

Abstract
The angiopoietin (Ang) ligands are potential therapeutic targets for lymphatic related diseases, which include lymphedema and cancer. Ang-1 and Ang-2 functions are established, but those of Ang-4 are poorly understood. We used intravital fluorescence microscopy to characterize Ang-4 actions on T241 murine fibrosarcoma-associated vessels in mice. The diameters of lymphatic vessels draining Ang-4- or VEGF-C (positive control)-expressing tumors increased to 123 and 135 μm, respectively, and parental, mock-transduced (negative controls) and tumors expressing Ang-1 or Ang-2 remained at baseline (∼60 μm). Ang-4 decreased human dermal lymphatic endothelial cell (LEC) monolayer permeability by 27% while increasing human dermal blood endothelial cell (BEC) monolayer permeability by 200%. In vivo, Ang-4 stimulated a 4.5-fold increase in tumor-associated blood vessel permeability compared with control when measured using intravital quantitative multiphoton microscopy. Ang-4 activated receptor signaling in both LECs and BECs, evidenced by tyrosine kinase with Ig and endothelial growth factor homology domains-2 (TIE2) receptor, protein kinase B, and Erk1,2 phosphorylation detectable by immunoblotting. These data suggest that Ang-4 actions are mediated through cell-type-specific networks and that lymphatic vessel dilation occurs secondarily to increased vascular leakage. Ang-4 also promoted survival of LECs. Thus, blocking Ang-4 may prune the draining lymphatic vasculature and decrease interstitial fluid pressure (IFP) by reducing vascular permeability.—Kesler, C. T., Pereira, E. R., Cui, C. H., Nelson, G. M., Masuck, D. J., Baish, J. W., Padera, T. P. Angiopoietin-4 increases permeability of blood vessels and promotes lymphatic dilation.
Keywords: endothelial, TIE2, lymphangiography, 2-photon microscopy
Lymphatic vessels preserve homeostatic fluid balance by reabsorbing fluid that has leaked into interstitial spaces from surrounding blood vessels. They also maintain immune function by serving as conduits for antigen and antigen-presenting cells to reach lymph nodes. Numerous growth factor pathways cooperate to develop and maintain blood and lymphatic vessels. Two main ligand-receptor pathways that mediate endothelial cell (EC) biology are VEGF/VEGF receptor (VEGFR) and Ang/tyrosine kinase with Ig and endothelial growth factor homology domains (TIE)2. VEGF/VEGFR signaling typically promotes new vessel formation, and TIE2 signaling is associated with vessel maturation and maintenance.
ANGs are secreted proteins that bind their targets as multimeric clusters (1). Differences in oligomerization properties distinguish the ANGs and are thought to contribute to the mechanistic basis for the distinct physiologic roles associated with each Ang. Ang-1 activates TIE2 and is considered a TIE2 agonist. Ang-1 production is constitutive, leading to low-concentration paracrine signaling that is attributed to keeping ECs quiescent in nonremodeling vessels. In cells that are engaged in vessel remodeling, Ang-1 maintains proliferative and prosurvival signaling (2). Ang-1 also promotes EC adhesion to other ECs, pericytes, and the extracellular matrix, all of which suppress vessel permeability (3, 4). These activities collectively counteract induction of angiogenesis when VEGF is present at low levels in a homeostatic state (5).
Conversely, Ang-2 competitively antagonizes Ang-1 and suppresses full activation of TIE2 in blood vessels (6). Its expression is typically autocrine and in response to proangiogenic stimuli. Although Ang-2 can activate TIE2, it is a weaker agonist than Ang-1 (7). Thus, when Ang-1 and Ang-2 are in competition, overall TIE2 signaling is lower than when Ang-1 is alone (6). Ang-2 is cooperative with VEGF in enabling EC migration, proliferation, permeability, and sprouting, but in the absence of VEGF, Ang-2 promotes loss of pericyte recruitment, EC death, and vessel regression (8). The ang2−/− mouse exhibits abnormal lymphatic patterning and postnatal chylous ascites along with abnormal mural cell recruitment to blood vessels (9). Interestingly, insertion of the ang1 gene into the ang2 locus restores a normal lymphatic phenotype—although it does not rescue the defective blood vascular phenotype—suggesting that the actions of Ang-2 toward lymphatic vessels are cooperative rather than antagonistic with those of Ang-1 in lymphatic vessels (9).
