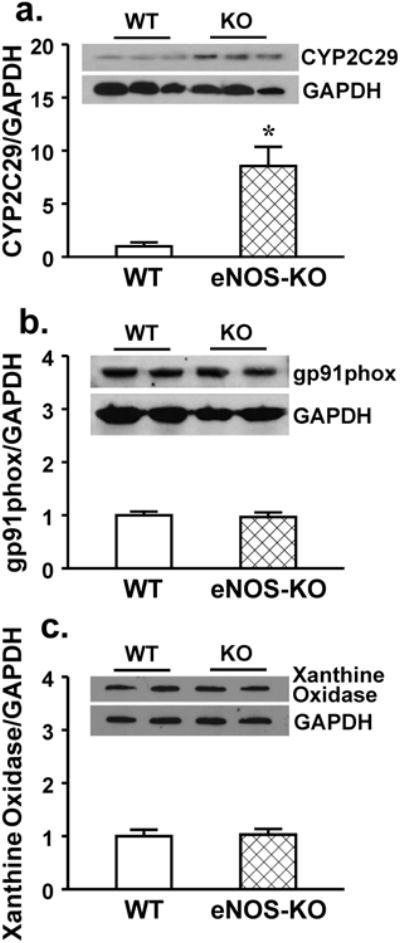
Protein expression of CYP2C29 (a), gp91phox (b, a subunit of NADPH oxidase), and xanthine oxidase (c) (3 blots per group) in mesenteric arteries of female WT and eNOS-KO mice. *Significant difference from WT mice.
Shear stress-stimulated, CYP2C29-derived superoxide in arteries of eNOS-KO mice
To provide evidence for the release of superoxide via activating CYP2C29, we used both qualitative and quantitative methods to localize intracellular superoxide and analyze its concentration in vessels. Confocal microscopy of DHE staining shows that in response to 10 dyne/cm2 shear stress, superoxide production was increased particularly in the endothelial layer of eNOS-KO compared to WT mice (fluorescence intensity: 1,750,000 vs 600,128, Figure 4a), but was comparable in smooth muscle layer of both groups of mice (fluorescence intensity: 2,540,146 vs 2,211,753, Figure 4b).
Figure 4.
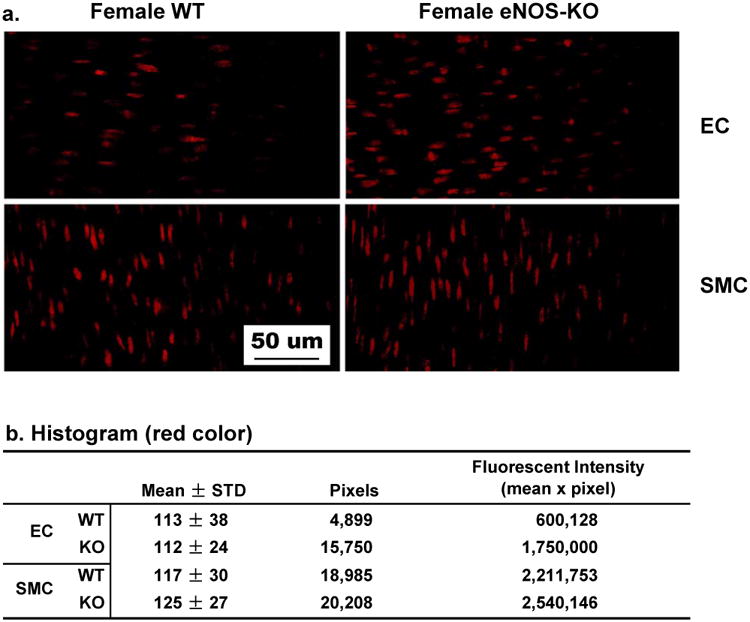
Shear stress-stimulated superoxide formation in mesenteric arteries of female WT and eNOS-KO mice. The red color in the cells represents the level of superoxide. Endothelial cells (EC) are aligned horizontally and smooth muscle cells (SMC) aligned vertically (a). Panel b summaries histogram data of the four shown in panel a. Only red color was analyzed and the fluorescent intensity (corresponding to superoxide formation) was calculated as the product of the mean intensity and total red pixels. ,
Figure 5 indicates that basal release (expressed as B) of superoxide was identical in vessels of WT and eNOS-KO mice. Shear stress-stimulated (expressed as SS) release of superoxide was four-fold higher in vessels of eNOS-KO than WT mice, in the latter, the level of shear stress-stimulated superoxide was comparable to its basal release. To determine the source responsible for the enhanced superoxide formation, vessels were pre-treated with PPOH or VAS2870 before exposure to shear stress. PPOH eliminated shear stress-stimulated superoxide release that however, was not influenced by VAS2870, suggesting that CYP2C29 enzyme is the responsible contributor to the shear stress-stimulated generation of superoxide in eNOS-KO vessels.
