

Abstract
Epidermal growth factor receptor (EGFR) plays pivotal roles in cell proliferation, differentiation, and tissue development, while EGFs protect neurons from toxic insults by binding EGFR and stimulating survival signaling. Furthermore, recent evidence implicates this receptor in neurometabolic disorders like Alzheimer disease and aging. Here we show that absence of presenilin 1 (PS1) results in dramatic decrease (>95%) of neuronal EGFR and that PS1-null (PS1−/−) brains have reduced amounts of this receptor. PS1−/− cortical neurons contain little EGFR and show no epidermal growth factor–induced survival signaling or protection against excitotoxicity, but exogenous EGFR rescues both functions even in absence of PS1. EGFR mRNA is greatly reduced (>95%) in PS1−/− neurons, and PS1−/− brains contain decreased amounts of this mRNA, although PS1 affects the stability of neither EGFR nor its mRNA. Exogenous PS1 increases neuronal EGFR mRNA, while down-regulation of PS1 decreases this mRNA. These effects are neuron specific, as PS1 affects the EGFR of neither glial nor fibroblast cells. In addition, PS1 controls EGFR through novel mechanisms shared with neither γ-secretase nor PS2. Our data reveal that PS1 functions as a positive transcriptional regulator of neuronal EGFR controlling its expression in a cell-specific manner. Severe downregulation of EGFR may contribute to developmental abnormalities and lethal phenotype found in PS1, but not PS2, null mice. Furthermore, PS1 may affect neuroprotection and Alzheimer disease by controlling survival signaling of neuronal EGFR.—Bruban, J., Voloudakis, G., Huang, Q., Kajiwara, Y., Al Rahim, M., Yoon, Y., Shioi, J., Gama Sosa, M. A., Shao, Z., Georgakopoulos, A., Robakis, N. K. Presenilin 1 is necessary for neuronal, but not glial, EGFR expression and neuroprotection via γ-secretase-independent transcriptional mechanisms.
Keywords: Alzheimer disease, EGFR mRNA, neurodegeneration, survival signaling
Mutations in genes encoding presenilin (PS) proteins PS1 and PS2 cause aggressive forms of early-onset familial Alzheimer disease (AD). Both PS proteins are found as important functional components of the proteolytic γ-secretase complexes that cleave many type I transmembrane proteins, including the amyloid precursor protein (APP), cadherins, Notch1, ErbB4, and EphB receptors (1). In addition, PS1 has γ-secretase-independent functions in cell signaling, intracellular trafficking, and neuronal survival (2, 3). Recently, several groups using artificially immortalized or cancer cells reported variable conclusions for the effects of PS1 and γ-secretase on the expression and function of epidermal growth factor receptor (EGFR) (4–7).
EGFR is a protein tyrosine kinase receptor that plays key roles in cell growth, cell differentiation, and tissue development and function, acting as an integrator where extracellular growth and survival signals converge and transform into intracellular outputs (8, 9). Furthermore, EGFR is known to play pivotal roles in human cell transformation and cancer (8, 9). Ligands to this receptor, known as epidermal growth factors (EGFs), are found in brain, where they regulate neuronal development, function, and survival (10–13). Binding of EGFs to their cognate receptor stimulates EGFR-dependent phosphorylation of survival kinases, thus increasing neuronal survival against toxic insults such as excitotoxicity, oxidative stress, and ischemia (11–16). Functions of EGFR have been implicated in a spectrum of neurometabolic disorders including diabetes, AD, and aging (8, 17). The crucial roles the EGF ligand–receptor system plays in development was shown by data that mouse embryos lacking either EGFR or its brain-enriched ligand heparin-binding EGF-like growth factor (Hb-EGF) die at birth (18–22), while surviving animals have cortical neurodegeneration (23). PS1-null mice also die at birth displaying severe neuronal abnormalities, highlighting the importance of both PS1 and EGFR in development and brain function. Importantly, although both PS1 and PS2 have γ-secretase activity, only the absence of PS1 results in severe developmental abnormalities and lethal phenotypes (24, 25).
Recently, we reported that PS1 is necessary for the neuroprotective functions of specific growth factors including brain-derived neurotrophic factor (BDNF) and ephrinB (3), but to our knowledge, no evidence for a PS1 role in the neuroprotective functions of EGFR or its ligands has been reported. In contrast, several groups have reported that PS1 suppresses expression of EGFR in immortalized mouse embryonic fibroblast (iMEF) cells, but proposed mechanisms are unclear because both pre- and posttranslational mechanisms of EGFR inhibition by PS1 were proposed (4–6). Furthermore, evidence for and against involvement of γ-secretase in the regulation of cellular levels of EGFR has been described in immortalized cells (4–7). Here we present data that, in contrast to results in iMEF cell lines, PS1 functions as a positive and specific regulator of neuronal EGFR. Thus, compared to wild-type (WT), PS1-null neurons contain very little EGFR and show no EGF-dependent signaling and neuroprotection against glutamate excitotoxicity. Absence of PS1, however, has no effect on the EGFR of primary glia or fibroblast cells, while PS1-null brain tissue shows a significant reduction of this receptor. Importantly, our data reveal that PS1, but not PS2, increases the levels of neuronal EGFR by transcriptional mechanisms independent of protein turnover and γ-secretase activity.
MATERIALS AND METHODS
Materials
Antibodies against EGFR were purchased from EMD Millipore (Billerica, MA, USA), tubulin from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and microtubule-associated protein 2 (MAP2) from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against actin, doublecortin, phospho-EGFR (Y1068), protein kinase B (AKT), phospho-AKT (S473 and T308), ERK, and phospho-ERK (T202/Y204) were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-PS1 monoclonal antibody 33B10 and R1 antiserum against C-terminal APP were described previously (26). Erlotinib and AG-1478 were from Selleck Chemicals (Houston, TX, USA). EGF, Hb-EGF, cycloheximide (CHX), U0126, wortmannin, and γ-secretase inhibitor L-645,458 were from Sigma-Aldrich. A Smartpool of Accell anti-PS1 small interfering RNA (siRNA; Dharmacon, Lafayette, CO, USA) containing a mixture of 4 siRNAs against PS1 and nontargeting Smartpool siRNAs (On-Targetplus for immortalized fibroblasts and Accell for neurons and primary fibroblasts) were from Dharmacon.
Cell cultures
WT and PS1 heterozygous (PS1+/−) knockout (KO) mouse colonies as well as production and genotyping of WT, PS1+/−, and PS1 homozygous KO (PS1−/−) mouse embryos were as described (3). In addition, Supplemental Fig. 1 shows increased accumulation of APP/carboxyl-terminal fragment 1 (CTF1) and N-cad/CTF1 fragments known to occur in the absence of PS1 (1, 27). All animal experiments were carried out according to regulations of Mount Sinai Medical Center. Primary cortical neuronal cultures (PCNC) were prepared from embryonic day (E) 15.5 mouse embryo brains, plated on poly-d-lysine-coated plates in Neurobasal medium as reported elsewhere (3, 28, 29) and kept in vitro before use as indicated in figure legends. At 9 d in vitro (DIV), postmitotic neurons represented more than 95% of cultured cells (Supplemental Fig. 2). Immortalized mouse embryonic fibroblast (MEF) cell lines were grown in DMEM plus 10% fetal bovine serum, penicillin-streptomycin (100 U/ml) in 5% CO2 at 37°C. Primary fibroblast (30) and primary glial cultures containing mostly astrocytes were prepared from mouse embryo brains as described (31). Glial cells were plated at 3 × 105 cells/cm2 in DMEM supplemented with 10% fetal bovine serum, penicillin-streptomycin (100 U/ml) in 5% CO2 at 37°C. Medium was replaced 24 h after plating, and cells were left growing for 8 d. Cells were then trypsinized and replated at a density of 3 × 104 cells/cm2 until desired confluence. MEF cells were isolated from trypsinized mouse tissue at E15 and immortalized using SV40 T antigen (pSV3neo), and clones were maintained in the presence of G418 (400–100 μg/ml). Single clones were selected at low density using cloning cylinders and individually propagated.