Ang-4 is the most recently identified of the human ANGs (10, 11). It is the human ortholog of murine Ang-3, though Ang-3 exhibits a divergent protein structure from that of Ang-4 (10). To date, knowledge about Ang-4 actions is derived almost exclusively from studies in blood ECs and vascular biology. Studies examining Ang-4 in blood ECs suggest that like Ang-2, Ang-4 expression is autocrine and in response to proangiogenic stimuli such as hypoxia (12, 13). Its oligomerization properties resemble those of Ang-2 (11); however, Ang-4 strongly activates TIE2 and downstream signaling (11). One study using human coronary ECs and human pulmonary aortic ECs has shown that like Ang-1, Ang-4 promotes survival, migration, and tube formation in vitro (13). Lee et al. has shown that Ang-4 promotes corneal angiogenesis in FVB mice (11). A contradictory study suggests that Ang-4 inhibits VEGF and basic fibroblast growth factor-induced human umbilical vein EC (HUVEC) migration in vitro as well as angiogenesis in vivo (14). In lymphatic vessels, systemically overexpressed Ang-4 increased the number of lymphatic filopodia in the trachea and the lymphatic density of a healing wound (15). However, the observed changes in lymphatics are thought to be an indirect response to blood vessel changes (15). Here we focus on the actions of Ang-4 on lymphatic vessels and LECs. We demonstrate that Ang-4 promotes lymphatic dilation, activates TIE2-dependent proliferative and survival signaling in LECs, and does not increase LEC monolayer permeability. We also show that Ang-4 increases blood endothelial permeability both in vitro and in vivo.
MATERIALS AND METHODS
Animal models
For lymphatic vessel measurements, 4- to 6-wk-old female athymic Nu/Nu mice were implanted subcutaneously with 1 × 105 T241 cells expressing the Ang-1, Ang-2, Ang-4, VEGF-C, or control vector. Tumors were grown to approximately 40 mm3. For blood vessel permeability measurements, 10-wk-old female athymic Nu/Nu mice were implanted dorsally with 1 × 105 T241 cells expressing the factors indicated above. Tumors were grown to 40 mm3. Dorsal skinfold chambers were then surgically implanted around the tumors as previously described (16). Mice were anesthetized by injection of ketamine/xylazine 100/10 mg/kg subcutaneously. All procedures were performed following the guidelines of the Institutional Animal Care and Use Committee of the Massachusetts General Hospital.
Intravital fluorescence microscopy and vessel diameters
Lymphangiography was performed in 8–16 mice per group by peritumoral injection of 3–5 μl of 2.5% FITC-dextran (MW = 2 million; Sigma-Aldrich, St. Louis, MO, USA) as described previously (17). Injections and imaging were performed 2–3×/wk beginning with the first appearance of a tumor and ending when the tumor reached approximately 40 mm3. To diminish variations within the lymphatic networks of individual mice, care was taken to image and quantify the same lymphatic networks within the mouse over the course of tumor growth. Mice were imaged using a Zeiss-West fluorescence microscope (Carl Zeiss AG, Irvine CA, USA). Images were processed for quantitation using ImageJ 1.45s (National Institutes of Health, Bethesda, MD, USA). Measurements of vessel diameters were made between lymphatic junctions and valves.
Multiphoton microscopy and vessel permeability quantitation
Mice were injected retro-orbitally with 100 μl of 5 mg/ml tetramethylrhodamine-bovine serum albumin (BSA) (Life Technologies, Grand Island, NY, USA). Image collection of vessels within the tumor margins began within 10 min of injection. Image stacks of 201.48 μm total thickness were taken as 2.76 μm slices from 2 fields per mouse continuously for 1 h using Fluoview software (v. 5.0). Four to 7 mice per experimental group were imaged using a custom-built multiphoton microscope based on an Olympus FV300 (Olympus, Tokyo, Japan) and BX61WI platform with a MaiTai HP Ti-Sapphire laser source (Spectra-Physics, Santa Clara, CA, USA). An Olympus XLumPlanFl 20X/0.95 water immersion objective was used. Images were analyzed using a custom program developed in MatLab (Mathworks Inc., Natick, MA, USA). The analysis program first created a 3-dimensional vascular map and calculated the distance of each voxel to its nearest corresponding vessels. The effective permeability is calculated based on mass balance of the extravascular space using
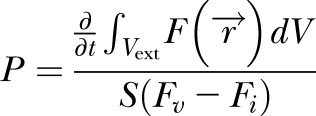 |
where 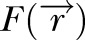 is the fluorescent intensity of a voxel at the distance dV from the vessel, S is the surface area of the vessels and Fv – Fi is the fluorescent intensity difference across the vessel wall. The integral of fluorescent intensity in extravascular space is evaluated numerically at each time point by
is the fluorescent intensity of a voxel at the distance dV from the vessel, S is the surface area of the vessels and Fv – Fi is the fluorescent intensity difference across the vessel wall. The integral of fluorescent intensity in extravascular space is evaluated numerically at each time point by
 |
Permeability was evaluated at a time interval when the fluorescent intensity difference across the vessel wall (Fv – Fi) stayed relatively constant. The calculation was made as an average over the entire field of interest.
