

Abstract
Serum- and glucocorticoid-regulated kinase (SGK)1 is associated with several important pathologic conditions and plays a modulatory role in adaptive immune responses. However, the involvement and functional role of SGK1 in innate immune responses remain entirely unknown. In this study, we establish that SGK1 is a novel and potent negative regulator of TLR-induced inflammation. Pharmacologic inhibition of SGK1 or suppression by small interfering RNA enhances proinflammatory cytokine (TNF, IL-12, and IL-6) production in TLR-engaged monocytes, a result confirmed in Cre-loxP-mediated SGK1-deficient cells. SGK1 inhibition or gene deficiency results in increased phosphorylation of IKK, IκBα, and NF-κB p65 in LPS-stimulated cells. Enhanced NF-κB p65 DNA binding also occurs upon SGK1 inhibition. The subsequent enhancement of proinflammatory cytokines is dependent on the phosphorylation of TGF-β-activated kinase 1 (TAK1), as confirmed by TAK1 gene silencing. In vivo relevance was established in a murine endotoxin model, in which we found that SGK1 inhibition aggravates the severity of multiple organ damage and enhances the inflammatory response by heightening both proinflammatory cytokine levels and neutrophil infiltration. These findings have identified an anti-inflammatory function of SGK1, elucidated the underlying intracellular mechanisms, and establish, for the first time, that SGK1 holds potential as a novel target for intervention in the control of inflammatory diseases.—Zhou, H., Gao, S., Duan, X., Liang, S., Scott, D. A., Lamont, R. J., Wang, H. Inhibition of serum- and glucocorticoid-inducible kinase 1 enhances TLR-mediated inflammation and promotes endotoxin-driven organ failure.
Keywords: SGK1, inflammatory cytokines, TAK1, LPS
TLR-mediated production of inflammatory cytokines plays a critical role in the progression of bacterial-driven sepsis, a significant cause of nosocomial mortality and morbidity (1, 2). Upon recognition of microbe-associated molecular patterns (MAMPs) on invading pathogens, innate immune cells such as macrophages and monocytes are activated and secrete inflammatory cytokines, chemokines, and reactive oxygen species. These inflammatory mediators, in concert, lead to inflammation and the activation of adaptive immunity (3). On the other hand, overzealous production of inflammatory mediators such as TNF, IL-1, IL-6, or IL-12 causes severe collateral damage, including hypertension, hemoconcentration, epithelium disruption, and blood coagulation that consequently leads to multiple organ failure (4, 5). MAMP-initiated inflammation through binding with TLRs is tightly regulated by an intricate hierarchical regulatory system that serves to both activate innate immunity while also avoiding the excessive production of inflammatory mediators. In this regard, the binding of specific microbe-derived components such as LPS, flagellin, double-stranded RNA, or cytosine-phosphate-guanine to TLRs leads to the recruitment of adaptor proteins, myeloid differentiation primary response gene 88 (MyD88), and/or Toll-IL-1 receptor-domain-containing adapter-inducing IFN-β (TRIF), which in turn phospho-activate other molecules, such as IL-1 receptor -associated kinase (IRAK), TNF receptor-associated factor (TRAF), TGF-β-activated kinase 1 (TAK1; by MyD88), or IFN-regulatory factor (IRF; by TRIF). The activation of these enzymes triggers multiple signaling pathways such as MyD88-mediated IKK-IκB-NF-κB and MAPK pathways, or the TRIF-mediated IRF-IFN-β pathway, ultimately inducing the expression of inflammatory cytokines. Endogenous molecular regulators such as myeloid differentiation 88 short (MyD88s), suppressor of cytokine signaling 1 (SOCS1), nucleotide-binding oligomerization domain-containing protein 2, PI3K, Toll-interacting protein, and TNF-α-induced protein 3 (A20), or transmembrane protein regulators like IL-1 receptor-like 1, single Ig IL-1 receptor-related molecule, and TNF-related apoptosis-inducing ligand receptor 1 (TRAILR) (6, 7), have been shown to play key roles in maintaining the homeostasis of immune responses. Among these regulators, the TLR-activated PI3K-Ak thymic lymphoma/PKB (Akt) pathway is critical for the control of immune responses through differentially regulating pro- and anti- inflammatory cytokine production (8, 9). Either inhibition or gene deficiency of PI3K results in the enhancement of proinflammatory cytokine production and exacerbates the severity of inflammatory responses (10, 11). Inhibition of Akt, a downstream kinase of PI3K, has also been demonstrated to dramatically aggravate the intensity of inflammation and reduce mouse survival in a mouse endotoxin model (9, 12, 13). Although a number of downstream molecules in the PI3K signaling pathway have been identified, the role of other PI3K-activated molecules in the regulation of TLR-mediated inflammation remains unknown.
Serum- and glucocorticoid-inducible kinases (SGKs) are a class of serine/threonine kinases belonging to the AGC kinase family (PKA, protein kinase G, and PKC family) (14). There are 3 isoforms of SGK, namely SGK1, SGK2, and SGK3. SGK1 is widely expressed and rapidly responds to a variety of stimuli such as follicle-stimulating hormone, osmotic shock, ischemia, TGF-β, glucocorticoids, and mineralocorticoids (14). Like Akt, SGK1 can be fully activated by PI3K through phosphorylation at Thr256 by 3-phosphoinositide-dependent protein kinase (PDK)-1 and Ser422 by PDK2. Activated SGK1 plays an important role in activating certain potassium, sodium, and chloride channels (15). Recent studies have shown that SGK1 activation is associated with apoptosis, cell cycle, and several serious medical conditions such as hypertension, diabetic nephropathy, and cancers (16, 17). In addition, SGK1 has been found to play an important role in the differentiation of T helper (Th) cells and the induction of pathogenic Th17 cells (18, 19). Activation of SGK1 has been reported to facilitate Th2 differentiation by negatively regulating degradation of the transcription factor JunB. Furthermore, gene deficiency of SGK1 enhances the production of IFN-γ and Th1 cell-mediated immunity upon viral infection (18). Sodium-mediated activation of SGK1 was shown to promote the differentiation of Th17 in vitro and in vivo through enhancing the expression of IL-23 receptor, thus accelerating the development of autoimmunity (19, 20). These studies revealed an essential regulatory role of SGK1 in adaptive immunity. However, the functional role of SGK1 in TLR-mediated inflammatory responses remains to be determined.
TAK1 was originally identified as a member of the MAPKK kinase family and is a critical mediator involved in TLR-induced inflammatory cytokine secretion (21). TAK1 forms a heterotrimeric complex with its activator proteins TGF-β-activated kinase 1-binding protein (TAB) 1 and TAB2 and, thus, functions as one of the most important upstream regulators for TLR-initiated NF-κB and MAPK pathways in different immune cells (22). The activity of TAK1 is regulated by multiple posttranslational modifications. Apart from TRAF6-mediated Lys63-linked polyubiquitination, phosphorylation of TAK1 has also been demonstrated as a key to the activation of TAK1 (23, 24). Recent studies reported that several protein kinases and phosphatases altered TAK1 activity through modifying phosphorylation and, consequently, regulated the transcriptional activity of its downstream inflammatory signaling pathways: NF-κB and MAPK (22, 25). One example is dual-specificity phosphatase 14, which was observed to dephosphorylate TAK1 at Thr187 negatively regulating TNF- and IL-1β-induced NF-κB activation (25). Nevertheless, considering the central role of TAK1 in the control of inflammatory signaling pathways, there are still substantial gaps in our understanding of this kinase, especially the regulation of TAK1 activity in TLR-initiated innate immune responses.
