

Abstract
The alcohol-induced depletion of hepatic retinoid stores correlates with the progression of liver injury; however, the mechanisms underlying alcohol’s effects have not been fully elucidated. Our goal was to gain a mechanistic understanding of alcohol-induced hepatic retinoid depletion. Wild-type and mutant mice were continuously fed alcohol through Lieber-DeCarli liquid diets, with matched control animals pair fed an isocaloric alcohol-free diet to ensure equal nutrient and calorie intake between groups. A systematic analysis of tissue retinol and retinyl ester levels was performed with HPLC, complemented by gene and protein expression analyses. Our results delineated 2 phases of alcohol-induced depletion of hepatic retinoid. Initially, ∼15% of hepatic retinoid content was mobilized from the liver, causing extrahepatic tissue retinoid levels to increase. Subsequently, there was a precipitous drop in hepatic retinoid content (>60%), without further retinoid accumulation in the periphery. Follow-up studies in mutant mice revealed roles for RBP, CRBP1, and CD36 in retinoid mobilization and extrahepatic retinoid uptake, as well as a role for CYP2E1 in the catabolism of hepatic retinoid. In summary, alcohol has a biphasic effect on hepatic retinoid stores, characterized by an initial phase of rapid mobilization to extrahepatic tissues followed by extensive catabolism within the liver.—Clugston, R. D., Huang, L.-S., Blaner, W. S. Chronic alcohol consumption has a biphasic effect on hepatic retinoid loss.
Keywords: liver, vitamin A, ethanol, retinoid mobilization, retinoid catabolism
Retinoids (vitamin A and its metabolites) are potent transcriptional regulators that modulate the expression of more than 500 genes (1). Retinoids function in controlling normal cell proliferation, differentiation, and apoptosis, thereby affecting numerous physiologic processes, including immunity, reproduction, and embryogenesis (2). Altered retinoid signaling has been implicated in the pathogenesis of several diseases, including those affecting the liver (3). Indeed, a growing body of literature highlights the importance of retinoid signaling in hepatic lipid metabolism and hepatic stellate cell (HSC) biology, which has important implications for improving our understanding of hepatic disease (2–5).
Chronic alcohol consumption is known to have a marked effect on hepatic retinoid homeostasis, as recently reviewed (6). A progressive alcohol-induced depletion of hepatic retinoid content in alcoholic patients, which correlates with worsening disease, was first reported by Leo and Lieber (7) more than 3 decades ago. More recently, it has been established that plasma retinol levels negatively correlate with the Child-Pugh and MELD (model for end-stage liver disease) scores of liver disease severity (8). The effects of alcohol on hepatic retinoid homeostasis in humans have been replicated in animal models of chronic alcohol consumption (6); however, a mechanistic understanding of how alcohol precipitates these effects has not been elucidated. The hallmarks of alcohol’s effect on whole-body retinoid homeostasis include the depletion of hepatic retinoid stores and an increase in extrahepatic retinoid content (6). Two hypotheses have been put forward to explain these effects: one proposes that alcohol induces catabolism of hepatic retinoid stores and a preferential accumulation of postprandial retinoid in extrahepatic tissues, and the second proposes that alcohol induces direct mobilization of retinoid stores out of the liver and its redistribution to extrahepatic tissues (6, 9–11).
In this study, our goal was to test those hypotheses and gain an improved mechanistic understanding of alcohol’s effects on retinoid homeostasis. To achieve this goal, we used wild-type (WT) and mutant mouse models of chronic alcohol consumption as a component of Lieber-DeCarli liquid diets (Bio-Serv, Frenchtown, NJ, USA). The use of mutant mice allowed us to perform a genetic dissection of alcohol’s effect on hepatic and extrahepatic tissue retinoid homeostasis. Specifically, we used mice that had 1) no hepatic retinyl ester stores caused by a null mutation in the gene encoding lecithin-retinol acyltransferase (LRAT), the sole enzyme responsible for hepatic retinyl ester synthesis (2); 2) an impaired ability to mobilize hepatic retinoid stores and reduced circulating retinol levels caused by a null mutation in retinol-binding protein (RBP), the sole specific transport protein for retinol in the circulation (2); 3) impaired cellular retinol homeostasis due to a null mutation in cellular retinol-binding protein (CRBP)-1, a cytoplasmic retinol-binding protein that facilitates retinol uptake and metabolism within cells (2); 4) a null mutation in cytochrome P450 2E1 (CYP2E1), an alcohol-induced enzyme that has been proposed to break down retinoids within the liver (12, 13); and 5) a null mutation in CD36, a well-studied plasma membrane free fatty acid uptake transporter (14). Although CD36 has not been implicated in hepatic retinoid metabolism, it was included here based on results in our recent study showing that these mice are resistant to alcohol-induced fatty liver (15) and on new data presented herein showing that alcohol’s effects on hepatic retinoid levels are blunted in Cd36−/− mice.
Our data demonstrate that the alcohol-induced loss of hepatic retinoid and its redistribution to peripheral tissues involves a biphasic process whereby there is initially a mobilization of hepatic retinoid stores and delivery to peripheral tissues, followed by a subsequent phase of alcohol-induced hepatic retinoid catabolism. We have synthesized our data into a model describing alcohol’s effects on hepatic retinoid homeostasis. This model serves as an important foundation for future work designed to better understand alcohol’s effect on hepatic retinoid metabolism and the impact of this effect on the development of alcoholic liver disease.
MATERIALS AND METHODS
Animal husbandry, mutant mice, and tissue dissection
In most of the experiments, we used 3-mo-old male WT C57BL/6 mice that were bred and maintained at Columbia University. Experiments were also performed in Lrat−/− and Cd36−/− mice in a congenic C57BL/6 background and Rbp−/−, Crbp1−/−, and Cyp2e1−/− mice in a mixed genetic background. All studies in mutant mice were performed with background-matched WT mice. The generation and genotyping of Lrat−/−, Cd36−/−, Rbp−/−, Crbp1−/−, and Cyp2e1−/− mice has been described (16–20). The genotype of all strains of mutant mice was confirmed by PCR genotyping. The mice were maintained on a 12-h light/dark cycle at ∼21°C. The Institutional Animal Care and Use Committee at Columbia University approved all animal experiments.
At the end of each experiment, plasma, liver, heart, lung, kidney, spleen, perigonadal white adipose tissue (WAT), and interscapular brown adipose tissue (BAT) were collected. All tissues were snap frozen in liquid N2 and stored at −80°C until further analysis. After a systematic analysis of extrahepatic tissue retinoid content, only data from the lung and WAT are typically presented because these tissues contain relatively high concentrations of retinol and retinyl esters in comparison with the other extrahepatic tissues. Lung and WAT also rely on different processes to store retinyl ester: the lung undergoes an LRAT-dependent process involving transesterification of an acyl group from phosphatidylcholine, whereas retinyl ester storage in WAT can occur independent of LRAT, involving an acyl-CoA-dependent process (21, 22).