Reagents and antibodies
Recombinant Ang-1, Ang-2 and Ang-4 were purchased from R&D Systems. Ang-1 was preclustered using anti-polyHis antibody (R&D Systems, Minneapolis, MN, USA) at a ratio of 1:4. Antibodies used for this study included phospho-Tyr 4G10 (EMD Millipore, Billerica, MA, USA), TIE2 (C-20; Santa Cruz Biotechnology, Dallas, TX, USA), phospho-protein kinase B (Akt) (Ser473), phospho-p44/42 MAPK (D13.14.4E), Akt, p44/42 MAPK (Erk1/2) (Cell Signaling Technology Inc., Danvers, MA, USA) and β-tubulin (EMD Millipore, Billerica, MA, USA).
Gene silencing and virus production
Lentiviral particles expressing scramble control short hairpin RNA (shRNA) (Sigma-Aldrich, St. Louis, MO, USA), or pLKO.1-shRNA-TEK (gene encoding for TIE2) (human tyrosine kinase with Ig and endothelial growth factor homology domains, Origene, Rockville, MD, USA) were used for gene silencing as previously described (18). For expression of ANGs, cDNA for human Ang-1, human Ang-2 (19), and human Ang-4 (Origene) were subcloned into pMCSV (Clontech, Mountain View, CA, USA). For viral packaging and production, human embryonic kidney 293 T (HEK293T) cells were transfected using the Fugene6 protocol (Roche Diagnostics Corp., Indianapolis, IN, USA).
Cell models
Human adult LECs and human adult BECs (passages 3–7, Lonza, Basel, Switzerland) were grown in EGM-2MV Microvascular Endothelial Cell Medium-2 (Lonza). LEC cell lines stably expressing RNA interference were grown and maintained under selection with 2 μg/ml puromycin (InvivoGen, San Diego, CA, USA). HEK293T and bEnd.3 cells (passages 6–12) were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA). T241 cells (passages 8–20) were described previously (20). HEK293T, bEnd.3, and T241 cells were cultured in DMEM supplemented with 5% fetal bovine serum and nonessential amino acids (Life Technologies). All cells were routinely tested as negative for mycoplasma. Cell authentication was performed by Idexx Radil (Columbia, MO, USA). T241 cells were maintained under selection with 0.9 mg/ml Geneticin (Life Technologies) or 2 μg/ml puromycin. To compare proliferation rates of T241 cell lines, 7.5 × 104 cells were seeded in triplicate. Cells were stained with Trypan Blue to exclude dead cells and counted using a hemocytometer every day for 5 d. Proliferation curves were determined by the formula:
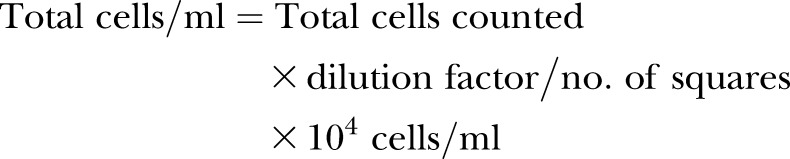 |
Survival assays
LECs were deprived of serum and growth factors for 24 h to promote apoptosis and cell death. The serum-free medium was then supplemented for 24 h with indicated factors. Cells were processed for detection of apoptosis using the TACS Annexin V Kit (Trevigen, Gaithersburg, MD, USA). The number of Annexin V-positive cells was measured by flow cytometry using the BD LSR II and BD FACSDiva software (BD Biosciences, San Jose, CA, USA).
Immunoprecipitation and immunoblotting
Protein A beads were preconjugated with 2 μg of indicated antibody or isotype control. LECs were harvested in RIPA buffer (1% Triton X-100, 1% deoxycholate, 0.1% SDS, 0.15 M NaCl, 50 M NaF, 5 mM EDTA, 50 mM Tris, pH 7.8) supplemented with protease and phosphatase inhibitor cocktails (Roche). Cell extracts were sonicated for 15 min in an ice water bath followed by centrifugation at 13,000 rpm at 4°C for 15 min. Preconjugated beads were rotated with cell supernatants at 4°C for 14–18 h. Bound proteins were eluted with 1× sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer. Eluates were heated at 95°C for 5 min prior to SDS-PAGE and immunoblotting for indicated proteins.
In vitro permeability assays
ECs were seeded in 0.5 ml growth medium to confluence in the upper chambers of transwell inserts containing a 0.4 μm polycarbonate membrane (Thermo Fisher Scientific, Waltham, MA, USA). Growth medium (0.5 ml) was placed into the bottom chambers. ECs were treated as indicated for 4 h followed by the addition of 1 μg/ml FITC-Dextran 40,000 to the upper chambers for 1 h. Permeability of the EC monolayer was measured by quantifying the fluorescence of the medium in the bottom chamber using the Fluoroskan Ascent fluorometer with Ascent 2.6 software (Thermo Electron Corp., Waltham, MA, USA).