In the present study, we have identified a negative regulatory role for SGK1 in TLR-mediated inflammatory responses through modifying the activity of TAK1. Suppression of SGK1 or CD11c-driven Cre-mediated deletion of loxP-flanked sgk1 enhanced the phosphorylation of TAK1 and increased the downstream activity of NF-κB and, thus, augmented the production of IL-12, TNF, and IL-6 in innate immune cells. Moreover, SGK1 inhibition heightened systemic proinflammatory cytokine levels, infiltration of neutrophils, and aggravated the tissue damage in multiple organs in a mouse endotoxin model. Collectively, our findings have identified a novel negative regulatory role of SGK1 in TLR-mediated inflammatory responses and characterized the role of the SGK1-TAK1 signaling pathway in TLR-mediated inflammatory responses, indicating that SGK1 may represent a novel target for intervention in the control of inflammatory diseases.
MATERIALS AND METHODS
Mice and reagents
cd11c-Cre/sgk1 fl/fl mice were generated by crossing cd11c-driven Cre C57BL/6 (purchased from The Jackson Laboratory, Bar Harbor, ME, USA) to loxP-flanked SGK1 mice (provided by Dr. Alexander, Dartmouth Medical School, Hanover, NH, USA). Control mice were negative littermates from this breeding. All the mice were housed in a specific pathogen-free facility at the University of Louisville, and the University of Louisville Institutional Animal Care and Use Committee approved all animal protocols. Pam3CSK4, flagellin, and ultrapure LPS from Escherichia coli 0111:B4 were purchased from InvivoGen (San Diego, CA, USA). Phospho-SGK1 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Total SGK1 and TAK1 antibodies were from Proteintech (Chicago, IL, USA). All other antibodies were from Cell Signaling Technology (Danvers, MA, USA). The SGK1 inhibitor EMD638683 was from MedChem Express (Monmouth Junction, NJ, USA) and has been characterized and shown to be specific for SGK1 without discernible effects on a panel of 68 other kinases (26). PI3K inhibitor LY294002 was from LC Laboratories (Woburn, MA, USA). Nontargeting pools of small interfering RNA (siRNA) and a mixture of 4 prevalidated siRNA duplexes specific for SGK1 or TAK1 (On-Targetplus) were from GE Healthcare Dharmacon (Fisher Scientific, Pittsburgh, PA, USA). The NF-κB p65 (Ser529/Ser536) inhibitory peptide set (p65 inhibitory peptide sequence, 5′-DRQIKIWFQNRRMKWKKNGLLSGDEDFSS-3′; control sequence, 5′-DRQIKIWFQNRRMKWKK-3′) was from Novus Biologicals (Littleton, CO, USA). Cytokine ELISA kits were from eBioscience (San Diego, CA, USA).
Cell preparation
Peripheral blood mononuclear cells were obtained from the venous blood of healthy donors as per protocols approved by the University of Louisville, Institutional Review Board, Human Subjects Protection Program (study number 12.0373). Monocytes were isolated by negative selection using the human Monocyte Isolation Kit II from Miltenyi Biotec Incorporated (San Diego, CA, USA). The purity of monocytes was routinely >90%, as determined by flow cytometry using an FITC-labeled anti-CD14 antibody. Bone marrow cells were isolated from wild-type or SGK1 Cre-loxP mice, and bone marrow-derived dendritic cells (BMDCs) were generated as previously described (27). Cells were cultured in Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal bovine serum, 50 μM 2-ME, 1 mM sodium pyruvate, 2 mM l-glutamine, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 50 U/ml penicillin, and 50 μg/ml streptomycin.
Transfection, cytokine detection, and Western blot
Transfection of human monocytes was carried out by electroporation using an Amaxa 4D-Nucleofector device (Lonza, Cologne, Germany) according to the manufacturer’s protocols. Briefly, purified monocytes (4 × 106) were resuspended in 100 μl Nucleofector Solution (Amaxa Human Monocyte Nucleofector Kit; Lonza) along with 2 μg of a green fluorescent protein (GFP)-coding plasmid (pCMV-GFP) and 2 μg siRNA duplexes or ectopic plasmids for each target. Immediately after electroporation, 400 μl prewarmed M-199 containing 10% fetal calf serum (FCS) was added to cells that were then transferred into culture plates containing prewarmed M-199 with 10% FCS. At 48 h posttransfection, cells were exposed to LPS (1 µg/ml) with or without SGK1 inhibitor. Cell lysates were prepared as previously described (28), and cell-free supernatants were assayed for cytokine levels by ELISA 20 h after the addition of LPS. The levels of total SGK1 and TAK1 were assessed by Western blot at 48 h posttransfection. Images were acquired using the ImageQuant LAS 100 (GE Healthcare BioScience, Pittsburgh, PA, USA). For experiments using inhibitors for PI3K (LY294002, 25 mM) or SGK1 (EMD638683, 10 μM), control cells were pretreated for 2 h with 0.01% DMSO (organic solvent control) prior to LPS (1 μg/ml) stimulation.
Endotoxin model and immunohistochemistry analysis
C57BL/6 mice were injected intraperitoneally with a sublethal dose of E. coli 0111:B4 LPS (1 mg/kg) with or without the pretreatment of EMD638683 (10 mg/kg) for 2 h. Control mice were injected with vehicle only. After 3- and 24-h intraperitoneal injection of vehicle or E. coli LPS, blood was collected, and plasma was isolated to measure the levels of TNF, IL-6, and IL-12P40. Livers and lung were harvested 6 and 24 h postinjection of LPS but from separate groups. Plasma levels of TNF, IL-6, and IL-12 were determined by ELISA. Liver and lung tissue samples from septic mice with or without treatment with EMD638683 were fixed in 4% formaldehyde, embedded in optimal cutting temperature compound (Fisher Scientific; Pittsburgh, PA, USA), and stored at −80°C. Serial sections (8 µm thick) were cut and stained with hematoxylin and eosin (H&E) to evaluate the inflammatory cell infiltration and tissue damage. Immunofluorescence staining was performed to assess infiltrated neutrophils. Slides were rehydrated, blocked, and incubated for 1 h at room temperature with primary FITC-conjugated antibodies to mouse Ly6G, a specific neutrophil marker (FITC conjugate; LifeSpan BioSciences, Incorporated, Seattle, WA, USA). The specificity of staining was confirmed by using appropriate FITC-conjugated isotype controls or normal rabbit IgG followed by Alexa Fluor 594 goat anti-rabbit IgG. Images were captured using a fluorescence microscope (Eclipse E800; Nikon, Tokyo, Japan) and processed by Neurolucida (MBF Bioscience, Williston, VT, USA).
RT-PCR and NF-κB p65 nuclear-binding assay
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Germantown, MD, USA), and real-time RT-PCR was performed using an Applied Biosystems 7500 system (Foster City, CA, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the endogenous control, and fold increase was calculated according to ΔΔCT method. Nuclear lysates were obtained from human monocytes using a nuclear/cytosolic isolation kit (Active Motif, Carlsbad, CA, USA). Nuclear lysates were analyzed for DNA-binding levels of NF-κB p65 using TransAMNF-κB p65 (Active Motif) and performed according to the manufacturer’s protocol.
Statistical analysis
Statistical significance between groups was evaluated by ANOVA and the Tukey multiple comparison test using the InStat program (GraphPad Software, San Diego, CA, USA). Differences between groups were considered significant at the level of P ≤ 0.05.
RESULTS
Activation of TLR4 leads to phosphorylation of SGK1
PI3K-mediated activation of SGK1 upon various stimuli such as sodium, follicle-stimulating hormone, and glucocorticoid has been reported in different cells. Because the function of SGK1 in TLR signaling is undetermined, and PI3K-activated Akt has been demonstrated to dampen TLR4-mediated immune responses, we initially sought to examine if SGK1 is involved in TLR-initiated signaling in innate immune cells. We found that LPS stimulation resulted in the phosphorylation of SGK1 both at Ser422 and Thr256 in human monocytes (Fig. 1). To further determine if TLR4-mediated phosphorylation of SGK1 was dependent on the activity of PI3K, we used the specific PI3K inhibitor LY294002 to suppress PI3K activity and examined phosphorylation of SGK1 upon LPS stimulation. As shown in Fig. 1, inhibition of PI3K abolished TLR4-induced phosphorylation of SGK1 in LPS-stimulated cells. These results establish that SGK1 is phosphorylated upon TLR4 activation and that this phosphorylation is dependent on the activity of PI3K.