Western blot analysis
Cell lysates were prepared in RIPA (50 mM Tris/HCl, pH7.4, 150 mM NaCl, 1 mM EDTA, 0.5% sodium deoxycholate, 1% Triton X-100, and 0.1% w/v SDS) supplemented with protease (Roche Diagnostics, Mannheim, Germany) and phosphatase inhibitors (20 mM NaF, 5 mM Na3VO4, 1 mM sodium pyrophosphate, and 100 nM microcystin-LR). Mouse brain lysates were prepared from E15.5 embryos by mechanical dissociation and sonication in RIPA buffer. Samples were centrifuged at 14,000 g, and protein in supernatants was measured using bicinchoninic acid assays (Pierce, Rockford, IL, USA). Aliquots were diluted with 3× Laemmli buffer and denatured in a boiling water bath; equal amounts of protein were resolved on SDS-PAGE, followed by Western blot (WB) analysis and protein detection with antibodies as described elsewhere (3).
Cell viability
Neuronal cell survival was evaluated using the 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay as previously described (3). Briefly, after incubation with growth factors and glutamate, MTT was added to neurons and incubated for 3 h at 37°C under 5% CO2. Cultures were then washed with PBS and isopropanol/HCl 1 N and incubated under shaking for 5 min; then 100 μl of supernatant was transferred into a fresh 96-well plate and absorbance measured at an optical density of 560 to 620 nm. Use of neuronal nuclear staining (Hoechst staining kit; Sigma-Aldrich), as previously described (3, 32), gave similar results. For convenience, MTT assays were used in this work.
Real-time PCR, DNA constructs, and transfections
Total RNA was isolated using the mirVana extraction kit and quantified according to manufacturer’s protocol (Ambion, Austin, TX, USA). cDNA was synthesized using 320 ng RNA, oligo(dT) primers, and Superscript III Reverse transcriptase according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). PCR primers were as follows: EGFR, 5′-gccatctgggccaaagatacc-3′ and 5′-gtcttcgcatgaataggccaat-3′; glyceraldehyde phosphate dehydrogenase (GAPDH), 5′-aggtcggttgtgaacggatttg-3′ and 5′-tgtagaccatgtagttgaggtca-3′. PCR amplification mixtures were prepared using QuantiFast SYBR Green PCR kit according to the manufacturer’s instructions (Qiagen, Germantown, MD, USA), and real-time PCR assay was performed on an ABI PRISM 7900HT sequence detection system (Applied Biosystems, Foster City, CA, USA). Mouse PS1 (FUGW–Mus musculus PS1)- or EGFR (FCbAIGW–Mus musculus EGFR)-expressing plasmids were constructed by cloning mouse PS1 or EGFR cDNA with upstream Kozak sequences into the FUGW or FCbAIGW lentiviral backbone vectors, respectively, and transduction of neurons was performed using Amaxa technology and the Nucleofector kit (Lonza, Basel, Switzerland) Primary Neurons (program O-005) according to the manufacturer’s instructions.
RESULTS
EGFR-mediated signaling and neuroprotection are abolished in PS1-null neurons
Recent literature reports that PS1 negatively regulates EGFR (4–6), suggesting that signaling by this receptor may increase in the absence of PS1. To investigate PS1 effects on survival signaling of neuronal EGFR, brain PCNC from WT, PS1 heterozygous (PS1+/−), and homozygous (PS1−/−) KO mice prepared as previously described (3) were treated with EGFR ligands EGF and Hb-EGF, and AKT and ERK phosphorylation signaling mediated by EGFR stimulation was analyzed (33). As expected, these treatments caused rapid increases in the phosphorylation of both kinases in WT and PS1+/− neurons. Surprisingly, however, the EGFs failed to stimulate phosphorylation of either kinase in PS1-null (PS1−/−) neurons, indicating that EGFR signaling is attenuated in the absence of PS1 (Fig. 1A). To ensure that activation of ERK and AKT was not delayed in PS1−/− neurons, we examined phosphorylation kinetics in response to Hb-EGF, an EGFR ligand abundantly expressed in the brain (13). Hb-EGF caused a sustained phosphorylation of both kinases in WT and PS1+/−, but not in PS1−/−, neurons (Fig. 1B). Thus, contrary to expectations, our data suggested that the absence of PS1 severely attenuates EGF-induced neuronal signaling and that presence of even one PS1 allele is sufficient for full signaling (Fig. 1A, B). In contrast, BDNF, a factor that signals to AKT and ERK through TrkB receptor, stimulated phosphorylation of these kinases in both WT and PS1−/− neurons, indicating that signaling of neuronal EGFR specifically decreases in the absence of PS1 (Fig. 1C). Because ERK and AKT mediate survival signaling of the neuroprotective system EGF/EGFR (11, 33), we asked whether EGF-dependent neuroprotection changes in the absence of PS1. To this end, neuronal cultures were treated with glutamate in the presence or absence of either EGF or Hb-EGF. Figure 2A shows that although these ligands decreased the glutamate-induced neuronal death in both PS1+/+ and PS1+/− cultures, they were unable to decrease neuronal death in PS1−/− cultures, indicating that PS1 is required for EGF-dependent neuroprotection. In contrast, progranulin rescues PS1-null neurons from excitotoxicity regardless of PS1 gene dosage (Fig. 2A) (32), confirming that EGF-dependent neuroprotection is specifically regulated by PS1. To examine which kinase is required for EGF neuroprotection, neuronal cultures were treated with either MEK/ERK1/2 inhibitor U0126 or PI3K/AKT inhibitor wortmannin followed by glutamate. Although these treatments decreased the Hb-EGF-induced activating phosphorylation of ERK and AKT, respectively (Fig. 2B), only inhibition of PI3K/AKT had a significant effect on the Hb-EGF-induced neuroprotection (Fig. 2C). Together, our data indicate that PS1 is necessary for EGF-induced phosphorylation of both AKT and ERK but that AKT activation is mainly responsible for EGF-dependent neuroprotection against excitotoxicity in vitro.
Figure 1.
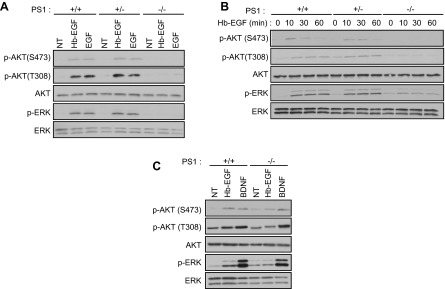
EGF-dependent activation of ERK and AKT survival signaling is abolished in PS1-deficient neurons. A) PCNC from WT (+/+), PS1 heterozygous (+/−), and PS1 homozygous (−/−) KO embryonic mouse brains cultured in 6-well plates were treated at 9 DIV with 20 ng/ml EGF or Hb-EGF for 15 min. After incubation, lysates were collected and probed on WBs for the indicated proteins. B) WT, PS1+/−, or PS1−/− PCNC prepared as above were treated at 9 DIV with 20 ng/ml Hb-EGF. After incubation, lysates were collected at different time points and assayed on WBs for the indicated proteins. Data were obtained from at least 3 separate experiments. C) WT or PS1−/− PCNC as above were treated at 9 DIV with 20 ng/ml Hb-EGF or 100 ng/ml BDNF for 15 min. Nontreated cultures (NT) were used as controls. After incubation, lysates were collected and assayed on WBs for the indicated proteins.
Figure 2.