Statistical analysis
Lymphatic diameters were plotted as a single diameter vs. tumor size. Least square linear regression was performed to obtain the optimal linear fit, and the standard error associated with each slope was determined and used to generate 95% confidence intervals. Statistical significance was determined by comparison of the slopes using 1-way ANOVA and Tukey’s honest significant difference (HSD) post hoc test. Nonlinear regression was performed to obtain the optimal linear fit for T241 tumor growth curves. The mean of the slopes for each tumor group was compared using 1-way ANOVA and post hoc testing using Tukey’s HSD post hoc test. Fisher’s exact probability and chi-square test were used for lymphatic metastasis. One-way ANOVA and post hoc testing using Tukey’s HSD post hoc test was performed for survival, in vitro and in vivo permeability assays.
RESULTS
Ang-4 induces lymphatic dilation
To study the actions of Ang-4 on lymphatic vessels, we generated murine fibrosarcoma T241 tumors that stably expressed Ang-4 and compared the effects to those conferred by tumors expressing human Ang-1 and human Ang-2. Tumors were validated for expression and secretion of these factors by qPCR and ELISA, respectively (Supplemental Fig. 1, Supplemental Table 1). Tumors were implanted subdermally into the ears of athymic nude mice. Previously, we showed that tumors expressing the lymphangiogenic growth factor VEGF-C induced dilation of lymphatic vessels (17). Using lymphangiography, we made multiple measurements of functional lymphatic vessel diameters in the tumor margins over time and compared between tumors of the same size (Fig. 1A, B). As expected, parental and mock-transduced tumors exhibited no apparent changes to lymphatic vessels with diameters remaining near the baseline of 60 μm as tumors approached 40 mm3 (Fig. 1A, Supplemental Fig. 2A, B). The VEGF-C-mediated increase on the diameters was the strongest of the factors examined with lymphatic diameters reaching 135.9 ± 24.7 μm (Fig. 1A, Supplemental Fig. 2C). Ang-1 and Ang-2 secretion did not change diameters relative to the mock-transduced control (Fig. 1A, B, Supplemental Fig. 2D, E). Ang-4 increased the lymphatic diameters to above 50 μm when tumors were approximately 1 mm3, and the lymphatic diameters continued to expand to 123.1 ± 16.1 μm (Fig. 1A, B, Supplemental Fig. 2F). Transgene expression did not affect T241 cell proliferation or tumor growth with the exception of Ang-2 overexpression, which caused a growth delay compared to the mock-transduced control group (Fig. 1C, D).
Figure 1.
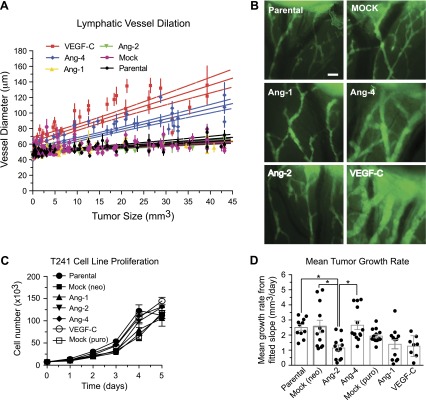
Ang-4 increases lymphatic vessel diameters. T241 tumors expressing indicated factors were implanted subdermally into the left ear. FITC-Dextran was injected into the edge of the tumor for lymphangiography and immunofluorescence imaging. A) Measurements of vessel diameters were made between lymphatic junctions and valves until the tumor reached approximately 40 mm3. Solid lines represent linear regression of the average change in diameter matched to tumor size of 8–16 mice per group. Dashed lines represent 95% confidence intervals. Statistical analysis was performed using 1-way ANOVA and Tukey’s HSD post hoc test. B) Representative images shown are of tumor-draining lymphatic vessels from each group taken when tumors were approximately 10 mm3, (scale bar, 250 μm). Live animals were imaged using a Zeiss-West Germany Axioskope, El Einsatz 451888 with a ×2.5/0.075 Plan-Neofluar air objective with a numerical aperture of 44 03 10. Images were acquired using Hamamatsu CCD Camera C2400 CX-77 (Hamamatsu Photonics, Hamamatsu, Japan) with Scion Image software, Release Alpha 4.0.3.2 (Scion Corp., Frederick, MD, USA). Images were processed for quantitation using ImageJ 1.45s (U.S. National Institutes of Health). C) In vitro proliferation curves for T241 cell lines. D) The mean of the slopes for each tumor group (column) is shown along with the slopes determined for the individual tumors (circles). A, B) Mock includes tumors transduced with virus produced from empty vectors with either neomycin (neo)- or puromycin (puro)-selection markers. *P < 0.05.