Figure 5.

Basal (B) and shear stress (SS, 10 dyme/cm2)-stimulated, with or without VAS2870 and PPOH, respectively, superoxide production in mesenteric arteries of female WT (N/n=5/12) and eNOS-KO (N/n=5/16) mice. *Significant difference from WT mice; and from basal and SS plus PPOH in eNOS-KO mice, respectively.
Changes in metabolic pathways for superoxide/hydrogen peroxide in vessels of eNOS-KO mice
To evaluate the pathophysiological relevance of enhanced superoxide in the regulation of oxidative/antioxidtive signaling, we evaluated vascular expression of SOD and catalase, enzymes that play crucial roles in the process of superoxide metabolism. Western blot analysis indicates that expression of catalase was significantly increased in eNOS-KO compared to WT controls, whereas, expressions of Mn- and Cu/Zn-SOD were comparable in both groups of mice (Figure 6). Interestingly, upregulation of catalase was specifically initiated in WT vessels when their expression of CYP2C29 were increased in response to L-NAME treatment (Figure 7), revealing a correlation between the two genes. Additionally, a possible role of H2O2 in shear stress-induced dilation was also evaluated. We found that treatment of eNOS-KO vessels with a sEH inhibitor (AUDA) significantly enhanced shear stress-induced dilations in a PPOH-sensitive manner, but had no effect on the superoxide production (Figure 8a-b). AUDA is able to increase systemic and cellular EET levels via the reduction of EET degradation [6], therefore it enhanced shear stress-induced dilation in an EET-dependent manner. We also found that PEG-CAT that had no effect on shear stress-induced dilation of WT vessels, significantly improved the attenuated dilation in eNOS-KO vessels (Figure 8c), responses that were similar to those initiated by tempol (Figure 2). Moreover, exposure of vessels to lower doses of H2O2 (10-9-10-8M) initiated minor dilator responses that became vasoconstrictions in response to higher doses of H2O2 (10-7-10-5M) (Figure 8d). Vessels were eventually damaged in response to 10-4M H2O2, characterized as loss of vascular tone, failure of constriction to phenylephrine, and the endothelial detachment (data not shown). These findings argue against the possibility that H2O2 is a dilator mediator for the shear stress-induced dilation, but rather it impairs vasodilations.
Figure 6.

Protein expression of catalase (a), CuZn-SOD (b) and Mn-SOD (c) in mesenteric arteries of female WT and eNOS-KO mice. *Significant difference from WT (3 blots per group).
Figure 7.

CYP2C29 and catalase mRNA in mesenteric arteries of female WT mice and WT mice treated with L-NAME. *Significant difference from untreated WT mice.
Figure 8.

Shear stress (5, 10 and 20 dyme/cm2)-induced dilation (a and c), shear stress (10 dyne/cm2)-stimulated superoxide (b), and H2O2 (10-9-10-5M)-induced responses (d) in mesenteric arteries of female eNOS-KO (N/n=8/18) mice, before and after administration of AUDA and AUDA plus PPOH (a), or PEG-CAT and PEG-CAT plus PPOH (c) respectively. *Significant difference from the control and the presence of PPOH.