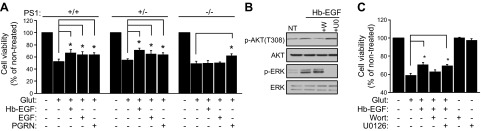
EGF neuroprotection against excitotoxicity is abolished in PS1-deficient neurons. A) WT, PS1+/−, and PS1−/− mouse PCNC were grown in 24-well plates in Neurobasal medium plus B27 supplement. At 9 DIV, neurons were treated with 20 ng/ml Hb-EGF or EGF or 35 nM progranulin (PGRN; a.k.a. GRN) overnight. The next day, the medium was switched to HBSS containing Hb-EGF, EGF, or PGRN for 30 min, followed by 50 μM glutamate incubation for 3 h, and cell viability was evaluated by MTT assay and normalized to nontreated cells, as previously described (3, 29). No significant effect compared to nontreated was observed when growth factors alone were added to cultures. B) WT PCNC in 6-well plates were treated at 9 DIV either with ERK inhibitor U0126 (U0, 5 μM) or PI3K/AKT inhibitor wortmannin (W, 50 nM) for 30 min before addition of 20 ng/ml Hb-EGF for 15 min in Neurobasal medium plus B27 supplement. After incubation, lysates were collected and assayed on WBs for the indicated proteins. C) Mouse PCNC grown as above [as in (A)] were treated at 9 DIV either with ERK inhibitor U0126 (5 μM) or PI3K/AKT inhibitor wortmannin (50 nM) for 30 min before addition of 20 ng/ml Hb-EGF. Three hours later, medium was switched to HBSS plus Hb-EGF and inhibitors for 30 min, followed by 50 μM glutamate incubation for 3 h. Cell viability was evaluated by MTT assay and normalized to nontreated cells. Results (means ± sem) were summarized from at least 4 independent experiments. In each experiment, each condition is the average of 4 identical wells. *P < 0.05 comparing between cultures treated with glutamate in the presence or absence of Hb-EGF and/or inhibitors (paired Student’s t test).
PS1 positively regulates neuronal EGFR
To determine the mechanism by which PS1 facilitates survival signaling of EGF ligands, we examined the PS1 effects on the expression of neuronal EGFR. Surprisingly, in contrast to reports that PS1 negatively regulates this receptor in iMEF cells (4–6), levels of neuronal EGFR were dramatically decreased (>95%) in PS1−/− neurons compared to WT (Fig. 3A). This outcome was replicated in several independent neuronal cultures prepared from different embryonic mouse brains derived from distinct pregnancies. Notably, PS1+/− neurons contain similar amounts of EGFR as WT neurons (Fig. 3A), showing that the absence of one PS1 allele has little effect on neuronal EGFR—an outcome consistent with similar signaling and neuroprotective activities of EGFs in WT and PS1+/− neurons (Fig. 2). Additional experiments showed that absence of PS1 causes a dramatic EGFR reduction in all neuronal cultures regardless of age (Fig. 3B) and that tissue from PS1−/− brains probed by either WB test (Fig. 3C) or immunohistochemistry (Supplemental Fig. 3) also contained reduced amounts of this receptor. Furthermore, examination of the neuronal markers doublecortin and MAP2 showed that the amounts of these proteins in our cultures and brain tissue were independent of PS1 (Fig. 3D), further supporting specific PS1 effects on EGFR. Because the absence of PS1 causes a smaller EGFR reduction in brain tissue than in PS1−/− neurons (40% vs. 95%, respectively; Fig. 3A, C), we asked whether PS1 deficiency exerts a less dramatic effect on EGFR of nonneuronal brain cells. Figure 3E shows that the levels of this receptor in primary glia from PS1−/− brains do not differ significantly from those in glia from WT brains, suggesting that the absence of PS1 results in specific reduction of neuronal EGFR. This outcome is consistent with data that absence of PS1 causes a more dramatic decrease in neuronal than brain EGFR, as total amount of brain EGFR is determined by the sum of its expression in all brain cells.
Figure 3.
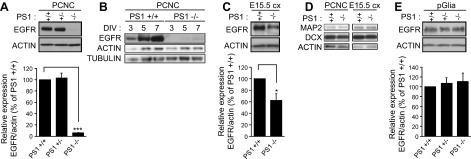
PS1 positively correlates with cellular levels of neuronal EGFR in vitro and in vivo. A) (Top) Lysates from WT, PS1+/−, or PS1−/− PCNC grown in 6-well plates were prepared at 9 DIV and probed on WBs for the indicated proteins. (Bottom) Densitometric analysis of the relative amounts of EGFR in PCNC is expressed as ratio of EGFR to actin. B) Lysates from WT (+/+) or PS1−/− PCNC grown as above were prepared at 3, 5, or 7 DIV and probed on WBs for indicated proteins. C) (Top) Lysates from embryonic brain cortex (E15.5 cx) from WT or PS1 homozygous KO were prepared as described in the Materials and Methods. (Bottom) Densitometric analysis of the relative amounts of EGFR in embryonic cortices is expressed as ratio of EGFR to actin. D) (Left) Lysates from WT or PS1−/− PCNC grown as above were prepared at 9 DIV and probed on WBs for the indicated proteins. (Right) Lysates from WT and PS1−/− embryonic brain cortex (E15.5 cx) were prepared and probed on WBs for indicated proteins. E) (Top) Primary glial cultures (pGlia) from WT, PS1 heterozygous, or homozygous KO were obtained as described in the Materials and Methods. Cells were cultured in 6-well plates. When cells reached about 80% confluence, lysates were collected and assayed on WBs for the proteins indicated. (Bottom) Densitometric analysis of the relative amounts of EGFR in primary glial cultures is expressed as ratio of EGFR to actin. Data were respectively obtained from at least 4 separate experiments. *P < 0.05; ***P < 0.001 (paired Student’s t test).
To examine whether acute down-regulation of PS1 has similar effects on expression and signaling of neuronal EGFR as chronic absence of PS1, we used anti-PS1 siRNAs. Because PS1+/− neurons have similar levels of EGFR and exhibit similar EGF signaling as WT neurons, and to ensure efficient down-regulation of EGFR by siRNAs, we used these neurons in our experiments. As shown in Fig. 4, acute knockdown of neuronal PS1 decreased both the amounts of EGFR (Fig. 4A) and the EGF-dependent neuroprotection against glutamate excitotoxicity (Fig. 4B). These data show that acute down-regulation of PS1 has similar effects on the expression of EGFR and EGF-dependent neuroprotection as those observed under conditions of chronic absence of PS1. Finally, reintroduction of mouse PS1 in PS1-null neurons caused a robust increase of EGFR (Fig. 4C), further supporting the conclusion that PS1 strongly stimulates the neuronal expression of EGFR.
Figure 4.

Acute knockdown of PS1 decreases EGFR and abolishes EGF neuroprotection while reintroduction of PS1 in PS1−/− neurons increases EGFR. A) (Top) PCNC from PS1 heterozygous KO (+/−) were cultured in 12-well plates and at 5 DIV were treated with 1 μM Accell Smartpool siRNA against PS1 for 72 h. Nontreated cultures were used as controls. After incubation, lysates were collected and assayed by WBs for the indicated proteins. WT neurons (+/+) were also included as control. (Bottom) Densitometric analysis of the relative amounts of EGFR and PS1–C-terminal fragment in PCNC is expressed as ratio to actin, then normalized as a percentage of EGFR or PS1–C-terminal fragment amount in the nontreated WT neurons. Data were respectively obtained from 4 separate experiments. *P < 0.05 (paired Student’s t test). No effect was observed on PS1 when neurons were treated with 1 μM Accell Smartpool scramble siRNA for 72 h (data not shown). B) PS1+/− PCNC were grown in 24-well plates with Neurobasal medium plus B27 supplement. At 5 DIV neurons were treated with 1 μM Accell Smartpool siRNA for 72 h and then incubated with 20 ng/ml Hb-EGF overnight. The next day, the medium was switched to HBSS containing Hb-EGF for 30 min, followed by 50 μM glutamate incubation for 3 h. Cell viability was evaluated by MTT assay and normalized to nontreated cells. Results (means ± sem) were summarized from 6 independent experiments; in each experiment, each condition is the average of 4 identical wells. *P < 0.05 comparing between cultures treated with glutamate in the presence or absence of Hb-EGF, PS1-SiRNA, or nontargeting siRNA (paired t test). C) (Top) PS1−/− PCNC were mock transfected (left lane) or transfected with either mouse PS1 in FUGW vector or vector alone. After incubation, lysates were collected at 8 DIV and assayed on WBs for the indicated proteins. (Bottom) Densitometric analysis of the amounts of EGFR in transfected PS1−/− neuronal cultures above is expressed as ratio of EGFR to actin and normalized to mock nucleofected neurons. Bars represent mean and error bars sem. Data were obtained from 4 independent experiments. *P < 0.05 (paired Student’s t test).