Ang-4 does not promote lymphatic metastasis
Expression of VEGF-C has been shown to promote metastasis to the sentinel lymph node in multiple tumor models, including T241 (17, 21–28). Although lymphatic vessel dilation by VEGF-C correlated with metastasis to the cervical lymph node in 84% of mice, Ang-4-mediated lymphatic dilation corresponded to only 25% (Table 1). Ang-4-secreting tumors exhibited statistically similar rates of lymph node metastasis to Ang-1 (13%), Ang-2 (17%), and parental and mock control tumors (23 and 10%, respectively).
TABLE 1.
Sentinel lymph node metastasis
| Tumor | + | − | Total | Positive (%) |
|---|---|---|---|---|
| Parental | 3 | 10 | 13 | 23 |
| Mock | 2 | 18 | 20 | 10 |
| Ang-1 | 1 | 7 | 8 | 13 |
| Ang-2 | 2 | 10 | 12 | 17 |
| Ang-4 | 5 | 15 | 20 | 25 |
| VEGF-C | 16 | 3 | 19 | 84* |
Animals were judged to have lymph node metastasis if they had palpable lymph nodes on or before 8 wk after primary tumor resection. Statistical analysis was performed using Fisher’s exact probability and chi-square test. Mock, transduced with empty vector; +, total number of mice with LN metastasis; −, total number of mice without LN metastasis.
P < 0.05.
Rapid activation of TIE2 and downstream signaling by Ang-4
To better understand how Ang-4 directly impacts LECs, we measured the effects of recombinant human Ang-4 on human adult dermal LECs. Activation of TIE2 was measured by immunoblotting for phosphorylation of the multiple tyrosine residues within the C-terminal cytoplasmic domain of TIE2. Ang-4 induced phosphorylation of TIE2, although the induction was not as strong as Ang-1 (Fig. 2A). Activation of TIE2 was detectable at 0.1 μg/ml for Ang-1 and 0.2 μg/ml for Ang-4 (Supplemental Fig. 3A, B). Activation of TIE2 by Ang-2 in LECs was not detectable at any concentration using our methods (data not shown). Activation of TIE-2 by Ang-4 occurred as early as 15 min, was sustained up to 16 h, and declined by 20 h (Fig. 2B). Ang-4 rapidly activated downstream effectors Akt and Erk1,2 (Fig. 2C). A mild proliferative induction was observed for Ang-4 that was similar to Ang-1 at low dose (62.5 ng/ml, Supplemental Fig. 3C). Because Ang-4-mediated lymphatic dilation was observed in mice, bEnd.3 murine ECs were treated with the recombinant human ANGs utilized in this study to show that murine TIE2 was also activated by human Ang-1 and Ang-4 (Supplemental Fig. 3D, E).
Figure 2.

Rapid and TIE2-dependent signaling by Ang-4. Activation of TIE-2 was measured by immunoblotting for phosphorylated tyrosine residues on immunoprecipitated TIE-2. A) LECs were treated for 30 min with 0.4 μg/ml Ang-1, Ang-2, or Ang-4 and (B) for indicated times with 0.4 μg/ml Ang-4. C) Activation of Akt and Erk1,2 with 0.4 μg/ml Ang-4. D) TIE2 expression in LECs transduced with lentivirus containing shSCR or shTEK (T1 and T2). E) Akt and Erk1,2 activation after incubation with 0.4 μg/ml Ang-4 for 15, 30, 45, 60, or 120 min in shSCR LECs are compared with shTEK T1 and F) shTEK T2 after serum and growth factor starvation overnight. G) Antagonism at TIE2 was determined by competing indicated concentrations of Ang-4 and with indicated concentrations of Ang-2 for 30 min. IgG, isotype control; ab, anti-polyHis antibody used to precluster Ang-1.
TIE2-dependence of Ang-4 signaling
TIE2-independent actions by ANGs have been documented for Ang-1 and Ang-2 in blood ECs (29, 30). To determine whether Ang-4 signaling was TIE2-mediated, we used shRNA to silence TEK. LECs stably expressing scrambled shRNA (shSCR) or 1 of 2 TEK shRNAs (shTEK) were generated (Fig. 2D). LECs were starved for 18 h and treated with Ang-4 for 15, 30, 45, 60, or 120 min. Although the Ang-4-mediated increases in phospho-Akt and phospho-Erk1,2 activation were maintained in the shSCR control cells, they were abolished by TIE2 knockdown (Fig. 2E, F). The shTEK (T1) cells demonstrated a higher baseline phospho-Akt and phospho-Erk1,2 (Fig. 2E). However, these cells did not demonstrate Ang-4-mediated increases in these signaling molecules even though activation of these mediators was robust when growth medium was added to the cells (Supplemental Fig. 3F).