Alcohol-feeding protocol
The mice were fed alcohol in a high-fat Lieber-DeCarli liquid diet formulation (Bio-Serv, Frenchtown, NJ, USA), a well-established nutritionally adequate model used to study the effects of alcohol on the liver that contains 4 IU/g vitamin A (23). We have used this alcohol-feeding paradigm to study the effects of alcohol on hepatic lipid and retinoid metabolism (15, 21, 24). A detailed description of our alcohol-feeding protocol, as well as the use of vitamin A-sufficient (VAS; 4 IU/g vitamin A) and vitamin A-deficient (VAD; 0 IU/g vitamin A) liquid diets has been published (21). The liquid diets (Bio-Serv) were control: F5937SP; alcohol: F5938SP; VAD control: F6360SP; and VAD alcohol: F6361SP. All mice were individually housed, and the control mice were pair fed according to the measured volume of diet consumed by alcohol-fed mice over the preceding 48 h. The alcohol-adaptation period consisted of 1 wk of consuming the alcohol-free control diet, 1 wk with 2.1% v/v alcohol, and 1 wk with 4.2% v/v alcohol. After the adaptation period, the animals were fed 6.4% v/v alcohol for up to 4 wk. Based on the results of our initial time course study, our subsequent studies focused on the alcohol-adaptation period. As shown in Supplemental Fig. 1A, there was no significant difference in average liquid diet consumption by all strains of mice studied. Moreover, WT, Lrat−/−, Rbp−/−, and Cd36−/− mice achieved comparable blood alcohol levels after acute administration of ethanol (Supplemental Fig. 1B). The effect of alcohol on body and liver weights in all strains studied was unremarkable and is presented in Supplemental Table 1.
Analysis of tissue retinoid content and plasma RBP concentration
Tissue retinoid content was analyzed by HPLC, according to our standard method (21, 25). Briefly, retinoids were extracted from homogenized tissues into hexane. After evaporation of the hexane, the residual lipids including retinoids were dissolved in benzene and separated on a Symmetry C18 column (Waters, Milford, MA, USA). Quantification of extracted retinoids was undertaken by measuring the area under the curve of absorbance peaks at 325 nm and adjusted based on the recovery of the internal standard (retinyl acetate; Sigma-Aldrich, St. Louis, MO, USA). Plasma RBP levels were measured with a commercially available mouse RBP ELISA kit according to the manufacturer’s instructions (AdipoGen, San Diego, CA, USA). The molar ratio of retinol to RBP was calculated based on a formula weight for RBP of 21 kDa. Hepatic secretion of retinyl ester was assessed by injecting unfed mice with the total lipase inhibitor P-407 (1 mg/g) (26).
Gene and protein expression profiling
RBP protein expression was determined by Western blot analysis, according to standard methods (25). In brief, 30 µg total hepatic protein was analyzed to determine the expression levels of RBP and the reference protein β-actin, by using a rabbit polyclonal anti-RBP (1:3000) (27) and a rabbit polyclonal anti-β-actin (1:1000, sc-10731; Santa Cruz Biotechnology, Santa Cruz, CA, USA) primary antibody, respectively. Quantitative analysis of Western blots was performed with ImageJ (28). Gene (mRNA) expression levels of Crbp1, Cyp26a1, Cyp26b1, and 18s (reference gene) were determined by standard quantitative (q)PCR (24). All PCR amplification was performed on a LightCycler480 (Roche Diagnostics, Indianapolis, IN, USA). The following primer sequences were used: Crbp1 forward, 5′-CTT ACT GTC CCT ACT GTG TGT CAA GCA CTA-3′; Crbp1 reverse, 5′-CCT GAG ATG AAC CTC CTG AGA TGG TTT A-3′; Crbp3 forward, 5-CCC GCT TGA GGC AAC TAC T-3′; Crbp3 reverse, 5′-GTT TCT CAT ACA GGC TGT GTG ACA T-3′; Cyp26a1 forward, 5′-GGC ACT GTG ATT GGC AGC TTC TAA-3′; Cyp26a1 reverse, 5′-TGC AGG GAG ATT GTC CAC AGG GTA-3′; Cyp26b1 forward, 5′-GCA GTA TAT GCT TAT GAC ATC TGA ATC-3′; and Cyp26b1 reverse, 5′-CCT GAC CAC TCA CCA ACA AA-3′. The primer sequences for 18s have been reported (24).
Assessment of hepatic triglyceride content
Frozen sections of liver tissue were stained with Oil Red O according to standard protocols in the Pathology Core Facility at the Columbia University Medical Center, and digital images were taken with an FSX100 microscope (Olympus, Center Valley, PA, USA). Biochemical measurement of hepatic triglyceride content was performed on lipids extracted from liver homogenates with a liquid stable triglyceride reagent (Thermo Fisher Scientific, Middleton, VA, USA) (24).
Statistical analyses
Data are presented as the means ± sd. Statistical analyses were performed with Prism 5 (GraphPad Software, La Jolla, CA, USA). Depending on the experimental design, the data were analyzed by Student’s t test, 1-way ANOVA with a Tukey’s post hoc test, or 2-way ANOVA with Bonferroni’s post hoc test, as specified. In all cases, P < 0.05 was considered statistically significant.
RESULTS
Alcohol consumption is associated with increased hepatic triglyceride content and decreased retinoid content
To assess the temporal effects of continuous alcohol consumption on hepatic retinol and retinyl ester levels, we collected tissues from control and alcohol-fed WT C57BL/6 mice immediately after the alcohol-adaptation period and after they consumed 6.4% alcohol for 1, 2, and 4 wk. Livers of alcohol-fed WT mice displayed more Oil Red O staining after 4 wk of consuming 6.4% alcohol, suggesting elevated lipid content (Fig. 1A). This finding was confirmed by quantitative measures showing significantly higher levels of triglyceride in the livers of alcohol- vs. control-fed mice (Fig. 1B). To increase the statistical power, the data presented in Fig. 1C, D were pooled from multiple alcohol-feeding tests in WT C57BL/6 mice, including a minority of hepatic retinyl ester measures reported by our group (21). These data show that hepatic retinol levels are maintained at control levels until wk 4 of consuming 6.4% alcohol, when they become significantly lower compared with those in control mice (Fig. 1C). In an interesting finding, there was a small but statistically significant decline in hepatic retinyl ester levels after the alcohol-adaptation period, with more quantitatively significant declines observed later in the alcohol-feeding protocol (Fig. 1D).
Figure 1.

Alcohol consumption is associated with increased hepatic triglyceride content and decreased retinoid content. A) Representative images of Oil Red O staining in the liver of control and alcohol-fed mice. Magnification ×20. B) Biochemical quantification of hepatic triglyceride (TG) content confirmed that chronic alcohol consumption is associated with hepatic steatosis (n = 6). Analysis of pooled data from multiple studies revealed significant declines in the hepatic concentrations of (C) retinol and (D) retinyl ester in alcohol-fed mice (control, n = 41; alcohol adaptation, n = 32; 1 wk 6.4% alcohol, n = 6; 2 wk 6.4% alcohol, n = 10; and 4 wk 6.4% alcohol, n = 4). B) *P < 0.05 vs. control; Student’s t test. C, D) P < 0.05; columns that do not share a common letter are significantly different; 1-way ANOVA.
Alcohol consumption is associated with an immediate increase in extrahepatic tissue retinoid levels
In addition to those in the liver, retinol and retinyl ester levels were measured in heart, lung, WAT, BAT, kidney, and spleen of WT mice (Fig 2). We observed elevated retinol levels in response to alcohol in all of these tissues, with an immediate and statistically significant increase in retinol levels during the alcohol-adaptation period and continued elevated levels with constant alcohol feeding. Moreover, we observed an alcohol-induced increase in retinyl ester levels in tissues that maintain retinyl ester stores, specifically the lungs, WAT, and BAT (data not shown) (21). When the mice were fed alcohol through the adaptation period and then returned to an alcohol-free diet for 1 mo, retinol and retinyl ester levels in all extrahepatic tissues studied returned to baseline (Supplemental Fig. 2).