Figure 1.

TLR4 activation induces PI3K-dependent phosphorylation of SGK1 in human monocytes. A) Western blot of cell lysates of human monocytes pretreated with LY294002 and stimulated with LPS. Blots were probed with antibodies to phospho-SGK1 (p-SGK1) and GAPDH as a loading control. B) Densitometric quantification of the mean (sd) ratio of phospho-to-total SGK1. *P < 0.05.
Pharmacologic inhibition of SGK1 enhances the production of TLR-mediated proinflammatory cytokines
Recent studies have demonstrated that SGK1 plays a key role in the induction of pathogenic Th17, and polarization of CD4+, Th cell subsets (17, 18). However, whether SGK1 functions in innate immune cells and regulates TLR-mediated inflammatory responses remains to be determined. Because we have observed that TLR4 activation resulted in the phosphorylation of SGK1, we next determined the effects of SGK1 inhibition on TLR4-mediated inflammatory cytokine production by innate immune cells. Purified human monocytes were used to determine the production of TLR-mediated inflammatory cytokines in the presence and absence of the SGK1-specific inhibitor EMD638683. Because EMD638683 is a novel SGK1 inhibitor and we are the first to assess its effect on TLR-mediated inflammatory cytokine production, a serial titration of EMD638683 was used to select the optimum concentration for the regulation of inflammatory cytokine production. We found that SGK1 inhibition with EMD638683 at the concentration of 10 μM significantly enhanced the production of IL-12, TNF, and IL-6 in LPS-stimulated human monocytes at both protein (Fig. 2A–C) and message levels (Fig. 2D). To determine if 10 μM EMD638683 indeed suppressed activity of SGK1, phosphorylation of the SGK1 physiologic substrate, NDRG-1 (N-myc downstream regulated gene 1), was assayed. Phosphorylation of NDRG-1 was completely suppressed by 10 μM EMD638683 in human monocytes upon LPS stimulation (Supplemental Fig. S1A). Moreover, the anti-inflammatory function of SGK1 was also found in Pam3Cys- and flagellin-stimulated cells. As shown in Fig. 2E, F, inhibition of SGK1 with 10 μM EMD638683 significantly enhanced TLR2- and TLR5-mediated production of IL-12, TNF, and IL-6 in human monocytes. These results identified the negative regulatory effect of SGK1 on TLR-mediated inflammatory cytokine production in human monocytes.
Figure 2.
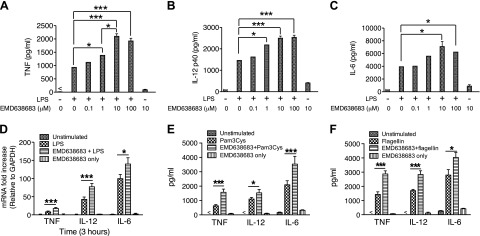
SGK1 inhibition enhances TLR-mediated inflammatory cytokine production in human monocytes. Human monocytes were pretreated with a series of concentrations of SGK1 inhibitor EMD638683 for 2 h and then stimulated with LPS, Pam3CSK4, or flagellin. After 24 h of stimulation, cell-free supernatants were collected, and LPS-stimulated production of (A) TNF, (B) IL-12, and (C) IL-6 was determined by ELISA. D) Message levels of TNF, IL-12, and IL-6 were detected by real-time PCR at 3 h after LPS stimulation. E and F) Production of TNF, IL-12, and IL-6 in human monocytes after SGK1 inhibition with EMD638683 and upon stimulation with Pam3CSK4 (E) and flagellin (F). Data represent the arithmetic means ± sd of 3 biologic replicates. *P < 0.05; ***P < 0.001.
siRNA knockdown or genetic deletion of SGK1 results in elevated proinflammatory cytokine production in LPS-stimulated innate immune cells
To exclude the possibility of nonspecific effects of the pharmacologic SGK1 inhibitor, we used prevalidated siRNA to knock down SGK1 in human monocytes. As shown in Fig. 3A, B, SGK1 silencing by specific siRNA resulted in a substantial decrease of total SGK1, as compared with the control. The suppressed activity of SGK1 was also confirmed by a robust decrease of phospho-NDRG-1 (Supplemental Fig. S1B). SGK1 deficiency led to a significant increase in TNF, IL-6, and IL-12 levels produced in LPS-stimulated human monocytes (Fig. 3C). These results corroborated the cytokine data obtained with the pharmacologic inhibitor EMD638683. Because neither EMD638683 nor siRNA can completely ablate SGK1 without effects on other kinases, we next used Cre-loxP-mediated sgk1 gene-deficient dendritic cells to determine the effect of SGK1 on TLR-mediated inflammatory cytokine production. We initially assessed the knockout efficiency of Cre-LoxP-mediated sgk1 in LPS-stimulated dendritic cells. As shown in Fig. 3D, dendritic cells from Cre-LoxP mice exhibited only trace amounts of SGK1, as compared with that of wild-type mice. Moreover, Cre-LoxP-mediated sgk1 deficiency resulted in a significant increase of TNF, IL-12, and IL-6 upon LPS stimulation (Fig. 3E–G). Taken together, these data demonstrate that SGK1 inhibition leads to the elevated proinflammatory cytokine production in LPS-stimulated innate immune cells.
Figure 3.
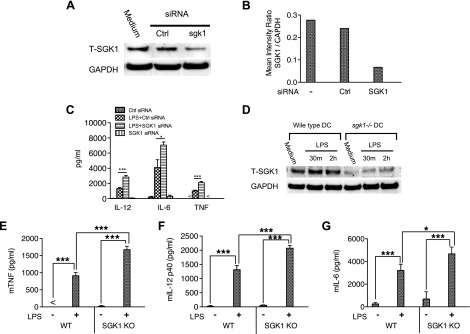
SGK1 deficiency leads to the elevated production of TNF, IL-12, and IL-6 in TLR4-stimulated innate immune cells. BMDCs with SGK1 deficiency were prepared by culturing bone marrow cells from cd11c-Cre-sgk1-fl/fl mice. Purified human monocytes were pretreated with nontarget or SGK1-specific siRNA and then stimulated with LPS. Whole-cell lysates and cell-free supernatants were collected to determine the transfection efficiency and cytokine levels, respectively. A) siRNA-mediated knockdown of SGK1 protein and total GAPDH levels were assessed by Western blot. Ctrl, control. B) The ratio of total SGK1-to-GAPDH was determined by densitometry. C) Production of TNF, IL-12, and IL-6 in LPS-stimulated cells following RNA silencing of SGK1. D) Western blot of whole-cell lysates from wild-type mice and Cre-LoxP-mediated SGK1-deficient mice probed for the total level of SGK1 and GAPDH. E) TNF, (F) IL-12, and (G) IL-6 levels in supernatants of LPS-stimulated (24 h) BMDCs harvested from wild-type (WT) and Cre-loxP-mediated SGK1 knockout (KO) mice. Data represent the arithmetic means ± sd of 3 biologic replicates. *P < 0.05; ***P < 0.001.