Discussion
The major findings of the present study are that in mesenteric arteries of female eNOS-KO mice, CYP2C29 epoxygenase is a superoxide generating enzyme that produces superoxide and consequently the formation of H2O2, to impair endothelial sensitivity to shear stress. Thus, the impact of superoxide/H2O2 on the EET-mediation of shear stress-induced dilation was for the first time, revealed. Moreover, findings that activation of CYP2C29 initiates an upregulation of catalase that in turn, protects against the superoxide/H2O2-induced endothelial dysfunction illustrate a profile of the interactions among EETs, superoxide and H2O2 during the process of shear stress-dependent regulation of vascular tone.
We previously demonstrated that mouse CYP2C29 and rat CYP2C7 are endothelial EET synthase, when these enzymes are activated by shear stress, they produce four isoforms of EETs to hyperpolarize vascular smooth muscle, leading to vasodilation [16]. This EDHF-dependent vasodilation functions as a back-up mechanism in the maintenance of shear stress-dependent regulation of vascular tone in NO deficient vessels, via an estrogen-dependent activation of phosphatidylinositol 3/Akt signaling [17]. Whereas the maintained flow-induced dilation in NO deficiency is associated with a reduced endothelial sensitivity to shear stress [15,18,40], questions were therefore, raised as whether superoxide plays a role in the responses, and if so, what is the superoxide generating source in the vessels.
CYP2C29-derived superoxide contributes to the impaired shear stress-induced dilation
We indicated that the attenuated shear stress-induced dilation in arteries of eNOS-KO mice was attributed to the action of superoxide, as evidenced by the fact that treatment of the vessels with a superoxide scavenger normalized the responses (Figures 1 and 2). Given that CYP2C29 epoxygenase is the major, if not only, endothelial enzyme responsible for the mediation of endothelium-dependent, shear stress-induced vasodilation in female eNOS-KO mice ([33] and Figure 2), it raises intriguing possibilities that CYP2C29 epoxygenase is the superoxide generating enzyme, and if so, the derived superoxide is mainly endothelium-located. Indeed, confocal microscopy indicates a predominant DHE staining in the endothelial layer of eNOS-KO vessels (Figure 4), suggesting an endothelium-specific superoxide formation. Inhibition of vascular NADPH and xanthine oxidase failed to affect shear stress-stimulated release of superoxide (Figure 5) and vasodilation (Figure 2), leaving CYP2C29 as the most probable candidate for the PPOH-sensitive formation of superoxide in the endothelium of eNOS-KO mice. All these functional (Figures 1 and 2) and biochemistry (Figures 4 and 5) results in combination with the molecular evidence for the upregulation of CYP2C29 (Figure 3) support our conclusion that CYP2C29 is a physiologically relevant source of ROS.
Unlike established mechanisms by which superoxide inactivates NO to impair shear stress-induced dilation [19-21,31,41,43], the mechanistic insight as to how superoxide directly interferes with EETs to reduce endothelial sensitivity to shear stress in the absence of NO, remains unexplored. Some possibilities including a direct action of ROS on vascular smooth muscle cells, or activation of inflammatory kinases to increase vascular tone have been suggested [3]. In the absence of NO, superoxide dismutation becomes a mayor metabolic process, leading to the formation of H2O2 that can cause endothelial dysfunction [36]. In agreement with that, PEG-CAT significantly improved shear stress-induced dilation in vessels of eNOS-KO mice (Figure 8c), revealing a specific role of H2O2 in the responses. More directly, the vasoconstrictor responses caused by H2O2 (Figure 8d) supported further that actions of H2O2 in vascular smooth muscle counteract vasodilator responses to shear stress. Thus, we draw the conclusion that both superoxide and H2O2 coordinately contribute to the impairment of endothelial sensitivity to shear stress in eNOS-KO mice.