Figure 2.

Alcohol consumption is associated with an immediate increase in retinol concentrations in multiple extrahepatic tissues, including (A) heart (B), lung, (C) WAT, (D) BAT, (E) kidney, and (F) spleen (control n = 12; alcohol-adaptation period, 1 wk 6.4% alcohol, and 2 wk 6.4% alcohol, n = 6; 4 wk 6.4% alcohol, n = 5). P < 0.05; columns that do not share a common letter are significantly different; 1-way ANOVA.
Retinoid accumulation in extrahepatic tissues originates from the liver
To determine the effect of alcohol on postprandial retinoid uptake into extrahepatic tissues, we fed control and alcohol-consuming WT mice VAS and VAD diets. If the hypothesis that alcohol stimulates the preferential accumulation of postprandial retinoid uptake into extrahepatic tissues is correct (6), then mice fed alcohol and a VAD diet would accumulate little or no retinol or retinyl ester in their extrahepatic tissues. We found that, after the alcohol-adaptation period, the alcohol-induced increase in lung and WAT retinol levels was almost identical in mice consuming the VAD and VAS diets (Fig. 3A, B). Similarly, retinyl ester levels increased equally in lung and WAT, regardless of dietary vitamin A content (data not shown) (21). Thus, the notion that dietary vitamin A intake accounts for the elevated total retinol concentrations observed in extrahepatic tissues with continuous alcohol consumption appears to be incorrect.
Figure 3.

The alcohol-induced increase in extrahepatic tissue retinol levels occur independent of dietary vitamin A intake and is blocked in Lrat−/− mice. Tissue retinol levels increased after the alcohol-adaptation period in (A) lung, and (B) WAT of alcohol-consuming mice fed a VAS (n = 4–6) or a VAD (n = 5–7) diet. Tissue retinol levels were increased in the (C) lung and (D) WAT of alcohol-consuming WT mice (n = 5), but this effect was blocked in Lrat−/− mice (n = 4). *P < 0.05 vs. animals of the same genotype; #P < 0.05 vs. animals consuming the same diet; 2-way ANOVA.
Next, we used Lrat−/− mice to test the alternate hypothesis that the additional retinoid present in the extrahepatic tissues of alcohol-fed mice originates in the liver. Because Lrat−/− mice have no hepatic retinoid stores (16) (Supplemental Table 2), we predicted that if the liver were the primary source of the retinoid accumulating in the extrahepatic tissues, then this effect would be absent in alcohol-fed Lrat−/− mice. As expected, alcohol-fed Lrat−/− mice had low levels of hepatic retinol and undetectable levels of retinyl ester (data not shown). Furthermore, although alcohol-consuming WT mice had significantly higher levels of retinol in the lung and WAT compared to levels in control WT mice after the alcohol-adaptation period, there was no significant alcohol-induced increase in the retinol levels in lung or WAT of the Lrat−/− mice (Fig. 3C, D). Similarly, the alcohol-induced increases in retinol levels observed in BAT, kidney, and spleen of the WT mice were absent in the Lrat−/− mice (data not shown). Retinyl esters are undetectable in the lungs of Lrat−/− mice, but the WAT retains its retinyl ester stores (16, 26); however, retinyl ester levels in WAT of alcohol-fed Lrat−/− mice did not change significantly in response to alcohol feeding (data not shown) (21). These observations are consistent with the hypothesis that continuous alcohol consumption causes hepatic retinoid stores to be redistributed to extrahepatic tissues.
Circulating retinol levels transiently increase in alcohol-consuming mice
To better understand the link between decreasing hepatic retinoid concentrations and increasing extrahepatic tissue retinoid levels in alcohol-fed mice, we measured circulating levels of retinol and RBP (Fig. 4). Plasma was collected from alcohol-fed mice every third day throughout the alcohol-adaptation period and after consuming a diet containing 6.4% alcohol for 1, 2, and 4 wk. Plasma retinol levels significantly increased during the alcohol-adaptation period and then returned to control levels at the onset of the 6.4% alcohol diet (Fig. 4A). This increase in plasma retinol levels during the alcohol-adaptation period was mirrored by a significant increase in plasma RBP levels (Fig. 4B). The molar ratio of retinol to RBP showed a slight upward trend throughout the alcohol-adaptation period, although the difference compared with levels in the control mice was significant only at the end of this period (Fig. 4C). Although secretion of retinol bound to RBP is the primary pathway for retinoid mobilization from the liver, retinyl ester is also secreted from the liver in very-low-density lipoprotein (VLDL) (2, 26). To determine the potential effects of alcohol on hepatic retinyl ester secretion, we measured plasma retinyl ester levels in unfed mice treated with the total lipase inhibitor P407 (26). Consistent with the known effect of P407, plasma triglyceride and retinyl ester levels were elevated, although alcohol had no significant effect on these parameters (Fig. 4D). Retinol is secreted from the liver bound to RBP; thus, we measured the hepatic expression level of RBP (Fig. 4E, F). Consistent with the observed change in circulating retinol, there was a significant increase in hepatic RBP expression during the alcohol-adaption period; however, no significant elevation in hepatic RBP expression level was observed at later time points (data not shown).
Figure 4.

A) Circulating levels of retinol and RBP and hepatic RBP expression transiently increased in alcohol-fed mice during the alcohol-adaptation period but returned to baseline levels with continuing alcohol consumption (control n = 26; 3 d 2.1% alcohol, n = 5; 6 d 2.1% alcohol, and 3 d 4.2% alcohol, n = 12; 6 d 4.2% alcohol, n = 14; 7 d 6.4% alcohol, n = 7; 14 d 6.4% alcohol, n = 11; and 21 d 6.4% alcohol, n = 5). The gray shaded area in (A) is expanded in (B) and plotted along with plasma RBP levels, showing that there was a parallel increase in plasma RBP (n = 6–12) and retinol during this period. C) The molar ratio of retinol to RBP, demonstrating a trend for increased RBP saturation during the alcohol-adaptation period (n = 6–12). D) Injection of unfed WT mice (n = 6–7) with the lipase inhibitor P407 elevated plasma triglyceride (TG) and retinyl ester (RE) levels equally in control and alcohol-fed mice. E) Representative Western blot of hepatic RBP expression for control and alcohol-consuming mice. F) Densitometric analysis of hepatic RBP expression revealed that this protein increased significantly during the alcohol-adaptation period, relative to its level in control mice (n = 4). A–C) *P < 0.05, retinol vs. d 0; #P < 0.05, RBP vs. d 0; 1-way ANOVA. F) *P < 0.05 vs. control; Student’s t test.
Mobilization of hepatic retinoid stores to extrahepatic tissues can occur independent of RBP and CRBP1
The data presented thus far suggest that the alcohol-induced mobilization of hepatic retinoid occurs via retinol bound to RBP. Thus, we hypothesized that alcohol-fed Rbp−/− mice would be unable to mobilize their hepatic retinoid stores and would not show an alcohol-induced increase in extrahepatic retinoid levels. As expected, alcohol consumption was associated with increased retinol levels in lung, WAT, kidney, and BAT of WT mice (Fig. 5A–D). The alcohol-induced increase in extrahepatic tissue retinol levels was blunted in alcohol-fed Rbp−/− mice, with a statistically significant increase observed only in WAT. In lung and kidney, the concentration of retinol in alcohol-fed WT mice was significantly higher than that in alcohol-fed Rbp−/− mice. Studies of mice consuming 6.4% ethanol for 2 wk revealed increased retinol levels in the extrahepatic tissues of alcohol-fed Rbp−/− mice and also showed that hepatic retinyl ester levels significantly declined in Rbp−/− mice, similar to the effect in WT mice (Supplemental Table 2). Circulating retinol levels are known to be significantly reduced in Rbp−/− mice (18) (Supplemental Table 3). Our analysis of circulating retinol in alcohol-fed Rbp−/− mice revealed a small but statistically significant increase in plasma retinol level during the alcohol-adaptation period. The magnitude of this increase was ∼0.2 µM, compared to an increase of ∼0.6 µM in the WT mice (Fig. 5E). Taken together, these data indicate that hepatic mobilization of retinol is blunted in Rbp−/− mice, although a small amount of mobilization still occurs during the alcohol-adaptation period.