SGK1 inhibition resulted in the enhancement of NF-κB activity upon TLR4 activation
We have demonstrated that inhibition of SGK1 enhances production of proinflammatory cytokines in TLR-stimulated innate immune cells, and thus, we next investigated the signaling pathways responsible for this effect. NF-κB is a major transcription factor controlling the production of inflammatory cytokines, and thus, we first investigated the effect of SGK1 inhibition on NF-κB activation in LPS-stimulated cells. As shown in Fig. 4A–C, inhibition of SGK1 by EMD638683 led to a substantial increase of phosphorylation on NF-κB p65 in LPS-stimulated human monocytes. Moreover, SGK1 inhibition also enhanced phosphorylation of IκBα and its kinase, IKK, which control the nuclear migration of NF-κB (Fig. 4A). To exclude the possible nonspecific influence of EMD638683, specific siRNA was used to knock down SGK1 (Fig. 4D), and the effects on the NF-κB signaling were examined. As shown in Fig. 4E–G, siRNA-mediated SGK1 deficiency resulted in the increased phosphorylation of IKK, IκBα, and NF-κB in LPS-stimulated cells. These results confirm the phosphorylation data pathway obtained with the pharmacologic inhibitor. Because NF-κB activity has been demonstrated to control proinflammatory cytokine production, we next determined whether SGK1 inhibition could affect the LPS-mediated DNA-binding activity of NF-κB. As expected, compared with LPS stimulation alone, SGK1 inhibition significantly enhanced the DNA-binding activity of NF-κB p65 (Fig. 4H). These results indicate that SGK1 inhibition enhances phosphorylation (Ser536) and DNA-binding activity of NF-κB in LPS-stimulated human monocytes.
Figure 4.

Inhibition of SGK1 enhances the phosphorylation and DNA-binding activity of NF-κB in LPS-stimulated human monocytes. A) Western blot of human monocyte lysates pretreated with EMD638683 and stimulated with LPS. Blots were probed for phospho (p)- and total IKK, IκBα, and NF-κB P65 and total GAPDH. B and C) Densitometric quantification of the mean (SD) ratio of phospho-to-total proteins for IKK and NF-κB P65 and phospho- and total IκB-to-GAPDH, *P < 0.05. D and E) Western blot of lysates of human monocytes pretreated with nontarget or SGK1-specific siRNA and then stimulated with LPS. Blots were probed with antibodies to SGK1, phospho- and total IKK, IκBα, and NF-κB p65, and total GAPDH as indicated. F and G) Densitometric quantification of the mean (sd) ratio of phospho-to-total proteins for IKK and NF-κB P65 and phospho- and total IκB-to-GAPDH. Data are representative of 3 biologic replicates, *P < 0.05. H) DNA binding of NF-κB in nuclear lysates of monocytes stimulated with LPS for 2 h in the presence or absence of EMD638683. Data represent the arithmetic means ± SD of 3 biologic replicates, ***P < 0.001.
SGK1 inhibition enhances NF-κB transcription activity through modification of TAK1 phosphorylation in LPS-stimulated human monocytes
Because we observed that inhibition of SGK1 enhanced phosphorylation of IKK and IκBαβ and in turn augmented NF-κB activity, we next sought to determine how SGK1 regulates this process in human monocytes upon TLR4 activation. An established downstream target of Akt, S6 kinase, has been shown to negatively regulate TAK1 activity and in turn control NF-κB-mediated proinflammatory cytokine production in different cell types (29). Because SGK1 is similar to Akt, we therefore determined the effect of SGK1 on the phosphorylation sites of TAK1. As shown in Fig. 5A, B, inhibition of SGK1 by EMD638683 enhanced the phosphorylation of TAK1 at Thr180/Thr187 and Ser412 in human monocytes upon LPS stimulation. As confirmatory evidence, specific siRNA-mediated SGK1 knockdown (Fig. 4D) also resulted in an increase of TAK1 phosphorylation in LPS-stimulated human monocytes (Fig. 5C, D). Unlike the influence of SGK1 inhibition on phosphorylation of TAK1, siRNA-mediated TAK1 knockdown (Fig. 5E) had no discernible effect on the phosphorylation level of SGK1 as compared with cells stimulated with LPS alone (Fig. 5F, G), which suggests that TAK1 is the downstream substrate of SGK1. Moreover, we found that TAK1 silencing suppressed the phosphorylation of NF-κB p65 at Ser536 (Fig. 5F). To assess if TAK1 inhibition affects the capability of SGK1 inhibition to increase the activation of NF-κB, we next used a DNA-binding activity assay to analyze the effects of TAK1 silencing on the activation of NF-κB. As expected, siRNA-mediated TAK1 knockdown abrogated the ability of SGK1 inhibition to elevate the DNA-binding activity of NF-κB (Ser536) in LPS-stimulated cells (Fig. 5H). Collectively, these results demonstrate that SGK1 inhibition enhances phosphorylation of TAK1 that then controls activation of NF-κB in LPS-stimulated innate immune cells.
Figure 5.

SGK1 inhibition enhances TLR4-mediated proinflammatory cytokine production dependent on the activity of TAK1. A) Western blot of human monocytes pretreated with EMD638683 for 2 h and then stimulated with LPS. Blots were probed with antibodies to phospho (p)- and total TAK1 and GAPDH as a loading control. B) Densitometric quantification of the mean (sd) ratio of phospho-to-total TAK1 upon LPS stimulation in the presence and absence of EMD638683. C) Western blots of lysates of human monocytes pretreated with nontarget siRNA or specific siRNA targeting SGK1 (as in Fig. 4D), then stimulated with LPS. Blots were probed with antibodies to SGK1, phospho- and total TAK1, and GAPDH as indicated. Data are representative of 3 biologic replicates, and densitometric quantification of the mean (sd) ratio of phospho-to-total TAK1 was performed (D). E) siRNA-mediated knockdown of TAK1 protein and total GAPDH levels were assessed by Western blots. F and G) The effects of siRNA-mediated TAK1 knockdown on the phosphorylation of SGK1 and NF-κB were monitored by Western blots. H) siRNA-mediated TAK1 knockdown and its effects on the DNA binding of NF-κB in nuclear lysates of LPS-stimulated monocytes in the presence and absence of EMD638683. Data represent the arithmetic means ± sd of 3 biologic replicates. *P < 0.05; ***P < 0.001.
SGK1 inhibition-mediated increased activity of TAK1 and NF-κB leads to elevated production of inflammatory cytokines
To determine whether SGK1 inhibition-enhanced activity of TAK1 and NF-κB was responsible for the increased production of proinflammatory cytokines, we initially employed specific siRNA to examine the effect of TAK1 inhibition on the production of LPS-induced proinflammatory cytokines in human monocytes. As shown in Fig. 6A–C, siRNA-mediated TAK1 inhibition (Fig. 5E) significantly reduced the production of TNF, IL-6, and IL-12 in LPS-stimulated monocytes. Furthermore, siRNA-mediated TAK1 inhibition abrogated the ability of EMD638683 to enhance the production of proinflammatory cytokines in LPS-stimulated monocytes (Fig. 6A–C). Because we have demonstrated that SGK1-mediated TAK1 phosphorylation modifies the activity of NF-κB p65 and proinflammatory cytokine production, we next determined if SGK1 inhibition enhanced NF-κB p65 phosphorylation and controlled the secretion of proinflammatory cytokines. Using an NF-κB inhibitory peptide, we found that inhibition of NF-κB significantly reversed the ability of EMD638683 to increase the production of TNF, IL-12, and IL-6 in LPS-stimulated human monocytes, as compared with cells treated with control peptide (Fig. 6D–F). These results establish that inhibition of SGK1 enhances the activity of TAK1 and NF-κB and thus augments the secretion of LPS-mediated inflammatory cytokines.
Figure 6.
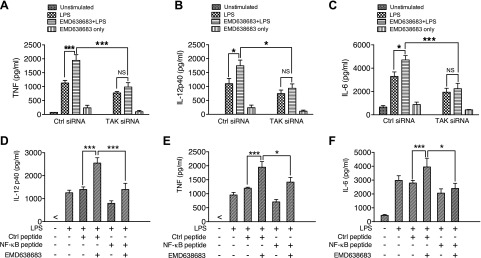
SGK1 inhibition-enhanced TAK1 phosphorylation and elevated activity of NF-κB heighten the secretion of inflammatory cytokines by LPS-stimulated human monocytes. A) TNF, (B) IL-12, and (C) IL-6 levels in the supernatants of monocytes pretreated with nontargeted or TAK1-specific siRNA and stimulated with LPS. D) IL-12, (E) TNF, and (F) IL-6 levels in LPS-stimulated monocytes pretreated with a control peptide or an NF-κB inhibitory peptide. Data represent the arithmetic means ± sd of 3 biologic replicates. *P < 0.05; ***P < 0.001.