In addition of CYP, endothelial NO synthase, COX, xanthine oxidase and NADPH oxidase can also generate ROS in amounts that may affect vascular function [27]. In this context, one can challenge our results by auguring why activations of eNOS and COX responsible for NO- and prostaglandin-mediated dilations of WT mice [15], are unassociated with increases in superoxide release (Figure 5). In this regard, we believe that although activation of eNOS/or COX may also initiate the release of superoxide, an oxidative balance is tightly maintained by NO. Superoxide reacts with NO three to four times faster than it reacts with SOD when especially, NO level is relatively high [38]. This explanation fits well with observations that the estrogen-dependent genomic and non-genomic potentiation of NO synthesis and bioavailability [14] in female WT mice benefit its antioxidant actions. Moreover, tempol and PEG-CAT did not affect shear stress-induced dilation in vessels of WT mice (Figures 2a and 8c), in which expression of nitrotyrisine, an indicative of peroxynitrite formed by the reaction of superoxide with NO, was very low [20,43], suggesting negligible impacts of superoxide in the modulation of vascular function in physiological conditions.
Adaptation of ROS metabolizing signaling in response to activation of CYP2C29
Intriguingly, the present study revealed a correlation between CYP2C29 and catalase in NO deficient vessels, characterized as the parallel changes in the expression of these two genes (Figures 3a, 6a and 7). We noted that when shear stress-induced dilation mediated by NO/prostaglandins (in WT) was switched to that mediated by EETs (in NO deficiency) [17,32,33], catalase expression was consequentially increased, emerging a relationship among EETs, superoxide and H2O2. Given that CYP2C29 produces superoxide, and that catalase is one of the key enzymes during the superoxide/H2O2 metabolizing process, it is plausible to speculate that increased superoxide per se, serves as a trigger to activate its degradation via perhaps, potentiation of catalase activity. On the other hand, CYP2C29 releases EETs that are capable of regulating catalase, for instance, exogenous administration of EETs dose- and time-dependently increases catalase expression and activity, accompanied by the attenuation of arsenic trioxide-stimulated superoxide/H2O2 production in cultured cells [23]. Moreover, the lack of NO-dependent inhibition of catalase [38] in eNOS-KO vessels may also benefit its expression.
Although H2O2 served as an EDHF in human [24,44] and porcine [1,13] coronary, as well as mouse mesenteric arteries [25] has been documented, the present study basically excludes the vasodilator property of H2O2 in the shear stress-induced dilation. We demonstrated that inhibition of sEH to increase vascular EETs [6,7] without effects on CYP2C29 activity and superoxide/H2O2 formation, significantly enhanced shear stress-induced dilations in a PPOH-sensitive manner (Figure 8a-b). This suggests that EETs, but not H2O2, are decisive mediator of the responses. In addition, elimination of H2O2 restored attenuated dilator responses (Figure 8c), but administration of it caused vasoconstrictions (Figure 8d), indicating that H2O2 prevent but not mediate, shear stress-induced dilation in NO deficient vessels. The discrepancy between our and other studies might be attributed to the differences in animal models, genders, experimental preparations, tissues/vasculatures and vasoactive stimuli selected, etc., leading to the lack of consensus in the definition of EDHFs. Specifically, an interaction between EETs and H2O2 has been proposed, characterized as the inhibition of CYP epoxygenases by H2O2 [22]. To this end, an upregulation of catalase in response to increases in superoxide/H2O2 may function as a compensatory mechanism to alleviate H2O2-induced inhibition of CYP and maintain EET-mediated dilator pathway.
Perspectives
Our results suggest that endothelial CYP2C29, expressed in mesenteric arteries, is not only an EET synthase but also a potential major source of ROS within the vessel wall. Taking into account the detrimental effect of activating CYP2C29 to release superoxide/H2O2 that in turn, alter shear stress sensitive mechanisms, our results seem somewhat gratifying given that an upregulation of catalase in these vessels may be a key step in the control of oxidative/antioxidative process in vivo. On the other hand, the net effects of CYP2C29 in terms of its beneficial verses detrimental actions on the cardiovascular system have not fully been clarified, leading to the conflicting results in response to therapeutic strategies that target to CYP genes/enzymes. In this context, the actions of sEH inhibitors in the regulation of EET degradation but not synthesis, to increase circulating and vascular EET levels have attracted considerable attention. Indeed, studies involving the specific role of sEH inhibitors in the regulation of blood pressure and potentiation of vascular functions (Figure 8a) have been carried out, and sEH inhibitors are currently under investigation in phase II clinical trials for the treatment of hypertension and type 2 diabetes [7], and also in the phase I single- and multiple-dose studies in healthy human volunteers [5].