To establish a direct link between reduced neuronal survival in the absence of PS1 and expression of EGFR, we expressed exogenous EGFR in PS1−/− neurons. Figure 5A shows that exogenous EGFR rescues the ability of EGF to increase neuronal survival even in the absence of PS1. Furthermore, expression of EGFR restores the ability of EGF to stimulate phospho-EGFR and activate its downstream targets AKT and ERK even in the absence of PS1 (cf. Fig. 5B and Fig. 1A). Importantly, Fig. 5C shows that in our neuronal cultures, EGFR dominates the survival signaling of both EGF and Hb-EGF, as treatment with EGFR inhibitor erlotinib blocks induced phosphorylation of AKT and ERK. These data reveal a direct link between EGFR and PS1-dependent phenotypes on neuronal survival and activation of AKT kinase.
Figure 5.
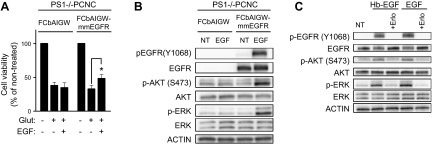
Expression of exogenous EGFR restores the PS1-dependent ability of EGF to rescue neurons from excitotoxicity and stimulate survival signaling. A) PS1−/− neurons were transfected with either empty vector (FCbAIGW) or an EGFR-expressing construct (FCbAIGW-EGFR), and at 8 DIV, neurons were treated with 20 ng/ml EGF overnight, as indicated. Next day, the medium was switched to HBSS containing EGF for 30 min, followed by 50 μM glutamate incubation for 3 h. Cell viability was then measured, counting healthy nuclei with a Hoechst 33342 kit (Materials and Methods) and normalized to nontreated cells as previously described (3). Results (mean ± sem) are from 3 independent experiments. *P < 0.05 (paired Student's t test). B) PS1−/− neurons were transfected with EGFR-expressing plasmid FCbAIGW-EGFR or vector alone (FCbAIGW), and at 8 DIV, cultures were treated with 20 ng/ml EGF for 15 min. Lysates were then collected and probed on WBs for indicated proteins. C) WT PCNC of 8 DIV were incubated with 10 μM erlotinib (Erlo) for 30 min and then treated with 20 ng/ml of either Hb-EGF or EGF. Cultures were incubated for an additional 15 min, and lysates were assayed for indicated proteins.
Neither γ-secretase nor PS2 regulates expression of neuronal EGFR
PS1 has been shown to have both γ-secretase-dependent and -independent functions (2, 3). However, use of immortalized cell systems to examine the effects of γ-secretase on the cellular levels of EGFR yielded inconsistent answers, as evidence has been reported both against (4, 6) and for (5, 7) involvement of γ-secretase activity in the regulation of this receptor. To examine the effects of γ-secretase on neuronal EGFR, we treated primary neuronal cultures with L-685,458, a potent γ-secretase inhibitor widely used in our and other laboratories (5, 26, 27). Figure 6A shows that although L-685,458 causes a robust accumulation of APP-derived γ-secretase substrates APP-CTFs, this treatment has no effect on neuronal EGFR, indicating that γ-secretase is not involved in the regulation of the expression of this receptor. To further explore this issue, we probed iMEF cells, which have been widely used to explore the role of γ-secretase in the regulation of EGFR (4–7). Figure 6B shows that, in agreement with data in primary neurons, inhibition of γ-secretase has no effect on the EGFR of iMEF cells. Because γ-secretase activity cleaves a large number of type I transmembrane proteins (1), we asked whether EGFR, a type I protein, may be processed by this activity. However, our efforts to detect EGFR-derived peptides expected from the γ-secretase processing of proteins (26, 34, 35) or complexes between PS1 and EGFR using coimmunoprecipitation protocols were unsuccessful (Georgakopoulos et al., unpublished data). Similar to PS1, its homolog PS2 also functions as a catalytic component of γ-secretase complexes that process substrates including APP and Notch1 (36, 37). We thus used PS2-null (PS2−/−) neurons to ask whether PS2 regulates neuronal EGFR. Figure 7 shows that absence of PS2 affects neither the levels of this receptor nor its neuroprotective activity, supporting the conclusion that PS1 controls EGFR through specific mechanisms independent of both γ-secretase and PS2.
Figure 6.

Inhibition of γ-secretase has no effect on the expression of EGFR. A) WT PCNC were grown in 6-well plates for 9 d and then treated with γ-secretase inhibitor L-685,458 (500 nM) overnight (+). WT control (−) cultures were treated with the same solution without the inhibitor. After incubation, lysates were collected and assayed on WBs for the indicated proteins. (Bottom) Densitometric analysis of the relative amounts of EGFR and APP-CTFs in PCNC normalized to actin and expressed as the percentage of the amounts in nontreated controls. Data were from 4 independent experiments. ***P < 0.001 (paired Student’s t test). B) Cultures of iMEF cells were treated overnight with γ-secretase inhibitor L-685,458 (+) as above or with medium without inhibitor as controls (−). After incubation, lysates were collected and assayed on WBs for indicated proteins. (Bottom) Densitometric analysis of the relative amounts of EGFR and APP-CTFs in iMEF cells as above. Data are from 4 separate experiments as above.
Figure 7.

PS2 affects neither EGFR expression nor EGF-dependent neuroprotection. A) (Top) PCNC from WT (+/+) and PS2 heterozygous (+/−) and homozygous (−/−) KO embryonic mouse brains were cultured in 6-well plates. At 9 DIV, lysates were collected and probed on WBs for the indicated proteins. (Bottom) Densitometric analysis of the relative amounts of EGFR shown above expressed as EGFR-to-actin ratio. B) WT, PS2+/−, and PS2−/− mouse PCNC were grown in 24-well plates. At 9 DIV, cultures were treated overnight with 20 ng/ml of either Hb-EGF or EGF. Next day, the medium was switched to HBSS containing Hb-EGF or EGF for 30 min, followed by 50 μM glutamate incubation for 3 h (Fig. 2A). Cell viability was evaluated by MTT assay and normalized to nontreated cells. Treatment with growth factors alone had no effect on neuronal viability compared to untreated cultures. Bars represent mean and error bars sem. Data were respectively obtained from at least 4 separate experiments. *P < 0.05 [unpaired Student’s t test for (A), paired Student’s t test for (B)].