Figure 5.

The alcohol-induced mobilization of hepatic retinoid stores can occur independent of RBP. Alcohol consumption was associated with significant increases in tissue retinol levels in both WT (n = 6–8) and Rbp−/− (n = 3–5) mice after the alcohol-adaptation period, in the (A) lung, (B) WAT, C) kidney, and (D) BAT. *P < 0.05 vs. animals of the same genotype; #P < 0.05 vs. animals consuming the same diet; 2-way ANOVA. E) Similar to WT mice, alcohol-fed Rbp−/− mice had transiently increased plasma retinol levels during the alcohol-adaptation period (n = 4 for Rbp−/− mice; WT data reproduced from Fig. 4B). A–D) *P < 0.05 vs. animals of the same genotype; #P < 0.05 vs. animals consuming the same diet; 2-way ANOVA. E) *P < 0.05 for WT mice vs. d 0; #P < 0.05 for Rbp−/− mice vs. d 0; 1-way ANOVA.
RBP transports retinol in the circulation, whereas CRBP1 functions in the cytoplasm, binding retinol and regulating retinol uptake and metabolism in cells (2, 3). To test the importance of CRBP1 in mediating alcohol-induced mobilization of retinol from the liver and uptake into extrahepatic tissues, we conducted an alcohol-feeding study in Crbp1−/− mice (Fig. 6). In agreement with the previously described phenotype (19), hepatic retinyl esters levels were lower in the Crbp1−/− mice; however, the response in those mice to alcohol was similar to that in the WT mice (Supplemental Table 2). We observed unexpected tissue-specific effects on extrahepatic tissue retinol levels in alcohol-fed Crbp1−/− mice. The alcohol-induced increase in retinol levels was blocked in lung, kidney, and spleen of Crbp1−/− mice, but was retained in WAT and BAT (Fig. 6A–E). Tissue retinyl ester levels showed a similar increase in adipose tissue of alcohol-fed Crbp1−/− mice, but not in lung (data not shown). This tissue-specific effect of Crbp1-deficiency closely corresponds to its known expression pattern in these tissues vs. other CRBPs. For example, kidney had relatively high levels of Crbp1 expression and low Crbp3 expression, whereas BAT had relatively low levels of Crbp1 expression and high levels of Crbp3 expression (Fig. 6F, Supplemental Table 4). Thus, mobilization of hepatic retinoid stores can occur independent of CRBPI, but CRBPI facilitates retinol uptake in certain extrahepatic tissues.
Figure 6.
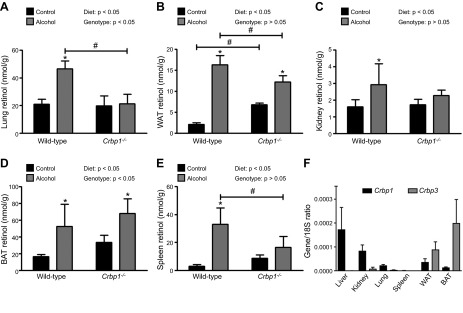
The alcohol-induced mobilization of hepatic retinoid stores can occur independent of CRBP1, but CRBP1-dependent uptake of extrahepatic retinol is tissue specific. Extrahepatic tissue retinol levels were measured in control and alcohol-fed WT (n = 3–6) and Crbp1−/− (n = 6–7) mice. The alcohol-induced increase in tissue retinol levels was blocked in the (A) lung, (C) kidney, and (E) spleen of Crbp1−/− mice, but was retained in (B) WAT and (D) BAT. F) Tissue expression levels of Crbp1 and Crbp3 measured in the lung, kidney, spleen, WAT, and BAT of WT mice ( n = 5-6; reference gene, 18s). *P < 0.05 vs. animals of the same genotype; #P < 0.05 vs. animals consuming the same diet; 2-way ANOVA.
The alcohol-induced mobilization of hepatic retinoid stores is blunted in Cd36−/− mice
We recently reported that Cd36−/− mice fed 5.1% alcohol for 6 wk are resistant to alcohol-induced hepatic steatosis (15). Although the data were not published as part of this earlier study, we also noted that the alcohol-induced decline in hepatic retinol and retinyl ester levels were blunted in Cd36−/− mice (Fig. 7A, B, Supplemental Table 2). To further investigate the possible role of CD36 in the alcohol-induced mobilization of hepatic retinoid stores, we performed a new study focused on the alcohol-adaptation period. We observed that retinol and retinyl ester levels in the lung increased significantly in WT but not in Cd36−/− mice (Fig. 7C, D). In WAT, alcohol consumption was associated with a significant increase in retinol levels in both strains of mice, although this effect was blunted in the Cd36−/− animals (Fig. 7E). Retinyl ester levels in WAT were significantly increased in WT mice, but not Cd36−/− mice (Fig. 7F). A survey of other extrahepatic tissues revealed a similar picture, with either diminished or completely blocked retinoid redistribution in response to alcohol consumption in kidney, spleen, and BAT of Cd36−/− mice (data not shown).
Figure 7.
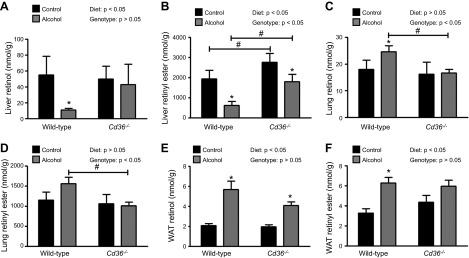
The alcohol-induced mobilization of hepatic retinoid stores to extrahepatic tissues is blunted in Cd36−/− mice. Hepatic (A) retinol and (B) retinyl ester levels were significantly decreased in WT mice consuming 5.1% alcohol for 6 wk (n = 2–10), but this effect was blunted in Cd36−/− mice (n = 6–7). Analysis of extrahepatic tissues after the alcohol-adaptation period revealed that the alcohol-induced increases in retinol and retinyl ester levels in (C, D) lung and (E, F) WAT in Cd36−/− mice (n = 4–5) was blunted in comparison to that in WT mice (n = 5). *P < 0.05 vs. animals of the same genotype; #P < 0.05 vs. animals consuming the same diet; 2-way ANOVA.