Acknowledgments
This work was supported by National Institute of Health (NIH HL070653 to A.H. and HL43023 to G.K).
Support provided by NIH HL070635 and HL43023
Abbreviations
- AUDA
12-(3-adamantan-1-yl-ureido)-dodecanoic acid
- CAT
catalase
- COX
cyclooxygenase
- CYP
cytochrome P450
- DHE
dihydroethidium
- DHETs
dithdroxyeicosatrienoic acids
- EDHF
endothelium-derived hyperpolarizing factor
- EETs
epoxyeicosatrienoic acids
- eNOS-KO
endothelial nitric oxide synthase knockout
- 2-EOH
2-hydroxyethidium
- H2O2
hydrogen peroxide
- HPLC
high-performance liquid chromatography
- L-NAME
NG-nitro-L-arginine-methyl ester
- O2-.
superoxide
- PEG-CAT
polyethylene glycol-catalase
- PPOH
6-(2-proparglyoxyphenel) hexanoic acid
- ROS
reactive oxygen species
- sEH
soluble epoxide hydrolase
- SOD
superoxide dismutase
- VAS2870
3-benzyl-7-(2-benzoxazolyl)thio-1,2,3-triazolo(4,5-d)pyrimidine
- WT
wild type
Footnotes
Conflict of Interest: None
References
- 1.Barlow RS, El-Mowafy AM, White RE. H(2)O(2) opens BK(Ca) channels via the PLA(2)-arachidonic acid signaling cascade in coronary artery smooth muscle. Am J Physiol Heart Circ Physiol. 2000;279:H475–H483. doi: 10.1152/ajpheart.2000.279.2.H475. [DOI] [PubMed] [Google Scholar]
- 2.Bodiga S, Zhang R, Jacobs DE, Larsen BT, Tampo A, Manthati VL, Kwok WM, Zeldin DC, Falck JR, Gutterman DD, Jacobs ER, Medhora MM. Protective actions of epoxyeicosatrienoic acid: dual targeting of cardiovascular PI3K and KATP channels. J Mol Cell Cardiol. 2009;46:978–988. doi: 10.1016/j.yjmcc.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Busse R, Fleming I. Regulation of endothelium-derived vasoactive autacoid production by hemodynamic forces. Trends Pharmacol Sci. 2003;24:24–29. doi: 10.1016/s0165-6147(02)00005-6. [DOI] [PubMed] [Google Scholar]
- 4.Campbell WB, Fleming I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch. 2010;459:881–895. doi: 10.1007/s00424-010-0804-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen D, Whitcomb R, MacIntyre E, Tran V, Do ZN, Sabry J, Patel DV, Anandan SK, Gless R, Webb HK. Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J Clin Pharmacol. 2012;52:319–328. doi: 10.1177/0091270010397049. [DOI] [PubMed] [Google Scholar]
- 6.Deng Y, Edin ML, Theken KN, Schuck RN, Flake GP, Kannon MA, DeGraff LM, Lih FB, Foley J, Bradbury JA, Graves JP, Tomer KB, Falck JR, Zeldin DC, Lee CR. Endothelial CYP epoxygenase overexpression and soluble epoxide hydrolase disruption attenuate acute vascular inflammatory responses in mice. FASEB J. 2011;25:703–713. doi: 10.1096/fj.10-171488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng Y, Theken KN, Lee CR. Cytochrome P450 epoxygenases, soluble epoxide hydrolase, and the regulation of cardiovascular inflammation. J Mol Cell Cardiol. 2010;48:331–341. doi: 10.1016/j.yjmcc.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dhanasekaran A, Gruenloh SK, Buonaccorsi JN, Zhang R, Gross GJ, Falck JR, Patel PK, Jacobs ER, Medhora M. Multiple antiapoptotic targets of the PI3K/Akt survival pathway are activated by epoxyeicosatrienoic acids to protect cardiomyocytes from hypoxia/anoxia. Am J Physiol Heart Circ Physiol. 2008;294:H724–H735. doi: 10.1152/ajpheart.00979.