Mechanisms of regulation of neuronal EGFR
Recent data obtained in iMEF cell lines indicate that PS1 regulates EGFR posttranslationally by promoting its degradation through the proteasomal and lysosomal systems (4, 6). In contrast, others have reported that PS1 negatively regulates transcription of EGFR in iMEF cell lines (5). We measured the turnover rate of EGFR in WT and PS1 KO neurons using the protein synthesis inhibitor CHX. Our data show that EGFR turnover in PS1−/− neurons is indistinguishable from that in WT neurons (Fig. 8A). To examine involvement of transcriptional controls, we measured the Egfr mRNA by quantitative real-time PCR using independent neuronal preparations. We found that the levels of this mRNA in PS1−/− neurons were reduced by more than 95% compared to WT neurons (Fig. 8B). Thus, protein and mRNA data indicate that PS1 positively regulates EGFR at the mRNA level. Measurements of brain mRNA also support a positive regulation of Egfr mRNA by PS1, as this mRNA is significantly decreased in PS1−/− embryonic brains compared to WT (Fig. 8C). Furthermore, similar to the relative decrease of EGFR protein in PS1−/− neurons and brain tissue (Fig. 3), reduction of Egfr mRNA in PS1-null brains is smaller than its reduction in PS1−/− neurons, in agreement with a specific decrease of neuronal mRNA in brain. Furthermore, similar to results in PS1−/− neurons, acute down-regulation of PS1 reduced the neuronal Egfr mRNA (Fig. 8D), supporting the conclusion that PS1 positively regulates this mRNA. To examine whether PS1 controls the stability of this mRNA, we used actinomycin to arrest transcription. Figure 8E shows that Egfr mRNA declines at similar rates in the presence or absence of PS1, indicating that PS1 regulates the Egfr mRNA at a step before synthesis of mature transcripts.
Figure 8.
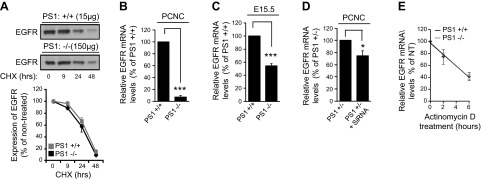
PS1 positively regulates expression of neuronal Egfr mRNA. A) (Top) WT (PS1+/+) or PS1 homozygous KO (PS1−/−) PCNC in Neurobasal medium plus B27 supplement were treated at 7 DIV with 50 μg/ml CHX for the indicated times. Nontreated (0 time) cultures were used as controls. After incubation, lysates (15 μg from WT or 150 μg from PS1−/−) were collected and probed on WBs for EGFR. (Bottom) Kinetic analysis of the relative amounts of EGFR in PCNC after CHX treatment expressed as a percentage of EGFR amount in time 0 samples. Data were obtained from 4 independent experiments. B, C) Total mRNA was isolated from WT (PS1+/+) or PS1-null (PS1−/−) mouse PCNC of 8 DIV (B) or embryonic cortex (E15.5) (C). After cDNA amplification using E15.5 cortical mRNA, quantitative real-time PCR assay was performed as described in Materials and Methods. Data were respectively obtained from at least 4 separate experiments. ***P < 0.001 (paired Student’s t test). D) PS1 heterozygous KO (PS1+/−) PCNC grown on 12-well plates were treated at 5 DIV with 1 μM Accell Smartpool siRNA against PS1 for 72 h. Nontreated cultures were used as controls. After incubation, total mRNA was isolated, followed by cDNA amplification and quantitative real-time PCR performed as described in the Materials and Methods. Data were respectively obtained from at least 4 separate experiments. *P < 0.05 (paired Student’s t test). E) PCNC of WT (PS1+/+) or PS1-null (PS1−/−) mice were treated with actinomycin D (2 μg/ml) to arrest transcription, and total RNA was isolated at 0, 2, or 6 h later. RNA samples were then subjected to cDNA amplification and quantitative real-time PCR, and relative amounts of Egfr mRNA were calculated using GAPDH as an internal control (ΔCt). Amounts of Egfr mRNA are expressed as a percentage of its amounts at time 0 and plotted against time using the 2−ΔΔCt method. The data were fitted to the linear regression model, and the slope of the curve was similar between genotypes: −0.159 in WT vs. −0.162 in PS1 KO (n = 3, 95% confidence interval).
In contrast to our data that PS1 positively regulates expression of neuronal EGFR, several groups have reported that PS1 inhibits expression of this receptor in nonneuronal systems. However, because evidence of a negative correlation between PS1 and EGFR was obtained using immortalized double KO (DKO) fibroblast cells line lacking both PS2 and PS1 (4–6), we examined the expression of EGFR in primary fibroblasts lacking only PS1. Similar to data obtained in primary glia cells, absence of either one or both alleles of PS1 had no effect on EGFR, a result consistently obtained in several independent preparations of primary fibroblasts (Fig. 9A). Importantly, measurements in distinct iMEF cell lines revealed clonal variability of the EGFR levels independent of PS1 expression or genotype (Fig. 9B), suggesting that clonal selection of iMEF cell lines used to examine PS1 effects on EGFR may contribute to disagreements in the literature on the role of PS1 on EGFR (4–7). We thus examined the effects of acute down-regulation of PS1 on the EGFR of iMEF cells using anti-PS1 siRNA. Figure 9C shows that down-regulation of PS1 in 2 independent PS1 heterozygous KO (PS1+/−) iMEF clones had no significant effect on the levels of EGFR. Furthermore, in contrast to data in neurons (Fig. 4C), reintroduction of PS1 in PS1 KO iMEF clones showed no effects on EGFR (Fig. 9D). These experiments indicate that PS1 may not regulate the EGFR of fibroblast cells, a conclusion strongly supported by absence of PS1 effects on the EGFR of primary fibroblasts (Fig. 9A). Together, our data show that PS1 positively regulates neuronal EGFR but has no effect on the expression of this receptor in glia or fibroblast cells. Our results, however, do not exclude the possibility that PS1 may regulate this receptor in other cell systems not examined here (38).
Figure 9.
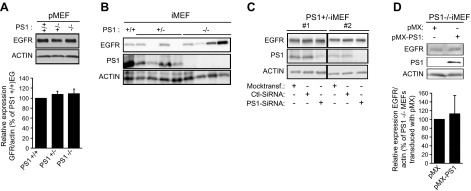
Absence of PS1 has no effect on EGFR expression of primary fibroblasts or iMEF cells. A) (Top) Primary mouse fibroblasts (pMEF) from WT (+/+), PS1+/−, and PS1−/− mouse embryos were obtained as described in the Materials and Methods. Cells were cultured in 6-well plates at 80% confluence, and lysates were prepared and assayed on WBs for the indicated proteins. (Bottom) Relative amounts of EGFR in pMEFs of the genotypes shown in the top image are expressed as EGFR-to-actin ratio (n = 6). B) Lysates from WT (+/+), PS1+/−, and PS1−/− MEF cell lines (iMEF) were assayed on WBs for the indicated proteins. Each cell line is derived from an independent iMEF clone. C) Immortalized PS1 heterozygous KO MEFs (PS1+/− iMEF) were transfected with 25 nM of anti-PS1 siRNA for 72 h. Mock-transfected and scrambled siRNA-transfected (Ctl-siRNA) cultures were used as controls. After incubation, lysates were collected and assayed by WBs for the indicated proteins. D) (Top) PS1-null iMEFs (PS1−/− iMEF) were stably transduced with either human PS1 in pMX vector or vector alone. Lysates were prepared and assayed on WBs for indicated proteins. (Bottom) Densitometric analysis of relative amounts of EGFR in 4 different PS1 KO iMEF clones transfected either with pMX vector or pMX-PS1. Amount of EGFR in each PS1-transfected clone is normalized to the EGFR of the vector-transfected clone. Bars represent mean and error bars sem.