Deletion of Cyp2e1 blocks the alcohol-induced decrease in hepatic retinol but not retinyl ester
CYP2E1 is induced in an alcohol-exposed liver (20). It has been proposed that CYP2E1 catalyzes the breakdown of retinoids within the liver, accounting for the alcohol-induced decline in hepatic retinoid content (9, 12, 13). We definitively tested this hypothesis in Cyp2e1−/− mice, with 2 predicted outcomes: Cyp2e1−/− mice would be resistant to the alcohol-induced decline in hepatic retinoid content, and the associated increase in extrahepatic tissue retinoid levels would be blocked. Tissues were collected from matched WT and Cyp2e1−/− mice after the alcohol-adaptation period and after the mice consumed a 6.4% alcohol diet for a further 2 wk (Fig. 8). Hepatic retinol levels did not change in alcohol-fed Cyp2e1−/− mice, whereas WT mice experienced a significant decline in hepatic retinol levels after 2 wk of consuming 6.4% alcohol (Fig. 8A, Supplemental Table 2). In contrast, hepatic retinyl ester levels decreased by a similar magnitude in alcohol-fed WT and Cyp2e1−/− mice (Fig. 8B, Supplemental Table 2). When we surveyed the retinoid content of extrahepatic tissues, we also obtained data contrary to our predicted results. After the alcohol-adaptation period (data not shown) and after 2 wk of a 6.4% alcohol diet, the retinol levels in lung, WAT, kidney, BAT, and spleen were all significantly increased in both WT and Cyp2e1−/− mice fed alcohol (Fig. 8C–G). Furthermore, in 4 of the 5 tissues surveyed, the average tissue retinol level was higher in Cyp2e1−/− mice than that in WT mice. The difference reached statistical significance in kidney and BAT. These data indicate that CYP2E1 does not have a major role in the alcohol-induced decline in hepatic retinoids.
Figure 8.

The alcohol-induced depletion of hepatic retinol is blocked in Cyp2e1−/− mice, but mobilization to extrahepatic stores is preserved. A) Hepatic retinol levels decreased significantly in WT mice consuming 6.4% alcohol for 2 wk (n = 6–10), but this effect was blocked in alcohol-consuming Cyp2e1−/− mice (n = 6–11). B) Hepatic retinyl ester levels decreased in alcohol-consuming WT and Cyp2e1−/− mice, although this effect was significant only in the Cyp2e1−/− mice. The tissue retinol levels in the (C) lung, (D) WAT, (E) kidney, (F) BAT, and (G) spleen increased significantly in alcohol-consuming WT (n = 5-6) and Cyp2e1−/− (n = 5-6) mice. A, B) #P < 0.05 vs. control; 1-way ANOVA; C–G) *P < 0.05 vs. animals of the same genotype; #P < 0.05 vs. animals consuming the same diet; 2-way ANOVA.
Sato and Lieber (29) reported that alcohol consumption increases total cytochrome P (CYP) 450 activity in the liver; we therefore measured gene and protein expression levels of Cyp26a1 and Cyp26b1—canonical retinoic acid-catabolizing enzymes that are expressed in hepatocytes and HSCs, respectively (25). In WT mice consuming 6.4% alcohol for 2 wk, there were significant increases in the mRNA levels of both Cyp26a1 and Cyp26b1 (Fig. 9A, Supplemental Table 5). When we studied the protein expression levels of these CYPs, we also observed markedly higher levels of CYP26A1 in the liver of alcohol-fed mice (Fig. 9B). In contrast, there was no apparent increase in the expression level of CYP26B, suggesting that the increase in mRNA levels for this gene do not correspond with a resultant increase in protein levels or that the Western blot assay was not as sensitive as the qPCR assay in detecting an increase. As a control, we also measured hepatic CYP2E1 levels and found this protein to be significantly increased by alcohol, consistent with the extensive literature on this protein.
Figure 9.

Alcohol consumption is associated with the induction of retinoic acid-catabolizing CYPs. A) Consumption of 6.4% alcohol for 2 wk was associated with increased mRNA expression levels of Cyp26a1 and Cyp26b1 in the liver (n = 8–11; reference gene, 18s). B) Consumption of 6.4% alcohol for 2 wk was also associated with increased protein expression levels of CYP2E1 and CYP26A1, but not CYP26B. *P < 0.05 vs. control; Student’s t test.
DISCUSSION
The goal of this study was to gain a better understanding of how chronic alcohol consumption induces loss of hepatic retinoid stores and increases extrahepatic retinoid levels. The literature proposes 2 different hypotheses to explain this effect. The first posits that alcohol precipitates CYP-mediated catabolism of retinoid within the liver, with a concurrent accumulation of postprandial retinoid in extrahepatic tissues. The second hypothesis proposes that alcohol consumption induces direct mobilization of retinoid from the liver to the extrahepatic tissues, with extrahepatic retinoid levels increasing in relation to decreasing hepatic retinoid levels (6, 9, 11, 29). As discussed below, our data indicate that each of these hypotheses has some validity, but neither is fully correct.
Based on our data, we propose a biphasic model that incorporates elements of both of these hypotheses to explain alcohol’s effect on hepatic and extrahepatic retinoid homeostasis (Fig. 10). The first phase of this model occurs at the onset of alcohol consumption (during the alcohol-adaptation period) and is characterized by mobilization of hepatic retinoid stores to extrahepatic tissues. During this phase, there is a relatively large increase (2- to 7-fold) in the retinoid content of extrahepatic tissues, which is mirrored by an ∼15% decline in hepatic retinoid content. We note that 80–90% of all retinoid present within the body is found in the liver (30), so even a modest decline in hepatic levels represents a relatively large gain in extrahepatic tissues. We propose that the major cause of the decline in hepatic retinoid content during this first phase is its mobilization and redistribution to extrahepatic tissues. The second phase, which ensues with continued alcohol consumption, is characterized by a quantitatively large decline in hepatic retinoid stores, with no further increase in extrahepatic tissue retinoid levels. We believe that the major reason for the decline in hepatic retinoid content during this second phase is CYP-mediated catabolism of hepatic retinoid.
Figure 10.

Chronic alcohol consumption has a biphasic effect on hepatic retinoid homeostasis, as is shown in this summary of our biphasic model describing alcohol’s effect on hepatic and extrahepatic retinoid homeostasis. The first phase is characterized by a relatively small but significant decline in hepatic retinoid stores, a relatively large increase in extrahepatic retinoid levels, and a transient increase in serum retinol, reflecting the mobilization of hepatic retinoid stores. The second phase, which commences with continued alcohol consumption, is characterized by a quantitatively large decline in hepatic retinoid stores reflecting CYP-mediated catabolism of retinoid within the liver, with no further mobilization to the extrahepatic tissues.
A key component of our model is the notion that during the alcohol-adaptation period, hepatic retinoid is mobilized from the liver and accumulates in extrahepatic tissues. Although this concept has been postulated (6), our data from Lrat−/− mice show that in the absence of hepatic retinoid stores, alcohol had no effect on extrahepatic tissue retinoid levels. These data provide definitive evidence that the retinol and retinyl ester accumulating in extrahepatic tissues of alcohol-fed mice originates in the liver. Indeed, based on the hepatic and extrahepatic retinoid levels we measured, we projected that the amount of retinoid that was exported from the liver during the alcohol-adaptation period would be quantitatively similar to the amount that accumulated in the extrahepatic tissues. Similarly, our data from alcohol-fed mice consuming a VAD diet did not support the alternate hypothesis that dietary retinoid preferentially accumulates in the extrahepatic tissues of alcohol-fed mice. In this regard, the magnitude of the alcohol-induced increase in WAT and lung retinoid levels was the same for mice consuming either a VAS or a VAD diet.