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edin ML, Wang Z, Bradbury JA, Graves JP, Lih FB, DeGraff LM, Foley JF, Torphy R, Ronnekleiv OK, Tomer KB, Lee CR, Zeldin DC. Endothelial expression of human cytochrome P450 epoxygenase CYP2C8 increases susceptibility to ischemia-reperfusion injury in isolated mouse heart. FASEB J. 2011;25:3436–3447. doi: 10.1096/fj.11-188300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fleming I. DiscrEET regulators of homeostasis: epoxyeicosatrienoic acids, cytochrome P450 epoxygenases and vascular inflammation. Trends Pharmacol Sci. 2007;28:448–452. doi: 10.1016/j.tips.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Fleming I, Michaelis UR, Bredenkotter D, Fisslthaler B, Dehghani F, Brandes RP, Busse R. Endothelium-derived hyperpolarizing factor synthase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res. 2001;88:44–51. doi: 10.1161/01.res.88.1.44. [DOI] [PubMed] [Google Scholar]
- 12.Granville DJ, Tashakkor B, Takeuchi C, Gustafsson AB, Huang C, Sayen MR, Wentworth P, Jr, Yeager M, Gottlieb RA. Reduction of ischemia and reperfusion-induced myocardial damage by cytochrome P450 inhibitors. Proc Natl Acad Sci U S A. 2004;101:1321–1326. doi: 10.1073/pnas.0308185100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayabuchi Y, Nakaya Y, Matsuoka S, Kuroda Y. Hydrogen peroxide-induced vascular relaxation in porcine coronary arteries is mediated by Ca2+-activated K+ channels. Heart Vessels. 1998;13:9–17. doi: 10.1007/BF02750638. [DOI] [PubMed] [Google Scholar]
- 14.Huang A, Kaley G. Gender-specific regulation of cardiovascular function: estrogen as key player. Microcirculation. 2004;11:9–38. doi: 10.1080/10739680490266162. [DOI] [PubMed] [Google Scholar]
- 15.Huang A, Sun D, Carroll MA, Jiang H, Smith CJ, Connetta JA, Falck JR, Shesely EG, Koller A, Kaley G. EDHF mediates flow-induced dilation in skeletal muscle arterioles of female eNOS-KO mice. Am J Physiol Heart Circ Physiol. 2001;280:H2462–H2469. doi: 10.1152/ajpheart.2001.280.6.H2462. [DOI] [PubMed] [Google Scholar]
- 16.Huang A, Sun D, Jacobson A, Carroll MA, Falck JR, Kaley G. Epoxyeicosatrienoic acids are released to mediate shear stress-dependent hyperpolarization of arteriolar smooth muscle. Circ Res. 2005;96:376–383. doi: 10.1161/01.RES.0000155332.17783.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang A, Sun D, Wu Z, Yan C, Carroll MA, Jiang H, Falck JR, Kaley G. Estrogen elicits cytochrome P450--mediated flow-induced dilation of arterioles in NO deficiency: role of PI3K-Akt phosphorylation in genomic regulation. Circ Res. 2004;94:245–252. doi: 10.1161/01.RES.0000111525.96232.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang A, Wu Y, Sun D, Koller A, Kaley G. Effect of estrogen on flow-induced dilation in NO deficiency: role of prostaglandins and EDHF. J Appl Physiol. 2001;91:2561–2566. doi: 10.1152/jappl.2001.91.6.2561. [DOI] [PubMed] [Google Scholar]
- 19.Huang A, Yan C, Suematsu N, Cuevas A, Yang YM, Kertowidjojo E, Hintze TH, Kaley G, Sun D. Impaired flow-induced dilation of coronary arterioles of dogs fed a low-salt diet: roles of ANG II, PKC, and NAD(P)H oxidase. Am J Physiol Heart Circ Physiol. 2010;299:H1476–H1483. doi: 10.1152/ajpheart.01250.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang A, Yang YM, Feher A, Bagi Z, Kaley G, Sun D. Exacerbation of endothelial dysfunction during the progression of diabetes: role of oxidative stress. Am J Physiol Regul Integr Comp Physiol. 