DISCUSSION
EGFR is a key tyrosine kinase growth factor receptor expressed in many cell types, where it regulates growth, migration, and differentiation (8, 39, 40). In addition, this receptor plays pivotal roles in tumor development and proliferation (8, 41), while other studies have found that EGFR signaling controls neuronal function and survival (10–13). Thus, ligands to this receptor, such as Hb-EGF and EGF, protect brain neurons from excitotoxicity, oxygen/glucose deprivation, and traumatic injuries—insults implicated in the pathogenesis of neurodegenerative disorders and stroke (12–16). Recently, we reported that PS1 is necessary for neuroprotective activities of BDNF and ephrinB, a function independent of γ-secretase activity (3). Here we show that in the absence of PS1, cortical neurons are unable to activate the EGFR neuroprotective signaling in response to EGFs, an outcome consistently obtained under both chronic absence of PS1 in PS1−/− neurons and acute down-regulation of neuronal PS1 using siRNAs. Thus, in contrast to WT neurons, neither EGF nor Hb-EGF showed any neuroprotective activity against excitotoxicity in PS1−/− neuronal cultures, suggesting that PS1 is indispensable for EGF-dependent neuroprotection. Furthermore, in the absence of PS1, EGFs are unable to stimulate activating phosphorylation of survival kinase AKT, an event critical to the neuroprotective function of EGF factors against excitotoxicity. Interestingly, in contrast to BDNF- and ephrinB-dependent neuroprotection, in which expression of both PS1 alleles is required (3), one PS1 allele is sufficient for full EGF-induced neuroprotective activity against excitotoxicity. This difference may reflect variations in the mechanisms by which PS1 mediates neuroprotective activities of specific ligand–receptor systems.
We found that lack of EGF-dependent neuroprotection correlates with decreased levels of neuronal EGFR. Furthermore, reintroduction of exogenous PS1 in PS1−/− neurons increased cellular EGFR, while acute down-regulation of neuronal PS1 decreased both the levels of EGFR and neuroprotective activity of EGF ligands. Together, these data show that PS1 is a critical positive regulator of the expression of neuronal EGFR and that as a result of a dramatic decrease of EGFR, PS1-null neurons are unable to use EGF factors to activate survival signaling. Importantly, expression of exogenous EGFR restores the ability of EGFs to phosphorylate EGFR, activates its targets, AKT and ERK, and rescues neurons from excitotoxicity even in absence of PS1, supporting the conclusion that the main role of PS1 in EGF neuroprotection is to regulate expression of EGFR. In contrast to the crucial role PS1 plays in the regulation of neuronal EGFR, PS1 has no effect on the EGFR of primary glia and fibroblast cells, indicating that PS1 specifically regulates the neuronal receptor. This conclusion is also supported by our finding that absence of PS1 causes a more dramatic drop in neuronal than brain EGFR, as the total amount of this receptor in brain is determined by its expression in all cells.
PS1 functions as catalytic component of γ-secretase complexes that process many type I transmembrane proteins, including receptors. Previous work on the role of γ-secretase on EGFR, however, yielded contradictory evidence for (5, 7) and against (4, 6) a role of this activity in the regulation of the receptor. Our data show that γ-secretase activity has no significant effect on neuronal EGFR. Furthermore, we found that PS1 has no effects on the stability of neuronal EGFR. We thus asked whether PS1 might stimulate expression of Egfr mRNA. Indeed, we found that this mRNA is greatly decreased in PS1−/− neurons, but this decrease is smaller in PS1-null brains—an observation consistent with our data that PS1 does not affect the EGFR levels of astrocytes. Acute down-regulation of neuronal PS1 resulted in a robust decrease of Egfr mRNA, providing strong evidence that PS1 directly regulates this mRNA. Importantly, our experiments showed that PS1 has no effect of the stability of Egfr mRNA, indicating that PS1 controls a step before synthesis of mature receptor mRNA. Together, our data show that PS1 functions as a critical positive regulator of neuronal Egfr mRNA. Interestingly, absence of PS2 affects neither the cellular levels of EGFR nor its neuroprotective functions, suggesting that a novel PS1-dependent transcriptional mechanism controls expression of neuronal EGFR independent of both PS2 and γ-secretase. Although the detailed mechanisms by which PS1 stimulates transcription of neuronal EGFR remain to be elucidated, it is tempting to speculate that recently described γ-secretase-independent functions of PS1 in cell signaling may be involved (2).
Our conclusion that PS1 stimulates neuronal, but not glial or fibroblast, EGFR mRNA was unexpected, as evidence obtained in iMEF cells indicate that PS1 negatively regulates cellular levels of EGFR. Although the source of this inconsistency is unclear, use of PS1 and PS2 DKO iMEF cells and tumor cells resulted in inconsistent conclusions for both the role of γ-secretase in the regulation of EGFR (4–7, 42) and the mechanisms by which PS1 suppresses this receptor (4–6). In addition, it has been found that DKO iMEF cells lacking both PS1 and PS2 have defects independent of presenilins (43), while conditional PS2/PS1 DKO mice may still express PS1 (44–46). Thus, transformed cells where gene expression may be distorted by immortalization, genomic rearrangements, and clonal selection may not be reliable indicators for in vivo effects of PS1 on EGFR. In support of this possibility, we observed large variations in the levels of EGFR among distinct iMEF cell lines, regardless of PS1 genotype. More importantly, in our experiments, neither down-regulation nor overexpression of PS1 had significant effects on the EGFR of iMEF cell lines—an outcome consistent with absence of PS1 effects on the EGFR of primary fibroblasts. Thus, our data bring into new focus previously inconsistent conclusions regarding the role of PS1 and γ-secretase in the expression of EGFR and reveal novel pathways by which PS1 regulates neuronal gene expression via γ-secretase-independent mechanisms.
In contrast to severe developmental abnormalities and lethal phenotypes caused by the absence of PS1 in transgenic mouse models (24, 47), PS2-null mice survive free of serious phenotypes (37, 48). It is currently believed that reduction of γ-secretase cleavage of the Notch1 receptor is responsible for the lethal phenotype of PS1-null mice (49). This suggestion, however, does not explain the absence of phenotypes in PS2-null mice, even though, similar to PS1, PS2 also catalyzes the γ-secretase processing of Notch1 (37). Our finding that PS1 is indispensible for the expression of neuronal EGFR, a function displayed neither by PS2 nor γ-secretase, raises the intriguing possibility that developmental abnormalities and lethal phenotype of PS1-null mice are, at least in part, due to reduced neuronal EGFR. Importantly, deficiencies in either EGFR or Hb-EGF cause early postnatal mortality with abnormalities similar to those observed in PS1-null mice (23), while recent evidence reveals that PS1 regulates proliferation of brain progenitor cells through EGFR (38). Recently it was reported that the tyrosine kinase activity of EGFR regulates biogenesis of miRNAs involved in cellular response to hypoxia (50). Thus, it is reasonable to infer that by controlling the levels of neuronal EGFR, PS1 tightly regulates and fine-tunes the functions of this receptor, including its roles in development, production of miRNAs, and neuronal survival and function. Importantly, emerging evidence indicates that the role of this receptor in the development of AD has been unappreciated. Recent analysis of genome-wide studies and protein–protein interaction modeling has identified EGFR as a significant risk factor for sporadic AD (51), while transcriptional profiling studies indicate that the AD-associated apoE4 allele changes the brain expression of EGFR compared to apoE3 (52). Furthermore, additional work indicates that EGFR functions mediate Aβ42-induced memory loss in experimental animal models (53). Thus, by controlling EGFR, PS1 may regulate the course of many diseases known to be modulated by this receptor, including cancer and sporadic AD.
Supplementary Material
Acknowledgments
This work was supported in part by U.S. National Institutes of Health (NIH) Grants AG-17926, AG-08200, and AG-05138 from the National Institute on Aging, and NIH NS047229 from the National Institute of Neurological Disorders and Stroke.