Inhibition of SGK1 aggravates inflammatory responses and neutrophil infiltration during endotoxic shock
Proinflammatory cytokines, especially TNF, play a central role in the initiation and development of LPS-induced endotoxic shock. Because we found that inhibition of SGK1 enhanced production of TNF, IL-6, and IL-12 in cultured cells, we next investigated the extent to which inhibition of SGK1 would enhance the production of these proinflammatory cytokines on a systemic level and thus exacerbate the tissue damage in an LPS-induced endotoxic shock model. As shown in Fig. 7A, mice pretreated with SGK1 inhibitor (EMD638683) exhibited higher levels of TNF, IL-6, and IL-12 in serum after 3-h stimulation with LPS, as compared with mice treated with LPS only. After the mice were stimulated with LPS for 24 h, both control mice and SGK1 inhibitor-treated mice all exhibited an enormous decrease of proinflammatory cytokine production (Fig. 7B). Moreover, the production of TNF and IL-12 did not display a significant difference between the mice with or without the pretreatment of EMD638683 at 24 h postinjection of LPS (Fig. 7B), suggesting that SGK1 might be involved only in the early stage of LPS-induced inflammatory responses. Because overwhelming neutrophil accumulation has been demonstrated to significantly contribute to the development of endotoxin-mediated multiple organ failure (30), we next examined if SGK1 inhibition affects organ damage and neutrophil infiltration upon LPS challenge. To this end, we collected mouse livers and lungs after 6- and 24-h challenge with LPS in the presence and absence of EMD638683. As shown in Fig. 7C, D, after 24-h LPS challenge, mice pretreated with EMD638683 displayed severe organ injury, including widespread disrupted tissue architecture, an extensive area of cell cytoplasmic vacuolization (Fig. 7C), and broad areas of inflammatory infiltration in lung (Fig. 7D). On the other hand, mild tissue injury and hemorrhage were found in the liver and lung of the mice upon LPS treatment only (Fig. 7C, D). Consistent with this result, SGK1-inhibited mice exhibited higher neutrophil infiltration in liver and lung tissues compared to the mice treated with LPS alone (Fig. 7C–H). These results suggest that inhibition of SGK1 systemically enhances the production of proinflammatory cytokine levels and exacerbates the severity of septic shock, which indicates that SGK1 could be a novel target for intervention in the control of inflammatory diseases.
Figure 7.
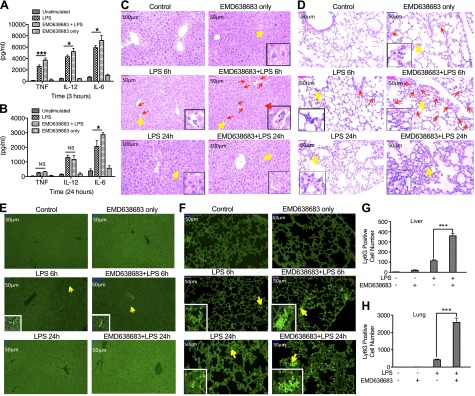
Inhibition of SGK1 aggravates inflammatory responses and enhances neutrophil infiltration in the murine model of endotoxic shock. TNF, IL-12, and IL-6 levels at 3 h (A) or 24 h (B) postinjection of LPS in C57BL/6 mice with or without EMD638683 pretreatment. H&E staining of serial sections of liver (C) and lung (D) from the mice injected with PBS (control), EMD638683, LPS, or LPS plus EMD638683 showing the infiltration of inflammatory cells (6 h; area with red arrows) and the severity of tissue damage (24 h) (×20 magnification; insets amplify the area indicated by yellow arrows). E and F) Polymorphonuclear neutrophil infiltration was assessed by staining tissue sections of liver and lung with FITC-conjugated anti-Ly6G (magnification, ×20). (A), (B), and (C)–(F) are separate, and each group has a total of 7 mice for analysis. G and H) Levels of infiltrated neutrophils presented by the average number of Ly6G-positive cells in 5 different views. Data represent the arithmetic means ± sd of 3 biologic replicates. *P < 0.05; ***P < 0.001.
DISCUSSION
TLR-induced proinflammatory cytokines play a central role in the development of endotoxic shock (1). Our and other previous studies have demonstrated that the PI3K-AKT pathway is involved in the regulation of inflammatory cytokine production in immune cells (9, 11, 31). Because diverse substrates are involved in PI3K-initiated signaling in different contexts (32, 33), a major aim of the current study was to identify novel downstream signaling components within the PI3K pathway that are capable of regulating inflammatory cytokine production, and we have now demonstrated that SGK1 is a critical negative regulator of inflammation (Fig. 8). Genetic deletion or siRNA-mediated knockdown of SGK1 resulted in the enhanced production of inflammatory cytokines in LPS-stimulated innate immune cells or in a murine endotoxin model. Analysis of the downstream targets of SGK1 revealed that the phosphorylation of TAK1 and the activity of prototypical inflammatory signaling pathway NF-κB were increased in SGK1 inhibitor- or siRNA-treated monocytes. TAK1 silencing attenuated activity of NF-κB and abrogated the ability of SGK1 inhibition to elevate production of inflammatory cytokines. Moreover, inhibition of SGK1 resulted in the enhancement of TNF, IL-6, and IL-12, higher recruitment of neutrophils, and aggravation of tissue damage in an LPS-induced murine endotoxin model. These findings establish, for the first time, a negative regulatory role of SGK1 in TLR4-mediated proinflammatory responses in vitro and in vivo, which positions SGK1 as a novel anti-inflammatory regulator in innate immune responses.
Figure 8.

Model of SGK1 regulation of proinflammatory cytokine production in LPS-stimulated monocytes. TLR4-mediated activation of PI3K phosphorylation activates SGK1 in a similar way to phosphorylation of Akt, converting phosphatidylinositol (PIP)2 to PIP3 that will recruit PDK1 and PDK2 and then fully phosphorylate SGK1. Inhibition of SGK1 enhances phosphorylation of TAK1 upon LPS stimulation and in turn elevates phosphorylation of IKK, IκB, and NF-κB, which cumulatively augment proinflammatory cytokine production in LPS-stimulated monocytes.
TLR recognition of MAMPs initiates the activation of several serine/threonine kinases such as IKK, MAPK family, and PI3K. These kinases relay information to a plethora of downstream signaling molecules, which then control the transcription and expression of target genes. Although IκB-mediated NF-κB and MAPK-mediated activator protein 1 (AP-1) signaling has been demonstrated as key in the induction of inflammation, PI3K-mediated activation of Akt has been shown to dampen TLR-induced inflammatory responses (11, 34, 35). Inhibition or gene deficiency of Akt caused increased production of inflammatory cytokines and produced a hyperinflammatory phenotype similar to that observed with PI3K inhibition (36, 37). Stimulation of innate immune cells with LPS has been demonstrated to induce phosphorylation of Akt both on Thr308 and Ser473, and blockade of PI3K reduced this site-specific phosphorylation (34). Similar to Akt, previous studies reported that SGK1 was activated by PI3K through hierarchical phosphorylation on Ser422 and Thr256 upon different stimulations (14, 15). Our current results confirm this conclusion in LPS-stimulated innate immune cells. As a serine/threonine kinase, activity of SGK1 would be suppressed by the expression of SGK1 and its phosphorylation. In our study, we found that siRNA and a Cre-loxP system reduce the expression of SGK1, and the chemical inhibitor EMD638683 robustly decreases the phosphorylation of NDRG-1, a direct substrate of SGK1, suggesting substantial inhibition of SGK1 activity. We also demonstrated that pharmacologic inhibition or siRNA-mediated SGK1 inhibition leads to an elevated inflammatory response upon LPS stimulation. By using Cre-loxP-mediated gene-deficient dendritic cells, we confirmed the effects of SGK1 on the production of inflammatory cytokine production in response to TLR stimulation. Interestingly, we noticed that cells treated with EMD638683 only exhibited slightly increased production of proinflammatory cytokines in the absence of LPS stimulation. Because cells were cultured in complete medium, elevated cytokine production may result from the presence of the amino acids and serum, which can activate signaling pathways such as PI3K-mammalian target of rapamycin complex (mTORC)1 (28, 38). Hence, inhibition of SGK1 can enhance the background cytokine level through these pathways. Taken together, our present findings underscore the regulatory function of SGK1 and its ability to mediate PI3K signaling as a fundamental process in controlling the LPS-mediated inflammatory response.