2012 doi: 10.1152/ajpregu.00699.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacobson A, Yan C, Gao Q, Rincon-Skinner T, Rivera A, Edwards J, Huang A, Kaley G, Sun D. Aging enhances pressure-induced arterial superoxide formation. Am J Physiol Heart Circ Physiol. 2007;293:H1344–H1350. doi: 10.1152/ajpheart.00413.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larsen BT, Gutterman DD, Sato A, Toyama K, Campbell WB, Zeldin DC, Manthati VL, Falck JR, Miura H. Hydrogen peroxide inhibits cytochrome p450 epoxygenases: interaction between two endothelium-derived hyperpolarizing factors. Circ Res. 2008;102:59–67. doi: 10.1161/CIRCRESAHA.107.159129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu L, Chen C, Gong W, Li Y, Edin ML, Zeldin DC, Wang DW. Epoxyeicosatrienoic acids attenuate reactive oxygen species level, mitochondrial dysfunction, caspase activation, and apoptosis in carcinoma cells treated with arsenic trioxide. J Pharmacol Exp Ther. 2011;339:451–463. doi: 10.1124/jpet.111.180505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Bubolz AH, Mendoza S, Zhang DX, Gutterman DD. H2O2 is the transferrable factor mediating flow-induced dilation in human coronary arterioles. Circ Res. 2011;108:566–573. doi: 10.1161/CIRCRESAHA.110.237636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, Kanaide H, Takeshita A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. 2000;106:1521–1530. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsunaga T, Watanabe K, Yamamoto I, Negishi M, Gonzalez FJ, Yoshimura H. cDNA cloning and sequence of CYP2C29 encoding P-450 MUT-2, a microsomal aldehyde oxygenase. Biochim Biophys Acta. 1994;1184:299–301. doi: 10.1016/0005-2728(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 27.Mueller CF, Laude K, McNally JS, Harrison DG. ATVB in focus: redox mechanisms in blood vessels. Arterioscler Thromb Vasc Biol. 2005;25:274–278. doi: 10.1161/01.ATV.0000149143.04821.eb. [DOI] [PubMed] [Google Scholar]
- 28.Nieto N, Friedman SL, Cederbaum AI. Stimulation and proliferation of primary rat hepatic stellate cells by cytochrome P450 2E1-derived reactive oxygen species. Hepatology. 2002;35:62–73. doi: 10.1053/jhep.2002.30362. [DOI] [PubMed] [Google Scholar]
- 29.Pokreisz P, Fleming I, Kiss L, Barbosa-Sicard E, Fisslthaler B, Falck JR, Hammock BD, Kim IH, Szelid Z, Vermeersch P, Gillijns H, Pellens M, Grimminger F, van Zonneveld AJ, Collen D, Busse R, Janssens S. Cytochrome P450 epoxygenase gene function in hypoxic pulmonary vasoconstriction and pulmonary vascular remodeling. Hypertension. 2006;47:762–770. doi: 10.1161/01.HYP.0000208299.62535.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seubert J, Yang B, Bradbury JA, Graves J, DeGraff LM, Gabel S, Gooch R, Foley J, Newman J, Mao L, Rockman HA, Hammock BD, Murphy E, Zeldin DC. Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ Res. 2004;95:506–514. doi: 10.1161/01.RES.0000139436.89654.c8. [DOI] [PubMed] [Google Scholar]