Glossary
- AD
Alzheimer disease
- AKT
protein kinase B
- APP
amyloid precursor protein
- BDNF
brain-derived neurotrophic factor
- CHX
cycloheximide
- CTF
carboxyl-terminal fragment
- DIV
days in vitro
- DKO
double knockout
- E15
embryonic day 15
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- GAPDH
glyceraldehyde phosphate dehydrogenase
- Hb-EGF
heparin-binding epidermal growth factor
- iMEF
immortalized mouse embryonic fibroblast
- KO
knockout
- MAP2
microtubule-associated protein 2
- MEF
mouse embryonic fibroblast
- MTT
3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide
- PCNC
primary cortical neuronal culture
- PS
presenilin
- siRNA
small interfering RNA
- WB
Western blot
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Barthet G., Georgakopoulos A., Robakis N. K. (2012) Cellular mechanisms of γ-secretase substrate selection, processing and toxicity. Prog. Neurobiol. 98, 166–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pimplikar S. W., Nixon R. A., Robakis N. K., Shen J., Tsai L. H. (2010) Amyloid-independent mechanisms in Alzheimer’s disease pathogenesis. J. Neurosci. 30, 14946–14954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barthet G., Dunys J., Shao Z., Xuan Z., Ren Y., Xu J., Arbez N., Mauger G., Bruban J., Georgakopoulos A., Shioi J., Robakis N. K. (2013) Presenilin mediates neuroprotective functions of ephrinB and brain-derived neurotrophic factor and regulates ligand-induced internalization and metabolism of EphB2 and TrkB receptors. Neurobiol. Aging 34, 499–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Repetto E., Yoon I.-S., Zheng H., Kang D. E. (2007) Presenilin 1 regulates epidermal growth factor receptor turnover and signaling in the endosomal–lysosomal pathway. J. Biol. Chem. 282, 31504–31516 [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y. W., Wang R., Liu Q., Zhang H., Liao F. F., Xu H. (2007) Presenilin/gamma-secretase-dependent processing of beta-amyloid precursor protein regulates EGF receptor expression. Proc. Natl. Acad. Sci. USA 104, 10613–10618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rocher-Ros V., Marco S., Mao J.-H., Gines S., Metzger D., Chambon P., Balmain A., Saura C. A. (2010) Presenilin modulates EGFR signaling and cell transformation by regulating the ubiquitin ligase Fbw7. Oncogene 29, 2950–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li T., Wen H., Brayton C., Das P., Smithson L. A., Fauq A., Fan X., Crain B. J., Price D. L., Golde T. E., Eberhart C. G., Wong P. C. (2007) Epidermal growth factor receptor and Notch pathways participate in the tumor suppressor function of gamma-secretase. J. Biol. Chem. 282, 32264–32273 [DOI] [PubMed] [Google Scholar]
- 8.Avraham R., Yarden Y. (2011) Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 12, 104–117 [DOI] [PubMed] [Google Scholar]
- 9.Berasain, C., Ujue Latasa, M., Urtasun, R., Goñi, S., Elizalde, M., Garcia-Irigoyen, O., Azcona, M., Prieto, J., and Avila, M. A. (2011) Epidermal growth factor receptor (EGFR) crosstalks in liver cancer. Cancers (Basel) 3, 2444–2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Enwere E., Shingo T., Gregg C., Fujikawa H., Ohta S., Weiss S. (2004) Aging results in reduced epidermal growth factor receptor signaling, diminished olfactory neurogenesis, and deficits in fine olfactory discrimination. J. Neurosci. 24, 8354–8365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farkas L. M., Krieglstein K. (2002) Heparin-binding epidermal growth factor-like growth factor (HB-EGF) regulates survival of midbrain dopaminergic neurons. J. Neural Transm. 109, 267–277 [DOI] [PubMed] [Google Scholar]
- 12.Hanke M., Farkas L. M., Jakob M., Ries R., Pohl J., Sullivan A. M. (2004) Heparin-binding epidermal growth factor-like growth factor: a component in chromaffin granules which promotes the survival of nigrostriatal dopaminergic neurones in vitro and in vivo. Neuroscience 124, 757–766 [DOI] [PubMed] [Google Scholar]
- 13.Opanashuk L. A., Mark R. J., Porter J., Damm D., Mattson M. P., Seroogy K. B. (1999) Heparin-binding epidermal growth factor-like growth factor in hippocampus: modulation of expression by seizures and anti-excitotoxic action. J. Neurosci. 19, 133–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casper D., Mytilineou C., Blum M. (1991) EGF enhances the survival of dopamine neurons in rat embryonic mesencephalon primary cell culture. J. Neurosci. Res. 30, 372–381 [DOI] [PubMed] [Google Scholar]
- 15.Peng H., Wen T. C., Tanaka J., Maeda N., Matsuda S., Desaki J., Sudo S., Zhang B., Sakanaka M. (1998) Epidermal growth factor protects neuronal cells in vivo and in vitro against transient forebrain ischemia- and free radical–induced injuries. J. Cereb. Blood Flow Metab. 18, 349–360 [DOI] [PubMed] [Google Scholar]
- 16.Sun D., Bullock M. R., Altememi N., Zhou Z., Hagood S., Rolfe A., McGinn M. J., Hamm R., Colello R. J. (2010) The effect of epidermal growth factor in the injured brain after trauma in rats. J. Neurotrauma 27, 923–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siddiqui S., Fang M., Ni B., Lu D., Martin B., Maudsley S. (2012) Central role of the EGF receptor in neurometabolic aging. Int. J. Endocrinol. 2012, 739428–739441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sibilia M., Wagner E. F. (1995) Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 269, 234–238 [DOI] [PubMed] [Google Scholar]
- 19.Miettinen P. J., Berger J. E., Meneses J., Phung Y., Pedersen R. A., Werb Z., Derynck R. (1995) Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 376, 337–341 [DOI] [PubMed] [Google Scholar]
- 20.Threadgill D. W., Dlugosz A. A., Hansen L. A., Tennenbaum T., Lichti U., Yee D., LaMantia C., Mourton T., Herrup K., Harris R. C (1995) Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science 269, 230–234 [DOI] [PubMed] [Google Scholar]
- 21.Kornblum H. I., Hussain R., Wiesen J., Miettinen P., Zurcher S. D., Chow K., Derynck R., Werb Z. (1998) Abnormal astrocyte development and neuronal death in mice lacking the epidermal growth factor receptor. J. Neurosci. Res. 53, 697–717 [DOI] [PubMed] [Google Scholar]
- 22.Iwamoto R., Yamazaki S., Asakura M., Takashima S., Hasuwa H., Miyado K., Adachi S., Kitakaze M., Hashimoto K., Raab G., Nanba D., Higashiyama S., Hori M., Klagsbrun M., Mekada E. (2003) Heparin-binding EGF-like growth factor and ErbB signaling is essential for heart function. Proc. Natl. Acad. Sci. USA 100, 3221–3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sibilia M., Steinbach J. P., Stingl L., Aguzzi A., Wagner E. F. (1998) A strain-independent postnatal neurodegeneration in mice lacking the EGF receptor. EMBO J. 17, 719–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen J., Bronson R. T., Chen D. F., Xia W., Selkoe D. J., Tonegawa S. (1997) Skeletal and CNS defects in presenilin-1-deficient mice. Cell 89, 629–639 [DOI] [PubMed] [Google Scholar]