Ang-2 antagonism of Ang-4
Ang-2 is a competitive antagonist of Ang-1 at TIE2 (6). In our studies, we did not detect Ang-2 activation of LEC TIE2. Because Ang-4 activates TIE2 in LECs, we asked whether Ang-2 antagonism was preserved against Ang-4. At a subsaturating concentration of Ang-4 (0.2 μg/ml), a weak activation of TIE2 was observed that was not reduced by a high concentration of Ang-2. However, at a higher concentration of Ang-4 (0.4 μg/ml), Ang-2 reduced Ang-4-induced phosphorylation of TIE2 when administered at equivalent (1:1) and excess (1:2) concentrations (Fig. 2G).
Ang-4 promotes LEC survival
We next examined whether TIE2 activation by Ang-4 translated into improved TIE2-dependent survival of LECs. Apoptosis was induced by starvation of LECs via removal of serum and growth factors for 24 h. To determine if Ang-4 protected LECs from apoptosis, Ang-4 was supplemented into the medium 24 h after starvation was initiated for an additional 24 h. Using flow cytometry to detect Annexin V, Ang-4-treated LECs were compared to LECs that had received Ang-1, Ang-2, or growth medium during the final 24 h of starvation. Ang-4 protected LECs against apoptosis to a similar extent as Ang-1, while Ang-2 offered no survival benefit (Fig. 3A, B). Protection by Ang-4 was abolished when TEK was silenced, but Ang-1 continued to promote survival, indicating that Ang-1 but not Ang-4 has a TIE2-independent mechanism for promoting LEC survival (Fig. 3C). Ang-1-mediated activation of Akt and Erk1,2 was diminished when TIE2 was knocked down, but it was not entirely abolished (Supplemental Fig. 3G).
Figure 3.

Ang-4 promotes TIE2-dependent LEC survival. LECs were starved for 24 h followed by treatments with 0.4 μg/ml of Angs or growth medium for 24 h. Apoptosis was assessed by measuring the number of Annexin V-positive cells using flow cytometry. Data are displayed as fold change relative to serum and growth factor-starved control (SF). Comparisons shown include (A) parental LECs, (B) shSCR LECs, and (C) shTEK T2 LECs. Statistical analysis was performed using 1-way ANOVA and Tukey’s HSD post hoc test. (ab), antibody used to precluster Ang-1; Asyn, asynchronously growing LECs that were not starved; GM, growth medium.
Ang-4 does not increase lymphatic monolayer permeability
To test whether lymphatic dilation is caused by an Ang-4-mediated decrease in LEC-LEC adhesions, a monolayer of LECs was grown in transwell chambers and the permeability was measured. As described previously using blood ECs (13), Ang-1 reduced LEC permeability by approximately 50% and Ang-2 trended toward an increase. Ang-4 demonstrated a trend toward reduction of the permeability of the LEC monolayer by approximately 30%, though the effect was not statistically significant between Ang-4 and the untreated control (Fig. 4A). As expected, VEGF-C induced a dose-dependent increase in permeability (Fig. 4B).
Figure 4.

Ang-4 and LEC permeability. LECs were seeded to confluence in the upper chambers of transwells and treated with 0.4 µg/ml Angs or indicated concentrations of VEGF-C for 4 h. FITC-Dextran 40,000 (1 μg/ml) was added to each chamber for 1 h. Permeability of the EC monolayer was determined by measuring the fluorescence of the medium in the bottom chamber using a Fluoroskan Ascent with Ascent 2.6 software for acquisition. Graphs represent the mean fold change relative to control from 8–10 combined experiments. Statistical analysis was performed using 1-way ANOVA and Tukey’s HSD post hoc test. (ab), antibody used to precluster Ang-1. *P < 0.05.
Ang-4 increases microvascular monolayer permeability
Because Ang-4 is known to exert actions on blood vessels and activate TIE2 (10–15) (Supplemental Fig. H, I), we pursued the possibility that Ang-4 may promote lymphatic dilation indirectly via its actions on blood ECs. Human adult dermal BECs were used to examine BEC monolayer permeability. In contrast to LECs, Ang-4 doubled the permeability of the BEC monolayer (P < 0.05), and Ang-1 reduced permeability by approximately 27% (P < 0.05, Fig. 5A). Moreover, Ang-4 and Ang-1 antagonized one another in this model (Fig. 5A). VEGF-C did not significantly affect BEC permeability in this assay (Fig. 5B).
Figure 5.