Accepting that alcohol induces mobilization of hepatic retinoid to extrahepatic tissues, the question of how this effect is mediated remains. We found that circulating levels of retinol and RBP and hepatic RBP expression transiently increased during the alcohol-adaptation period, coinciding with the large increase in extrahepatic retinoid content and a modest decrease in hepatic retinoid stores. In contrast, we found no evidence for increased hepatic retinyl ester secretion via VLDL in alcohol-fed mice. Together, these data suggest that alcohol stimulates the mobilization of hepatic retinoid stores via retinol bound to RBP. RBP is the sole specific transport protein for retinol in the fasting circulation, facilitating the mobilization of retinol stores from HSCs to extrahepatic tissues (2, 3). The rise in circulating retinol levels that we observed is not a large one (∼20%), but this is undoubtedly sufficient over time to allow for the observed increase in extrahepatic tissue retinoid levels. In agreement with our data, a transient increase in serum retinol and RBP levels has been reported in alcohol-fed baboons (31). To definitively test the hypothesis that hepatic retinol is mobilized to the periphery via RBP, we conducted an alcohol-feeding study in WT vs. Rbp−/− mice. In agreement with our hypothesis, the mobilization of hepatic retinoid stores was blunted in the Rbp−/− mice; however, it was apparent that some retinol was still being mobilized from the liver. Thus, the secretion of retinol bound to RBP is the primary mechanism that mediates the alcohol-induced mobilization of hepatic retinoids stores to the extrahepatic tissues during the alcohol-adaptation period.
We believe that the small amount of hepatic retinoid mobilization that occurs in Rbp−/− mice reflects a compensatory mechanism unique to these mutant mice. In this regard, the literature describes other proteins capable of transporting retinol in the serum, most notably albumin (18, 32). Our original description of Rbp−/− mice established that the relatively low level of retinol present in their circulation is bound to a 68 kDa protein that we propose is albumin (18). Our current data show that in the context of chronic alcohol consumption, Rbp−/− mice can export small amounts of retinol from the liver, which we suspect is bound to albumin. This finding supports the notion that, at least under these genetically altered conditions, other circulating proteins can mediate alcohol-induced hepatic retinol export, independent of RBP.
Within the aqueous environment of the cell, retinol is bound to CRBP1, which functions to facilitate retinol uptake and efflux via coupling with the RBP receptor STRA6, retinol esterification via LRAT, and retinol oxidation to retinoic acid via retinol and retinal dehydrogenases (2, 3, 19). Our data from alcohol-fed Crbp1−/− mice show that CRBP1 is dispensable for the mobilization of hepatic retinoid stores, but is essential for the uptake of retinol by the lung, spleen, and kidney, but not by WAT and BAT. This finding is consistent with a report establishing that the alcohol-induced increase in retinol levels in testes are blocked in Crbp1−/− mice (33). These tissue-specific effects of Crbp1 deficiency reflect the known expression pattern of this gene. For example, in agreement with Eriksson et al. (34), we found Crbp1 expression to be highest in liver, with progressively lower levels in kidney, lung, and spleen. Although we also detected relatively high expression of Crbp1 in WAT, it is known that this mRNA is exclusively expressed in the stromovascular fraction, whereas Crbp3 is primarily expressed in adipocytes (35, 36). In this regard, we also showed relatively high levels of Crbp3 expression in WAT and BAT. Thus, although Crbp1 deficiency had no effect on uptake of retinol into WAT and BAT, we predicted that alcohol-fed Crbp3−/− mice would display diminished retinol uptake into their adipose depots. Our observation that uptake of retinol into adipose tissue was unaffected in alcohol-fed Crbp1−/− mice is consistent with a recent report that adipocyte-specific knockdown of Stra6 has no effect on cellular retinol uptake (37), suggesting that coupling of STRA6 and CRBP1 is not necessary for the uptake of retinol into adipose tissue. In summary, the alcohol-induced mobilization of hepatic retinoid stores can occur independent of CRBP1, but CRBP1-faciliated retinol uptake into extrahepatic tissues occurs in a tissue-specific manner.
CD36 has been extensively studied as a free fatty acid transporter and its role in cellular fatty acid uptake is well established (14). CD36 can also catalyze cholesterol efflux from hepatocytes and macrophages, as well as facilitate proretinoid carotenoid uptake in the intestine and adipose tissue (14). Recently, CD36 has been shown to regulate triglyceride hydrolysis within adipocytes, where it exerts negative feedback regulation on lipolysis, primarily through its effects on hormone-sensitive lipase (38). However, as far as we are aware, there is no published evidence that CD36 has a role in either hepatic or extrahepatic retinoid uptake or metabolism. Thus, our finding that mice lacking CD36 are protected against alcohol-induced hepatic retinoid redistribution to peripheral tissues is surprising. Given that CD36 is known to be expressed in HSCs—the major site of retinoid storage in the liver (39, 40)—and that we and others have shown that CD36 expression is increased in the alcohol-exposed liver (15, 24), we speculate that CD36 has a direct role in alcohol-induced retinoid efflux from the liver. The precise mechanism through which CD36 mediates this effect is currently unknown. We hypothesize that CD36 mediates transfer of retinol from HSC retinoid stores to the apo-RBP present in the extracellular space surrounding the HSCs. This effect would be similar to the proposed role of CD36 in facilitating cholesterol efflux from cells (14). As an alternative, CD36 may act to regulate lipolytic activity within HSCs, as has been shown for triglyceride lipolysis in adipocytes (38), thereby increasing retinyl ester hydrolysis to retinol, which is subsequently exported bound to RBP. This too could account for why alcohol consumption fails to increase hepatic retinoid efflux from Cd36-deficient mice, as there would be less free retinol to mobilize. Studies are continuing in our laboratory into the role that CD36 plays in hepatic retinoid metabolism.
During the alcohol-adaption period, we observed a moderate (∼15%) decline in hepatic retinoid content that was redistributed to extrahepatic tissues. However, with continuing alcohol consumption, hepatic retinoid levels precipitously dropped (>60%) without further accumulation in the periphery. We propose that the major factor driving this second phase of decline was catabolism. Indeed, increased CYP activity has been described in the alcoholic liver and has been proposed to contribute to the breakdown and loss of hepatic retinoid (10). Moreover, it has been hypothesized that the alcohol-inducible enzyme CYP2E1 is primarily responsible for this effect (12, 13). We tested this hypothesis in alcohol-fed Cyp2e1−/− mice, observing that although the alcohol-induced loss of hepatic retinol was blocked in Cyp2e1−/− mice, there was still a significant decline in hepatic retinyl esters. It is known that >98% of the retinoid present in HSCs is stored as retinyl ester and that these cells contain low levels of retinol (30). Consequently, we believe that retinol measured in whole liver extracts primarily reflects the hepatic pool of retinoid within hepatocytes, whereas retinyl ester levels reflect the hepatic pool of retinoid within HSCs. It is known that CYP2E1 is primarily expressed in hepatocytes (41); thus, we can rationalize our observations in Cyp2e1−/− mice by concluding that CYP2E1 contributes to the breakdown of retinol within hepatocytes, but not the decrease in retinyl ester in HSCs. This interpretation of our data accounts for why retinol levels were unaffected in the liver of alcohol-fed Cyp2e1−/− mice, but retinyl ester levels still declined. This observation in Cyp2e1−/− mice has added significance because it is the first to suggest that alcohol has distinct effects on retinoid metabolism in hepatocytes and HSCs.
As discussed above, alcohol consumption is known to increase hepatic total CYP activity (10). The observation that hepatic retinyl ester levels continue to decline in alcohol-fed Cyp2e1−/− mice suggests that other factors contribute to this effect. In this respect, several CYPs have been shown to catabolize retinoic acid, with CYP26A1 and CYP26B1 thought to be the most important (2, 3). Our analysis of hepatic Cyp26a1 and Cyp26b1 expression shows that these retinoic acid–catabolizing enzymes are induced at the mRNA level in the liver of alcohol-fed mice. Our analysis of CYP26A1 protein confirms the significant increase in the expression of this enzyme; however, we did not observe a corresponding increase in CYP26B expression levels. Alcohol’s effect on the expression level and activity of retinoic acid in catabolizing CYPs is the focus of continuing studies in our laboratory. It is also unclear how alcohol stimulates hepatic Cyp26a1 and Cyp26b1 expression, another question that will be the focus of future work.