- 25.Donoviel D. B., Hadjantonakis A. K., Ikeda M., Zheng H., Hyslop P. S., Bernstein A. (1999) Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 13, 2801–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marambaud P., Shioi J., Serban G., Georgakopoulos A., Sarner S., Nagy V., Baki L., Wen P., Efthimiopoulos S., Shao Z., Wisniewski T., Robakis N. K. (2002) A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 21, 1948–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barthet G., Shioi J., Shao Z., Ren Y., Georgakopoulos A., Robakis N. K. (2011) Inhibitors of γ-secretase stabilize the complex and differentially affect processing of amyloid precursor protein and other substrates. FASEB J. 25, 2937–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baki L., Neve R. L., Shao Z., Shioi J., Georgakopoulos A., Robakis N. K. (2008) Wild-type but not FAD mutant presenilin-1 prevents neuronal degeneration by promoting phosphatidylinositol 3-kinase neuroprotective signaling. J. Neurosci. 28, 483–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xuan Z., Barthet G., Shioi J., Xu J., Georgakopoulos A., Bruban J., Robakis N. K. (2013) Presenilin-1/γ-secretase controls glutamate release, tyrosine phosphorylation, and surface expression of N-methyl-d-aspartate receptor (NMDAR) subunit GluN2B. J. Biol. Chem. 288, 30495–30501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greber B., Lehrach H., Adjaye J. (2007) Fibroblast growth factor 2 modulates transforming growth factor beta signaling in mouse embryonic fibroblasts and human ESCs (hESCs) to support hESC self-renewal. Stem Cells 25, 455–464 [DOI] [PubMed] [Google Scholar]
- 31.Kim H. J., Magrané J. (2011) Isolation and culture of neurons and astrocytes from the mouse brain cortex. Methods Mol. Biol. 793, 63–75 [DOI] [PubMed] [Google Scholar]
- 32.Xu J., Xilouri M., Bruban J., Shioi J., Shao Z., Papazoglou I., Vekrellis K., Robakis N. K. (2011) Extracellular progranulin protects cortical neurons from toxic insults by activating survival signaling. Neurobiol. Aging 32, 2326.e5–2326.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin K., Mao X. O., Del Rio Guerra G., Jin L., Greenberg D. A. (2005) Heparin-binding epidermal growth factor–like growth factor stimulates cell proliferation in cerebral cortical cultures through phosphatidylinositol 3′-kinase and mitogen-activated protein kinase. J. Neurosci. Res. 81, 497–505 [DOI] [PubMed] [Google Scholar]
- 34.Georgakopoulos A., Litterst C., Ghersi E., Baki L., Xu C., Serban G., Robakis N. K. (2006) Metalloproteinase/presenilin1 processing of ephrinB regulates EphB-induced Src phosphorylation and signaling. EMBO J. 25, 1242–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Litterst C., Georgakopoulos A., Shioi J., Ghersi E., Wisniewski T., Wang R., Ludwig A., Robakis N. K. (2007) Ligand binding and calcium influx induce distinct ectodomain/gamma-secretase-processing pathways of EphB2 receptor. J. Biol. Chem. 282, 16155–16163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xia W., Zhang J., Kholodenko D., Citron M., Podlisny M. B., Teplow D. B., Haass C., Seubert P., Koo E. H., Selkoe D. J. (1997) Enhanced production and oligomerization of the 42-residue amyloid beta-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J. Biol. Chem. 272, 7977–7982 [DOI] [PubMed] [Google Scholar]
- 37.Steiner H., Duff K., Capell A., Romig H., Grim M. G., Lincoln S., Hardy J., Yu X., Picciano M., Fechteler K., Citron M., Kopan R., Pesold B., Keck S., Baader M., Tomita T., Iwatsubo T., Baumeister R., Haass C. (1999) A loss of function mutation of presenilin-2 interferes with amyloid beta-peptide production and Notch signaling. J. Biol. Chem. 274, 28669–28673 [DOI] [PubMed] [Google Scholar]
- 38.Gadadhar A., Marr R., Lazarov O. (2011) Presenilin-1 regulates neural progenitor cell differentiation in the adult brain. J. Neurosci. 31, 2615–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peus D., Hamacher L., Pittelkow M. R. (1997) EGF-receptor tyrosine kinase inhibition induces keratinocyte growth arrest and terminal differentiation. J. Invest. Dermatol. 109, 751–756 [DOI] [PubMed] [Google Scholar]
- 40.Hudson L. G., McCawley L. J. (1998) Contributions of the epidermal growth factor receptor to keratinocyte motility. Microsc. Res. Tech. 43, 444–455 [DOI] [PubMed] [Google Scholar]
- 41.Normanno N., De Luca A., Bianco C., Strizzi L., Mancino M., Maiello M. R., Carotenuto A., De Feo G., Caponigro F., Salomon D. S. (2006) Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366, 2–16 [DOI] [PubMed] [Google Scholar]
- 42.Song X., Xia R., Cui Z., Chen W., Mao L. (2012) Presenilin 1 is frequently overexpressed and positively associates with epidermal growth factor receptor expression in head and neck squamous cell carcinoma. Head Neck Oncol. 4, 47–54 [Google Scholar]
- 43.Watanabe H., Smith M. J., Heilig E., Beglopoulos V., Kelleher R. J. III, Shen J. (2009) Indirect regulation of presenilins in CREB-mediated transcription. J. Biol. Chem. 284, 13705–13713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feng R., Rampon C., Tang Y. P., Shrom D., Jin J., Kyin M., Sopher B., Miller M. W., Ware C. B., Martin G. M., Kim S. H., Langdon R. B., Sisodia S. S., Tsien J. Z. (2001) Deficient neurogenesis in forebrain-specific presenilin-1 knockout mice is associated with reduced clearance of hippocampal memory traces. Neuron 32, 911–926 [DOI] [PubMed] [Google Scholar]
- 45.Yu H., Saura C. A., Choi S. Y., Sun L. D., Yang X., Handler M., Kawarabayashi T., Younkin L., Fedeles B., Wilson M. A., Younkin S., Kandel E. R., Kirkwood A., Shen J. (2001) APP processing and synaptic plasticity in presenilin-1 conditional knockout mice. Neuron 31, 713–726 [DOI] [PubMed] [Google Scholar]
- 46.Saura C. A., Choi S. Y., Beglopoulos V., Malkani S., Zhang D., Shankaranarayana Rao B. S., Chattarji S., Kelleher R. J. III, Kandel E. R., Duff K., Kirkwood A., Shen J. (2004) Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42, 23–36 [DOI] [PubMed] [Google Scholar]
- 47.Wong P. C., Zheng H., Chen H., Becher M. W., Sirinathsinghji D. J., Trumbauer M. E., Chen H. Y., Price D. L., Van der Ploeg L. H., Sisodia S. S. (1997) Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature 387, 288–292 [DOI] [PubMed] [Google Scholar]
- 48.Herreman A., Hartmann D., Annaert W., Saftig P., Craessaerts K., Serneels L., Umans L., Schrijvers V., Checler F., Vanderstichele H., Baekelandt V., Dressel R., Cupers P., Huylebroeck D., Zwijsen A., Van Leuven F., De Strooper B. (1999) Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc. Natl. Acad. Sci. U. S. A. 96, 11872–11877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Strooper B., Annaert W., Cupers P., Saftig P., Craessaerts K., Mumm J. S., Schroeter E. H., Schrijvers V., Wolfe M. S., Ray W. J., Goate A., Kopan R. (1999) A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522 [DOI] [PubMed] [Google Scholar]
- 50.Shen J., Xia W., Khotskaya Y. B., Huo L., Nakanishi K., Lim S.-O., Du Y., Wang Y., Chang W.-C., Chen C.-H., Hsu J. L., Wu Y., Lam Y. C., James B. P., Liu X., Liu C.-G., Patel D. J., Hung M.-C. (2013) EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature 497, 383–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Talwar P., Silla Y., Grover S., Gupta M., Agarwal R., Kushwaha S., Kukreti R. (2014) Genomic convergence and network analysis approach to identify candidate genes in Alzheimer’s disease. BMC Genomics 15, 199–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Conejero-Goldberg C., Hyde T. M., Chen S., Dreses-Werringloer U., Herman M. M., Kleinman J. E., Davies P., Goldberg T. E. (2011) Molecular signatures in post-mortem brain tissue of younger individuals at high risk for Alzheimer’s disease as based on APOE genotype. Mol. Psychiatry 16, 836–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang L., Chiang H.-C., Wu W., Liang B., Xie Z., Yao X., Ma W., Du S., Zhong Y. (2012) Epidermal growth factor receptor is a preferred target for treating amyloid-β-induced memory loss. Proc. Natl. Acad. Sci. USA 109, 16743–16748 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.