Ang-4 and BEC permeability. BECs were seeded for permeability measurements as described in Fig. 4. Cells were treated with 100 ng/ml VEGF, 0.4 µg/ml Angs, or indicated concentrations of VEGF-C. Graphs represent the mean fold change relative to control from 7–10 combined experiments. Statistical analysis was performed using 1-way ANOVA and Tukey’s HSD post hoc test. (ab), antibody used to precluster Ang-1. *P < 0.05.
Ang-4 promotes extravasation from blood vessels
With the increase in BEC permeability in vitro, we tested whether Ang-4 affected the local microvasculature in vivo. T241 tumors were grown subdermally, and tumor-associated vessels were visualized by multiphoton microscopy using the dorsal skinfold chamber model (31). We assessed vessel permeability by measuring the transport kinetics of fluorescently labeled BSA across vessel walls. By evaluating the fluorescent intensity change over time outside the vessel, we were able to calculate the effective permeability of blood vessels in a given volume. Parental, mock, Ang-1-, and Ang-2-expressing tumors did not produce highly permeable vessels (4.4 × 0−7 ± 0.9 × 10−7 cm/s, 2.0 × 10−7 ± 0.6 × 10−7 cm/s, 4.2 × 10−7 ± 1.1 × 10−7 cm/s, and 5.0 × 10−7 ± 1.7 × 10−7 cm/s, respectively, Fig. 6), and VEGF-C-expressing tumors exhibited elevated vascular permeability (8.9 × 10−7 ± 1.8 × 10−7 cm/s, Fig. 6). Like VEGF-C, Ang-4 significantly increased the permeability of tumor-associated blood vessels (8.6 × 10−7 ± 1.2 × 10−7 cm/s, Fig. 6), suggesting that Ang-4 promotes fluid extravasation.
Figure 6.

Ang-4 promotes vascular permeability in vivo. Blood vessel permeability was measured in T241 tumor-associated vessels using multiphoton microscopy and dorsal skinfold chambers. Ang-4- and VEGF-C-expressing tumors showed marked elevations in vascular permeability. Four to 7 live animals were imaged using multiphoton microscopy. Statistical analysis was performed using 1-way ANOVA and Tukey’s HSD post hoc test. *P < 0.05.
DISCUSSION
We began our investigation into Ang-4 effects on lymphatic vessels using the T241 murine fibrosarcoma cell line to express Ang-4 in vivo. The T241 model was chosen for its moderate growth rate and lack of basal metastasis formation. This allowed us to perform multiple, size-matched measurements of lymphatic diameters over time and look for lymphatic metastasis. We found that the diameters of lymphatic vessels draining the control tumors increased only slightly when these tumors approached maximum sizes of 40 mm3. Under these conditions, it became noteworthy that only Ang-4 and VEGF-C increased lymphatic diameters while tumors were still small (<2 mm3). These observations are consistent with those made by Kim et al., who used systemic expression of Ang-4 through adenoviral delivery to show lymphatic vessel enlargement and formation of lymphatic sprouts and filopodia in the microvasculature of ear skin wounds and in the trachea (15). However, our methods did not reproduce their observations with systemic Ang-1 and Ang-2, which promoted lymphatic vessel enlargement in ear skin wounds of the previous study (15).
The proliferation rates of the T241 cell lines used for this study were similar in vitro; however, the T241-Ang-2 tumors exhibited a growth delay in vivo. Ang-2 exhibits a strong dependence on the local cytokine milieu where angiogenic cytokines cooperate with Ang-2 to promote angiogenesis and tumor growth. In the absence of angiogenic cytokines, Ang-2 promotes vessel regression, which is predicted to slow tumor growth (8, 32). Similarly, VEGF-C has shown an inhibitory effect on tumor growth (22). It is likely that the T241-Ang-2 tumor microenvironment promoted the observed growth delay.
Ang-4 and lymphatic vessels
Several studies have correlated lymphatic vessel formation and dilation in the tumor margin with lymphatic metastasis (21, 22). VEGF-C, which causes peritumor lymphatic dilation, has been identified as a driver of lymphatic metastasis (17, 21–28). We expected to observe a similar outcome from Ang-4-mediated lymphatic dilation. When Ang-4 did not promote metastasis, it became evident that the mechanism of Ang-4-mediated lymphatic dilation was distinct from that of VEGF-C.
With little known about the direct effects of Ang-4 on LECs compared with VEGF-C, Ang-1, and Ang-2, it became essential to test the effects of Ang-4 directly on LECs. Ang-4-mediated effects were remarkably similar to those of Ang-1 in most assays examining LECs. Rapid activation of TIE2 signaling, moderate LEC proliferation, LEC survival, and an impermeable LEC monolayer were observed for both Ang-1 and Ang-4. The antagonism between Ang-2 and Ang-1 was also preserved between Ang-2 and Ang-4 when TIE2 signaling was examined. Although all Ang-4 effects examined here were TIE2-dependent, Ang-1 exhibited a TIE2-independent ability to promote LEC survival. This observation is consistent with studies showing that Ang-1 transduces signals through integrins at the cell membrane (29, 30). Whether Ang-4 engages other receptors besides TIE2 to affect other facets of lymphatic biology remains an open question.