- 31.Sun D, Huang A, Yan EH, Wu Z, Yan C, Kaminski PM, Oury TD, Wolin MS, Kaley G. Reduced release of nitric oxide to shear stress in mesenteric arteries of aged rats. Am J Physiol Heart Circ Physiol. 2004;286:H2249–H2256. doi: 10.1152/ajpheart.00854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun D, Jiang H, Wu H, Yang Y, Kaley G, Huang A. A novel vascular EET synthase: role of CYP2C7. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1723–R1730. doi: 10.1152/ajpregu.00382.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun D, Yang YM, Jiang H, Wu H, Ojaimi C, Kaley G, Huang A. Roles of CYP2C29 and RXR gamma in vascular EET synthesis of female mice. Am J Physiol Regul Integr Comp Physiol. 2010;298:R862–R869. doi: 10.1152/ajpregu.00575.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsao CC, Coulter SJ, Chien A, Luo G, Clayton NP, Maronpot R, Goldstein JA, Zeldin DC. Identification and localization of five CYP2Cs in murine extrahepatic tissues and their metabolism of arachidonic acid to regio- and stereoselective products. J Pharmacol Exp Ther. 2001;299:39–47. [PubMed] [Google Scholar]
- 35.Tsao CC, Wester MR, Ghanayem B, Coulter SJ, Chanas B, Johnson EF, Goldstein JA. Identification of human CYP2C19 residues that confer S-mephenytoin 4′-hydroxylation activity to CYP2C9. Biochemistry. 2001;40:1937–1944. doi: 10.1021/bi001678u. [DOI] [PubMed] [Google Scholar]
- 36.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 37.Wind S, Beuerlein K, Armitage ME, Taye A, Kumar AH, Janowitz D, Neff C, Shah AM, Wingler K, Schmidt HH. Oxidative stress and endothelial dysfunction in aortas of aged spontaneously hypertensive rats by NOX1/2 is reversed by NADPH oxidase inhibition. Hypertension. 2010;56:490–497. doi: 10.1161/HYPERTENSIONAHA.109.149187. [DOI] [PubMed] [Google Scholar]
- 38.Wolin MS. Reactive oxygen species and the control of vascular function. Am J Physiol Heart Circ Physiol. 2009;296:H539–H549. doi: 10.1152/ajpheart.01167.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu D, Cederbaum AI. Oxidative stress mediated toxicity exerted by ethanol-inducible CYP2E1. Toxicol Appl Pharmacol. 2005;207:70–76. doi: 10.1016/j.taap.2005.01.057. [DOI] [PubMed] [Google Scholar]
- 40.Wu Y, Huang A, Sun D, Falck JR, Koller A, Kaley G. Gender-specific compensation for the lack of NO in the mediation of flow-induced arteriolar dilation. Am J Physiol Heart Circ Physiol. 2001;280:H2456–H2461. doi: 10.1152/ajpheart.2001.280.6.H2456. [DOI] [PubMed] [Google Scholar]
- 41.Yan C, Huang A, Wu Z, Kaminski PM, Wolin MS, Hintze TH, Kaley G, Sun D. Increased superoxide leads to decreased flow-induced dilation in resistance arteries of Mn-SOD-deficient mice. Am J Physiol Heart Circ Physiol. 2005;288:H2225–H2231. doi: 10.1152/ajpheart.01036.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang B, Graham L, Dikalov S, Mason RP, Falck JR, Liao JK, Zeldin DC. Overexpression of cytochrome P450 CYP2J2 protects against hypoxia-reoxygenation injury in cultured bovine aortic endothelial cells. Mol Pharmacol. 2001;60:310–320. doi: 10.1124/mol.60.2.310. [DOI] [PubMed] [Google Scholar]
- 43.Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol. 2009;297:H1829–H1836. doi: 10.1152/ajpheart.00230.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang DX, Borbouse L, Gebremedhin D, Mendoza SA, Zinkevich NS, Li R, Gutterman DD. H2O2-Induced Dilation in Human Coronary Arterioles: Role of Protein Kinase G Dimerization and Large-Conductance Ca2+-Activated K+ Channel Activation. Circ Res. 2012;110:471–480. doi: 10.1161/CIRCRESAHA.111.258871. [DOI] [PMC free article] [PubMed] [Google Scholar]