Although SGK1 is sequentially phosphorylated by PDK1 and mTORC2 in a similar manner to Akt, and the 2 kinases share similar targets (15), some stress stimuli activate SGK1 but are unable to phosphorylate AKT, suggesting separate rather than redundant roles for these 2 protein kinases (39). Recent studies have also demonstrated that SGK1 was involved in the induction and differentiation of Th cells. SGK1 gene deficiency resulted in a selective defect of pathogenic Th17 through suppressing expression of the receptor for IL-23 (20). Moreover, SGK1 has been reported to promote Th2 differentiation by preventing ubiquitination and degradation of the transcription factor JunB. In contrast, loss of SGK1 in T cells led to higher levels of IFN-γ and promoted Th1 polarization (18). These studies suggest that SGK1 not only functions in metabolism but is also essential for the homeostasis of immune responses. Novel functions of SGK1 in adaptive immunity also highlight a requirement for investigating the possible function of SGK1 in innate immunity. Our results show, for the first time, that SGK1 is activated upon TLR engagement and negatively regulates LPS-induced innate immune responses. In conjunction with the role of SGK1 in adaptive immunity, SGK1 could thus be a potent immunomodulator that can be exploited in order to control multiple inflammatory diseases.
We have shown that inhibition of SGK1 enhanced the activity of NF-κB and increased proinflammatory cytokine production in innate immune cells. Similarly, a study by Heikamp et al. (18) demonstrated the ability of SGK1 to promote a Th2-type immune response and concurrently suppress Th1 cytokine production, and sgk1 deficiency substantially enhanced production of the Th1 cytokine, IFN-γ. Our findings are consistent with these results in that they highlight the anti-inflammatory properties of SGK1. Interestingly, SGK1 has also been reported to be regulated by NF-κB upon LPS stimulation, suggesting the existence of a possible negative feedback loop (40). Apart from the effect of SGK1 on inflammatory cytokine production, recent reports show that inhibition of SGK1 counteracts antiapoptotic signals in neutrophils, and thus, SGK1 is involved in the resolution of inflammation at late stages of infection, which indicates an extensive role of SGK1 in inflammatory immune responses (41). SGK1 is thus emerging as a potent regulator of the differentiation of CD4+ T-cell subsets, the apoptosis of neutrophils, and inflammatory cytokine production by innate immune cells.
In our in vivo study, we found that the plasma cytokines drastically subside, and inhibition of SGK1 exerts no significant effects on the production of TNF and IL-12 at 24 h after administration of LPS. Although LPS or other MAMP-activated proinflammatory pathways are increasingly characterized, more and more anti-inflammatory pathways are also identified. Anti-inflammatory pathways can be crudely divided into cell-intrinsic and cell-extrinsic mechanisms. Examples of cell-intrinsic pathways mainly include TRAILR, MyD88s, sterile-α and armadillo motif-containing protein, A20, TNF-α-induced protein 3-binding inhibitor of NF-κB, TRAF4, β-arrestin, SOCS1, SH-2 containing protein tyrosine phosphatase-2, and some endogenous protein kinases such as IRAK-M, PI3K, Akt, MAPK phosphatase 1 (MKP-1), and newly synthesized IκBα (42, 43). Our recent studies have added mTORC1, p70S6K1, and Janus kinase-3 to this list (28, 31). Inhibition or gene deficiency of each of these factors in mice leads to complex inflammatory diseases due to failure to control the intensity of inflammatory mediators such as TNF. Cell-extrinsic mechanisms include anti-inflammatory cytokines such as IL-10, TGF-β, and IL-1 receptor a and soluble decoy receptors such as soluble TLRs and TNF receptor (42, 43). Additionally, several neuroimmune feedbacks, including the hypothalamo-pituitary-adrenal axis and the acetylcholine-mediated cholinergic anti-inflammatory reflex, are also involved in endogenously constraining the intensity of inflammation in response to LPS and other TLR agonists (44–47). Our previous study has demonstrated that the acetylcholine-activated PI3K-AKT pathway will control the production of LPS-induced proinflammatory cytokines through modifying activity of the downstream serine/threonine kinase: glycogen synthase kinase 3-β (48). Our in vivo results showed that inhibition of SGK1 significantly enhances production of TNF, IL-12, and IL-6 at 3 h rather than 24 h. Considering that inhibition of SGK1 enhances phosphorylation of NF-κB at a very early stage, it is highly possible that SGK1 inhibition exerts its effect mainly on the early stage of the LPS-induced endotoxic process.
TAK1 has been shown to play an essential role in innate and adaptive immune responses through controlling the activity of NF-κB and MAPK pathways (22, 49). Many studies have demonstrated that TAK1 is a positive regulator of innate immune signaling and apoptosis. Inhibition of TAK1 was reported to suppress TLR-mediated signaling and in turn reduce proinflammatory cytokine production in different cells (49, 50). Systemic administration of selective chemical inhibitors or specific siRNA targeting on TAK1 was reported to significantly suppress inflammatory responses in different animal models (51, 52). Conversely, a recent study reported that TAK1 negatively regulates cell development and activation of proinflammatory signaling pathways in neutrophils, which suggests that the anti-inflammatory property of TAK1 is cell-type specific (53). Our findings in this study confirmed the proinflammatory role of TAK1 in LPS-stimulated monocytes. We found that SGK1 inhibition enhanced activation of TAK1, which was required for the LPS-mediated proinflammatory cytokine production in human monocytes.
TAK1-regulated NF-κB and MAPK/AP-1 pathways play key roles in the immune system. Tight regulation of TAK1 following stimuli-dependent activation is important for immune homeostasis. Although it was demonstrated that activation of TAK1 was triggered by TRAF6-mediated Lys63-linked polyubiquitination reactions and further fully activated through TAB1- and TAB2-mediated ubiquitination and autophosphorylation (54), the regulation of TAK1 phosphorylation and its significance to the TAK1 activation are less understood. Our results demonstrated that SGK1 is one of the regulators of TAK1 in response to TLR-mediated signaling. Inhibition of SGK1 promotes the phosphorylation of TAK1 in LPS-stimulated innate immune cells. Moreover, the increased phosphorylation of TAK1 promotes the activity of downstream NF-κB. Although we found that inhibition of SGK1 enhances the phosphorylation of TAK1 at different residues, it remains to be determined if TAK1 and SGK1 physically interact. Because SGK1 deficiency led to a decrease of MKP-1 expression in TLR-stimulated dendritic cells (Supplemental Fig. S2), it is possible that enhancement of TAK1 phosphorylation may result from an SGK1-mediated decrease of MKP-1 phosphatase activity, although further study is required to resolve the matter.
In summary, we have identified SGK1 as a negative regulator of TLR-mediated immune responses that suppresses the production of proinflammatory cytokines, and we have characterized the molecular mechanism by which this occurs. Inhibition of SGK1 increases the inflammatory response via enhancing the activity of TAK1, which subsequently augments the activity of NF-κB in LPS-stimulated innate immune cells. Considering the recently reported role of SGK1 in the polarization of Th1 responses, our findings identifying the influence of SGK1 on innate immune response highlight the possibility to develop exogenous SGK1 to be a possible novel target for intervention in the control of inflammatory diseases.