The increased CYP-mediated catabolism of hepatic retinoids is likely to be a key factor in the transition between the mobilization phase and the catabolism phase of our model (Fig. 10). We recently reported that the acyl composition of retinyl ester undergoes a significant change during the alcohol-adaptation period (21). Specifically, retinyl palmitate levels in alcohol-fed mice precipitously drop, whereas levels of usually less abundant retinyl esters, such as retinyl oleate, increase significantly. We believe that this change in retinyl ester composition reflects an alcohol-induced, futile cycle of retinyl ester hydrolysis and re-esterification. As can be seen in Supplemental Fig. 3, all of the different strains of mutant mice used in this study underwent an identical switch in their retinyl ester acyl composition during the alcohol-adaptation period. The biochemical underpinning of this phenomenon is extensively discussed in our recent article (21).
In the context of our model, we believe that, during the mobilization phase, alcohol stimulates the hydrolysis of hepatic retinyl esters, with a small fraction of the liberated retinol exported to the extrahepatic tissues, whereas most is converted back into retinyl ester. As indicated above, we believe that a breakdown in this cycle precipitates the quantitatively large declines in hepatic retinoid characteristic of the catabolism phase. We speculate that the loss of retinol from this system governs the switch between the mobilization phase and the catabolism phase—that is, retinol mobilization and re-esterification stops because it is no longer available. How this retinol is lost from the system is the subject of continuing studies in our laboratory, but we propose the following 2 working hypotheses: first, retinol is directly catabolized by hepatic CYPs; second, CYP-mediated breakdown of retinoic acid drives increased retinol metabolism to retinoic acid. In the second scenario, the continued catabolism of retinoic acid exerts negative pressure on retinol, as it keeps physiologic levels of retinoic acid constant in the liver. In addition to our continuing studies of alcohol-inducible CYPs that can catabolize retinol and retinyl ester, we are also studying the ability of alcohol to stimulate retinyl ester hydrolysis.
In summary, we have established the time course of events responsible for alcohol-induced hepatic retinoid depletion. We have delineated the existence of an initial phase of retinoid mobilization from the liver to the extrahepatic tissues, followed by a phase of hepatic retinoid catabolism. Our data show that mobilization is a quantitatively minor contributor toward alcohol-induced hepatic retinoid depletion, but can account for the increase in extrahepatic tissue retinoid levels. The second phase, characterized by catabolism, is responsible for most of the hepatic retinoid loss. Our investigations establish that there are significant changes in hepatic retinoid homeostasis that occur before the onset of alcohol-induced liver injury and are consistent with the hypothesis that alcohol’s impact on retinoid homeostasis and signaling contribute to disease development.
Supplementary Material
Acknowledgments
This study was supported by U.S. National Institutes of Health (NIH) National Institute on Alcohol Abuse and Alcoholism Grants RC2 AA019413 and R21 AA021336 (to W.S.B.), R21 AA020561 (to L.S.H.), and K99 AA022652 (to R.D.C.), and NIH National Institute of Diabetes and Digestive and Kidney Diseases Grants R01 DK068437 and R01 DK079221 (to W.S.B.).
Glossary
- BAT
brown adipose tissue
- CRBP1
cellular retinol binding protein 1
- CYP
cytochrome P450
- HSC
hepatic stellate cell
- LRAT
lecithin-retinol acyltransferase
- MELD
model for end-stage liver disease
- qPCR
quantitative PCR
- RBP
retinol-binding protein
- VAD
vitamin A-deficient
- VAS
vitamin A-sufficient
- VLDL
very low-density lipoprotein
- WAT
white adipose tissue
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
References
- 1.Balmer J. E., Blomhoff R. (2002) Gene expression regulation by retinoic acid. J. Lipid Res. 43, 1773–1808 [DOI] [PubMed] [Google Scholar]
- 2.D’Ambrosio D. N., Clugston R. D., Blaner W. S. (2011) Vitamin A metabolism: an update. Nutrients 3, 63–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shirakami Y., Lee S. A., Clugston R. D., Blaner W. S. (2012) Hepatic metabolism of retinoids and disease associations. Biochim. Biophys. Acta 1821, 124–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee Y. S., Jeong W. I. (2012) Retinoic acids and hepatic stellate cells in liver disease. J. Gastroenterol. Hepatol. 27(Suppl 2), 75–79 [DOI] [PubMed] [Google Scholar]
- 5.Bechmann L. P., Canbay A. (2014) The hunt for treatment options of fatty liver continues: effects of retinoic acid on hepatic steatosis reveal novel transcriptional interactions of nuclear receptors. Hepatology 59, 1662–1664. [DOI] [PubMed] [Google Scholar]
- 6.Clugston R. D., Blaner W. S. (2012) The adverse effects of alcohol on vitamin A metabolism. Nutrients 4, 356–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leo M. A., Lieber C. S. (1982) Hepatic vitamin A depletion in alcoholic liver injury. N. Engl. J. Med. 307, 597–601 [DOI] [PubMed] [Google Scholar]
- 8.Ouziel R., Trepo E., Cremer A., Moreno C., Degre D., Chaouni M., Vercruysse V., Quertinmont E., Deviere J., Lemmers A., Gustot T. (2013) Correction of all-trans retinoic acid deficiency in alcoholic cirrhosis lessens the excessive inflammatory monocyte response: a translational study. Liver Int. 34, 343-352. [DOI] [PubMed] [Google Scholar]
- 9.Wang X. D. (1999) Chronic alcohol intake interferes with retinoid metabolism and signaling. Nutr. Rev. 57, 51–59 [DOI] [PubMed] [Google Scholar]
- 10.Sato M., Lieber C. S. (1982) Increased metabolism of retinoic acid after chronic ethanol consumption in rat liver microsomes. Arch. Biochem. Biophys. 213, 557–564 [DOI] [PubMed] [Google Scholar]
- 11.Leo M. A., Kim C., Lieber C. S. (1986) Increased vitamin A in esophagus and other extrahepatic tissues after chronic ethanol consumption in the rat. Alcohol. Clin. Exp. Res. 10, 487–492 [DOI] [PubMed] [Google Scholar]
- 12.Liu C., Russell R. M., Seitz H. K., Wang X. D. (2001) Ethanol enhances retinoic acid metabolism into polar metabolites in rat liver via induction of cytochrome P4502E1. Gastroenterology 120, 179–189 [DOI] [PubMed] [Google Scholar]
- 13.Liu C., Chung J., Seitz H. K., Russell R. M., Wang X. D. (2002) Chlormethiazole treatment prevents reduced hepatic vitamin A levels in ethanol-fed rats. Alcohol. Clin. Exp. Res. 26, 1703–1709 [DOI] [PubMed] [Google Scholar]