Supplementary Material
Acknowledgments
This research was supported by Grants DE023633 (to H.W.), DE017921 and DE011111 (to R.J.L.), and DE017680 (to D.A.S.) from the U.S. National Institutes of Health, National Institute of Dental and Craniofacial Research.
Glossary
- A20
TNF-α-induced protein 3
- Akt
Ak thymic lymphoma/PKB
- AP-1
activator protein 1
- BMDC
bone marrow-derived dendritic cell
- FCS
fetal calf serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GFP
green fluorescent protein
- H&E
hematoxylin and eosin
- IRAK
IL-1 receptor-associated kinase
- IRF
IFN-regulatory factor
- MAMP
microbe-associated molecular pattern
- MKP-1
MAPK phosphatase 1
- mTORC
mammalian target of rapamycin complex
- MyD88
myeloid differentiation primary response gene 88
- MyD88s
myeloid differentiation 88 short
- NDRG-1
N-myc downstream regulated gene 1
- PDK
3-phosphoinositide-dependent protein kinase
- SGK
serum- and glucocorticoid-inducible kinase
- siRNA
small interfering RNA
- SOCS1
suppressor of cytokine signaling 1
- TAB
TGF-β-activated kinase 1-binding protein
- TAK1
TGF-β-activated kinase 1
- Th
T helper
- TRAF
TNF receptor-associated factor
- TRAILR
TNF-related apoptosis-inducing ligand receptor 1
- TRIF
Toll-IL-1 receptor-domain-containing adapter-inducing IFN-β
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Schulte W., Bernhagen J., Bucala R. (2013) Cytokines in sepsis: potent immunoregulators and potential therapeutic targets—an updated view. Mediators Inflamm. 2013, 165974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kellum J. A., Kong L., Fink M. P., Weissfeld L. A., Yealy D. M., Pinsky M. R., Fine J., Krichevsky A., Delude R. L., Angus D. C.; GenIMS Investigators (2007) Understanding the inflammatory cytokine response in pneumonia and sepsis: results of the Genetic and Inflammatory Markers of Sepsis (GenIMS) Study. Arch. Intern. Med. 167, 1655–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawai T., Akira S. (2007) TLR signaling. Semin. Immunol. 19, 24–32 [DOI] [PubMed] [Google Scholar]
- 4.Lee M. S., Kim Y. J. (2007) Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu. Rev. Biochem. 76, 447–480 [DOI] [PubMed] [Google Scholar]
- 5.O’Neill L. A. (2008) ‘Fine tuning’ TLR signaling. Nat. Immunol. 9, 459–461 [DOI] [PubMed] [Google Scholar]
- 6.Coll R. C., O’Neill L. A. (2010) New insights into the regulation of signalling by toll-like receptors and nod-like receptors. J. Innate Immun. 2, 406–421 [DOI] [PubMed] [Google Scholar]
- 7.Li L. (2004) Regulation of innate immunity signaling and its connection with human diseases. Curr. Drug Targets Inflamm. Allergy 3, 81–86 [DOI] [PubMed] [Google Scholar]
- 8.Fukao T., Tanabe M., Terauchi Y., Ota T., Matsuda S., Asano T., Kadowaki T., Takeuchi T., Koyasu S. (2002) PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat. Immunol. 3, 875–881 [DOI] [PubMed] [Google Scholar]
- 9.Martin M., Rehani K., Jope R. S., Michalek S. M. (2005) Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 6, 777–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukao T., Yamada T., Tanabe M., Terauchi Y., Ota T., Takayama T., Asano T., Takeuchi T., Kadowaki T., Hata Ji J., Koyasu S. (2002) Selective loss of gastrointestinal mast cells and impaired immunity in PI3K-deficient mice. Nat. Immunol. 3, 295–304 [DOI] [PubMed] [Google Scholar]
- 11.Martin M., Schifferle R. E., Cuesta N., Vogel S. N., Katz J., Michalek S. M. (2003) Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. J. Immunol. 171, 717–725 [DOI] [PubMed] [Google Scholar]
- 12.Zhang W. J., Wei H., Hagen T., Frei B. (2007) Alpha-lipoic acid attenuates LPS-induced inflammatory responses by activating the phosphoinositide 3-kinase/Akt signaling pathway. Proc. Natl. Acad. Sci. USA 104, 4077–4082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schabbauer G., Tencati M., Pedersen B., Pawlinski R., Mackman N. (2004) PI3K-Akt pathway suppresses coagulation and inflammation in endotoxemic mice. Arterioscler. Thromb. Vasc. Biol. 24, 1963–1969 [DOI] [PubMed] [Google Scholar]
- 14.Tessier M., Woodgett J. R. (2006) Serum and glucocorticoid-regulated protein kinases: variations on a theme. J. Cell. Biochem. 98, 1391–1407 [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi T., Cohen P. (1999) Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem. J. 339, 319–328 [PMC free article] [PubMed] [Google Scholar]
- 16.Lang F., Cohen P. (2001) Regulation and physiological roles of serum- and glucocorticoid-induced protein kinase isoforms. Sci. STKE 2001, re17. [DOI] [PubMed] [Google Scholar]
- 17.Lu X., Li M., Zhou L., Jiang H., Wang H., Chen J. (2014) Serum- and glucocorticoid-inducible kinase SGK1 as a deleterious mediator of IgA nephropathy. [E-pub ahead of print] Nephrology (Carlton) 19, 307–317 [DOI] [PubMed] [Google Scholar]
- 18.Heikamp E. B., Patel C. H., Collins S., Waickman A., Oh M. H., Sun I. H., Illei P., Sharma A., Naray-Fejes-Toth A., Fejes-Toth G., Misra-Sen J., Horton M. R., Powell J. D. (2014) The AGC kinase SGK1 regulates TH1 and TH2 differentiation downstream of the mTORC2 complex. Nat. Immunol. 15, 457–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kleinewietfeld M., Manzel A., Titze J., Kvakan H., Yosef N., Linker R. A., Muller D. N., Hafler D. A. (2013) Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496, 518–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu C., Yosef N., Thalhamer T., Zhu C., Xiao S., Kishi Y., Regev A., Kuchroo V. K. (2013) Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496, 513–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee J., Mira-Arbibe L., Ulevitch R. J. (2000) TAK1 regulates multiple protein kinase cascades activated by bacterial lipopolysaccharide. J. Leukoc. Biol. 68, 909–915 [PubMed] [Google Scholar]
- 22.Shim J. H., Xiao C., Paschal A. E., Bailey S. T., Rao P., Hayden M. S., Lee K. Y., Bussey C., Steckel M., Tanaka N., Yamada G., Akira S., Matsumoto K., Ghosh S. (2005) TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 19, 2668–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ajibade A. A., Wang H. Y., Wang R. F. (2013) Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 34, 307–316 [DOI] [PubMed] [Google Scholar]
- 24.Fan Y., Yu Y., Shi Y., Sun W., Xie M., Ge N., Mao R., Chang A., Xu G., Schneider M. D., Zhang H., Fu S., Qin J., Yang J. (2010) Lysine 63-linked polyubiquitination of TAK1 at lysine 158 is required for tumor necrosis factor alpha- and interleukin-1beta-induced IKK/NF-kappaB and JNK/AP-1 activation. J. Biol. Chem. 285, 5347–5360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng H., Li Q., Chen R., Zhang J., Ran Y., He X., Li S., Shu H. B. (2013) The dual-specificity phosphatase DUSP14 negatively regulates tumor necrosis factor- and interleukin-1-induced nuclear factor-κB activation by dephosphorylating the protein kinase TAK1. J. Biol. Chem. 288, 819–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ackermann T. F., Boini K. M., Beier N., Scholz W., Fuchss T., Lang F. (2011) EMD638683, a novel SGK inhibitor with antihypertensive potency. Cell. Physiol. Biochem. 28, 137–146 [DOI] [PubMed] [Google Scholar]
- 27.Lutz M. B., Kukutsch N., Ogilvie A. L., Rössner S., Koch F., Romani N., Schuler G. (1999) An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 223, 77–92 [DOI] [PubMed] [Google Scholar]
- 28.Wang H., Brown J., Gu Z., Garcia C. A., Liang R., Alard P., Beurel E., Jope R. S., Greenway T., Martin M. (2011) Convergence of the mammalian target of rapamycin complex 1- and glycogen synthase kinase 3-β-signaling pathways regulates the innate inflammatory response. J. Immunol. 186, 5217–5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S. Y., Baik K. H., Baek K. H., Chah K. H., Kim K. A., Moon G., Jung E., Kim S. T., Shim J. H., Greenblatt M. B., Chun E., Lee K. Y. (2014) S6K1 negatively regulates TAK1 activity in the toll-like receptor signaling pathway. Mol. Cell. Biol. 34, 510–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagner J. G., Roth R. A. (1999) Neutrophil migration during endotoxemia. J. Leukoc. Biol. 66, 10–24 [DOI] [PubMed] [Google Scholar]
- 31.Wang H., Brown J., Gao S., Liang S., Jotwani R., Zhou H., Suttles J., Scott D. A., Lamont R. J. (2013) The role of JAK-3 in regulating TLR-mediated inflammatory cytokine production in innate immune cells. J. Immunol. 191, 1164–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blunt M. D., Ward S. G. (2012) Targeting PI3K isoforms and SHIP in the immune system: new therapeutics for inflammation and leukemia. Curr. Opin. Pharmacol. 12, 444–451 [DOI] [PubMed] [Google Scholar]
- 33.Troutman T. D., Bazan J. F., Pasare C. (2012) Toll-like receptors, signaling adapters and regulation of the pro-inflammatory response by PI3K. Cell Cycle 11, 3559–3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guha M., Mackman N. (2002) The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J. Biol. Chem. 277, 32124–32132 [DOI] [PubMed] [Google Scholar]
- 35.Luyendyk J. P., Schabbauer G. A., Tencati M., Holscher T., Pawlinski R., Mackman N. (2008) Genetic analysis of the role of the PI3K-Akt pathway in lipopolysaccharide-induced cytokine and tissue factor gene expression in monocytes/macrophages. J. Immunol. 180, 4218–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wrann C. D., Tabriz N. A., Barkhausen T., Klos A., van Griensven M., Pape H. C., Kendoff D. O., Guo R., Ward P. A., Krettek C., Riedemann N. C. (2007) The phosphatidylinositol 3-kinase signaling pathway exerts protective effects during sepsis by controlling C5a-mediated activation of innate immune functions. J. Immunol. 178, 5940–5948 [DOI] [PubMed] [Google Scholar]
- 37.Yu Y., Nagai S., Wu H., Neish A. S., Koyasu S., Gewirtz A. T. (2006) TLR5-mediated phosphoinositide 3-kinase activation negatively regulates flagellin-induced proinflammatory gene expression. J. Immunol. 176, 6194–6201 [DOI] [PubMed] [Google Scholar]
- 38.Bar-Peled L., Sabatini D. M. (2014) Regulation of mTORC1 by amino acids. Trends Cell Biol. 24, 400–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waldegger S., Klingel K., Barth P., Sauter M., Rfer M. L., Kandolf R., Lang F. (1999) h-sgk serine-threonine protein kinase gene as transcriptional target of transforming growth factor beta in human intestine. Gastroenterology 116, 1081–1088 [DOI] [PubMed] [Google Scholar]
- 40.De Seigneux S., Leroy V., Ghzili H., Rousselot M., Nielsen S., Rossier B. C., Martin P. Y., Féraille E. (2008) NF-kappaB inhibits sodium transport via down-regulation of SGK1 in renal collecting duct principal cells. J. Biol. Chem. 283, 25671–25681 [DOI] [PubMed] [Google Scholar]
- 41.Burgon J., Robertson A. L., Sadiku P., Wang X., Hooper-Greenhill E., Prince L. R., Walker P., Hoggett E. E., Ward J. R., Farrow S. N., Zuercher W. J., Jeffrey P., Savage C. O., Ingham P. W., Hurlstone A. F., Whyte M. K., Renshaw S. A. (2014) Serum and glucocorticoid-regulated kinase 1 regulates neutrophil clearance during inflammation resolution. J. Immunol. 192, 1796–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown J., Wang H., Hajishengallis G. N., Martin M. (2011) TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J. Dent. Res. 90, 417–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murray P. J., Smale S. T. (2012) Restraint of inflammatory signaling by interdependent strata of negative regulatory pathways. Nat. Immunol. 13, 916–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bethin K. E., Vogt S. K., Muglia L. J. (2000) Interleukin-6 is an essential, corticotropin-releasing hormone-independent stimulator of the adrenal axis during immune system activation. Proc. Natl. Acad. Sci. USA 97, 9317–9322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borovikova L. V., Ivanova S., Zhang M., Yang H., Botchkina G. I., Watkins L. R., Wang H., Abumrad N., Eaton J. W., Tracey K. J. (2000) Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405, 458–462 [DOI] [PubMed] [Google Scholar]
- 46.Hosoi T., Okuma Y., Matsuda T., Nomura Y. (2005) Novel pathway for LPS-induced afferent vagus nerve activation: possible role of nodose ganglion. Auton. Neurosci. 120, 104–107 [DOI] [PubMed] [Google Scholar]
- 47.Perlstein R. S., Whitnall M. H., Abrams J. S., Mougey E. H., Neta R. (1993) Synergistic roles of interleukin-6, interleukin-1, and tumor necrosis factor in the adrenocorticotropin response to bacterial lipopolysaccharide in vivo. Endocrinology 132, 946–952 [DOI] [PubMed] [Google Scholar]
- 48.Rehani K., Scott D. A., Renaud D., Hamza H., Williams L. R., Wang H., Martin M. (2008) Cotinine-induced convergence of the cholinergic and PI3 kinase-dependent anti-inflammatory pathways in innate immune cells. Biochim. Biophys. Acta 1783, 375–382 [DOI] [PubMed] [Google Scholar]
- 49.Sato S., Sanjo H., Takeda K., Ninomiya-Tsuji J., Yamamoto M., Kawai T., Matsumoto K., Takeuchi O., Akira S. (2005) Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 6, 1087–1095 [DOI] [PubMed] [Google Scholar]
- 50.Gu M., Ouyang C., Lin W., Zhang T., Cao X., Xia Z., Wang X. (2014) Phosphatase holoenzyme PP1/GADD34 negatively regulates TLR response by inhibiting TAK1 serine 412 phosphorylation. J. Immunol. 192, 2846–2856 [DOI] [PubMed] [Google Scholar]
- 51.Courties G., Seiffart V., Presumey J., Escriou V., Scherman D., Zwerina J., Ruiz G., Zietara N., Jablonska J., Weiss S., Hoffmann A., Jorgensen C., Apparailly F., Gross G. (2010) In vivo RNAi-mediated silencing of TAK1 decreases inflammatory Th1 and Th17 cells through targeting of myeloid cells. Blood 116, 3505–3516 [DOI] [PubMed] [Google Scholar]
- 52.Ninomiya-Tsuji J., Kajino T., Ono K., Ohtomo T., Matsumoto M., Shiina M., Mihara M., Tsuchiya M., Matsumoto K. (2003) A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J. Biol. Chem. 278, 18485–18490 [DOI] [PubMed] [Google Scholar]
- 53.Ajibade A. A., Wang Q., Cui J., Zou J., Xia X., Wang M., Tong Y., Hui W., Liu D., Su B., Wang H. Y., Wang R. F. (2012) TAK1 negatively regulates NF-κB and p38 MAP kinase activation in Gr-1+CD11b+ neutrophils. Immunity 36, 43–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakurai H. (2012) Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol. Sci. 33, 522–530 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.