- 14.Silverstein R. L., Febbraio M. (2009) CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2, re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clugston R. D., Yuen J. J., Hu Y., Abumrad N. A., Berk P. D., Goldberg I. J., Blaner W. S., Huang L. S. (2014) CD36-deficient mice are resistant to alcohol- and high-carbohydrate-induced hepatic steatosis. J. Lipid Res. 55, 239–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Batten M. L., Imanishi Y., Maeda T., Tu D. C., Moise A. R., Bronson D., Possin D., Van Gelder R. N., Baehr W., Palczewski K. (2004) Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J. Biol. Chem. 279, 10422–10432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Febbraio M., Abumrad N. A., Hajjar D. P., Sharma K., Cheng W., Pearce S. F., Silverstein R. L. (1999) A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J. Biol. Chem. 274, 19055–19062 [DOI] [PubMed] [Google Scholar]
- 18.Quadro L., Blaner W. S., Salchow D. J., Vogel S., Piantedosi R., Gouras P., Freeman S., Cosma M. P., Colantuoni V., Gottesman M. E. (1999) Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 18, 4633–4644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghyselinck N. B., Båvik C., Sapin V., Mark M., Bonnier D., Hindelang C., Dierich A., Nilsson C. B., Håkansson H., Sauvant P., Azaïs-Braesco V., Frasson M., Picaud S., Chambon P. (1999) Cellular retinol-binding protein I is essential for vitamin A homeostasis. EMBO J. 18, 4903–4914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee S. S., Buters J. T., Pineau T., Fernandez-Salguero P., Gonzalez F. J. (1996) Role of CYP2E1 in the hepatotoxicity of acetaminophen. J. Biol. Chem. 271, 12063–12067 [DOI] [PubMed] [Google Scholar]
- 21.Clugston R. D., Jiang H., Lee M. X., Berk P. D., Goldberg I. J., Huang L. S., Blaner W. S. (2013) Altered hepatic retinyl ester concentration and acyl composition in response to alcohol consumption. Biochim. Biophys. Acta 1831, 1276–1286 [PubMed] [Google Scholar]
- 22.O’Byrne S. M., Wongsiriroj N., Libien J., Vogel S., Goldberg I. J., Baehr W., Palczewski K., Blaner W. S. (2005) Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT). J. Biol. Chem. 280, 35647–35657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De la M. Hall P., Lieber C. S., DeCarli L. M., French S. W., Lindros K. O., Järveläinen H., Bode C., Parlesak A., Bode J. C. (2001) Models of alcoholic liver disease in rodents: a critical evaluation. Alcohol. Clin. Exp. Res. 25(5 Suppl ISBRA), 254S–261S [DOI] [PubMed] [Google Scholar]
- 24.Clugston R. D., Jiang H., Lee M. X., Piantedosi R., Yuen J. J., Ramakrishnan R., Lewis M. J., Gottesman M. E., Huang L. S., Goldberg I. J., Berk P. D., Blaner W. S. (2011) Altered hepatic lipid metabolism in C57BL/6 mice fed alcohol: a targeted lipidomic and gene expression study. J. Lipid Res. 52, 2021–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.D’Ambrosio D. N., Walewski J. L., Clugston R. D., Berk P. D., Rippe R. A., Blaner W. S. (2011) Distinct populations of hepatic stellate cells in the mouse liver have different capacities for retinoid and lipid storage. PLoS ONE 6, e24993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wongsiriroj N., Jiang H., Piantedosi R., Yang K. J., Kluwe J., Schwabe R. F., Ginsberg H., Goldberg I. J., Blaner W. S. (2014) Genetic dissection of retinoid esterification and accumulation in the liver and adipose tissue. J. Lipid Res. 55, 104–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muto Y., Goodman D. S. (1972) Vitamin A transport in rat plasma. Isolation and characterization or retinol-binding protein. J. Biol. Chem. 247, 2533–2541 [PubMed] [Google Scholar]
- 28.Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato M., Lieber C. S. (1982) Changes in vitamin A status after acute ethanol administration in the rat. J. Nutr. 112, 1188–1196 [DOI] [PubMed] [Google Scholar]
- 30.Blaner W. S., O’Byrne S. M., Wongsiriroj N., Kluwe J., D’Ambrosio D. M., Jiang H., Schwabe R. F., Hillman E. M., Piantedosi R., Libien J. (2009) Hepatic stellate cell lipid droplets: a specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta 1791, 467–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato M., Lieber C. S. (1981) Hepatic vitamin A depletion after chronic ethanol consumption in baboons and rats. J. Nutr. 111, 2015–2023 [DOI] [PubMed] [Google Scholar]
- 32.Futterman S., Heller J. (1972) The enhancement of fluorescence and the decreased susceptibility to enzymatic oxidation of retinol complexed with bovine serum albumin, -lactoglobulin, and the retinol-binding protein of human plasma. J. Biol. Chem. 247, 5168–5172 [PubMed] [Google Scholar]
- 33.Kane M. A., Folias A. E., Wang C., Napoli J. L. (2010) Ethanol elevates physiological all-trans-retinoic acid levels in select loci through altering retinoid metabolism in multiple loci: a potential mechanism of ethanol toxicity. FASEB J. 24, 823–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eriksson U., Das K., Busch C., Nordlinder H., Rask L., Sundelin J., Sällström J., Peterson P. A. (1984) Cellular retinol-binding protein: quantitation and distribution. J. Biol. Chem. 259, 13464–13470 [PubMed] [Google Scholar]
- 35.Tsutsumi C., Okuno M., Tannous L., Piantedosi R., Allan M., Goodman D. S., Blaner W. S. (1992) Retinoids and retinoid-binding protein expression in rat adipocytes. J. Biol. Chem. 267, 1805–1810 [PubMed] [Google Scholar]
- 36.Vogel S., Mendelsohn C. L., Mertz J. R., Piantedosi R., Waldburger C., Gottesman M. E., Blaner W. S. (2001) Characterization of a new member of the fatty acid-binding protein family that binds all-trans-retinol. J. Biol. Chem. 276, 1353–1360 [DOI] [PubMed] [Google Scholar]
- 37.Zemany L., Kraus B. J., Norseen J., Saito T., Peroni O. D., Johnson R. L., Kahn B. B. (2014) Downregulation of STRA6 in adipocytes and adipose stromovascular fraction in obesity and effects of adipocyte-specific STRA6 knockdown in vivo. Mol. Cell. Biol. 34, 1170–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vroegrijk I. O., van Klinken J. B., van Diepen J. A., van den Berg S. A., Febbraio M., Steinbusch L. K., Glatz J. F., Havekes L. M., Voshol P. J., Rensen P. C., van Dijk K. W., van Harmelen V. (2013) CD36 is important for adipocyte recruitment and affects lipolysis. Obesity (Silver Spring) 21, 2037–2045 [DOI] [PubMed] [Google Scholar]
- 39.Schneiderhan W., Schmid-Kotsas A., Zhao J., Grünert A., Nüssler A., Weidenbach H., Menke A., Schmid R. M., Adler G., Bachem M. G. (2001) Oxidized low-density lipoproteins bind to the scavenger receptor, CD36, of hepatic stellate cells and stimulate extracellular matrix synthesis. Hepatology 34, 729–737 [DOI] [PubMed] [Google Scholar]
- 40.Da Silva Morais A., Abarca-Quinones J., Horsmans Y., Stärkel P., Leclercq I. A. (2007) Peroxisome proliferated-activated receptor gamma ligand: pioglitazone, does not prevent hepatic fibrosis in mice. Int. J. Mol. Med. 19, 105–112 [PubMed] [Google Scholar]
- 41.Forkert P. G., Massey T. E., Jones A. B., Park S. S., Gelboin H. V., Anderson L. M. (1991) Distribution of cytochrome CYP2E1 in murine liver after ethanol and acetone administration. Carcinogenesis 12, 2259–2268 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.