An important difference between Ang-1 and Ang-4 involves their multimeric quaternary structures (11). Ang-1 functions as a high-order multimer, consisting of clusters of dimers and trimers (1). This property has been linked to its ability to promote cell-cell and cell-matrix adhesions that decrease vessel permeability (3, 4). Ang-2 forms lower-order multimers consisting mainly of unclustered dimers, which are presumably unable to sustain the same types of TIE2 contacts as Ang-1 (11). Given the similarity in structure between Ang-2 and Ang-4, which we independently verified (data not shown), we expected Ang-4 to promote a more permeable LEC monolayer than Ang-1. However, Ang-4 did not change monolayer permeability, indicating that the mechanisms affecting lymphatic cell-cell contacts are more complex (33). More importantly, this result revealed that Ang-4-mediated lymphatic dilation was likely due to factors beyond direct effects on LECs. As predicted, VEGF-C increased the permeability of LEC monolayers, in contrast to Ang-4. This differential response may explain why VEGF-C promotes lymphatic metastasis and Ang-4 does not, even though overexpression of both molecules induces hyperplasia of tumor margin lymphatic vessels.
Ang-4 and blood vessels
Because lymphatic vessels function alongside blood vessels, we hypothesized that Ang-4-mediated signaling to blood vessels was causing the lymphatic vessel phenotype apparent in our animal model. Ang-4 has previously been shown to lower tumor IFP in a subcutaneous model of non-small cell lung cancer tumor (14) and to reduce permeability in vitro in BECs of the pulmonary artery (13). These observations suggested that Ang-4 normalizes tumor-associated blood vessels in the lung microenvironment without promoting angiogenesis. However, our examination of human dermal BECs revealed that Ang-4 increases the permeability of the BEC monolayer and that Ang-1 and Ang-4 antagonize one another. This finding was preserved in vivo, where Ang-4 promoted vascular permeability of the blood vessels in the tumor margin, while Ang-1 and Ang-2 had no such effects. The difference between Ang-1 and Ang-4 effects suggests that the Ang-4-induced lymphatic dilation occurs secondarily to extravasation of fluid from surrounding blood vessels. That Ang-4 has exhibited different outcomes on ECs and vessels in different studies indicates that the organ microenvironment contributes to phenotypic response to Ang-4 by ECs.
Ang-4 in human health and disease
When assessing noncancerous tissue, ang-4 expression is found almost exclusively in human lung tissue (10). Expression studies of isolated aortic ECs have shown autocrine expression of ang-4 by ECs that is inducible by hypoxia, similarly to ang-2 (12, 13, 34). These studies suggest that the role of Ang-4 may be specific to hypoxia in the lung and, possibly, to conditions elsewhere that promote local hypoxia, such as cancer. The redundancies observed in this study of the Ang-1 and Ang-4 ligands toward LECs may involve this expression dynamic, whereby ang-4 is up-regulated to function as an inducible ang-1 to maintain the lymphatic endothelium as it manages local edema associated with hypoxia-triggered angiogenesis and remodeling.
The Ang/TIE2 family has generated profound interest for the development of anti-angiogenic cancer therapies (18, 35). Although the ang-4 connection to cancer is still in its infancy, recent studies have correlated ang-4 expression in ovarian cancer and non-small cell lung carcinoma to poor patient prognosis (36, 37). Expression of ang-4 has also been correlated with estrogen receptor positivity in human breast cancers (38). Ang-4 expression by human glioblastoma cells has been shown to promote intracranial growth by increasing angiogenesis in a mouse model (39). As factors within the Ang/Tie2 family are targeted for ablation, dissecting Ang-4 signaling will become increasingly important to understand potential toxicities from these therapies. We have shown that Ang-4 promotes survival in LECs and increases blood vessel permeability. Thus, blocking Ang-4 may cause the loss of the protective effect of Ang-4 on lymphatic vessels while lowering tumor blood vessel permeability, both of which may be beneficial in limiting the spread of cancer and improving tumor drug delivery by reducing IFP.
Supplementary Material
Acknowledgments
This work was funded by U.S. National Institutes of Health (NIH) National Cancer Institute (NCI) Grant R00CA137167 (to T.P.P.), NIH Office of the Director Grant DP2OD008780 (to T.P.P.), and NCI Federal Share/Massachusetts General Hospital Proton Beam Income Grant C06 CA059267 (to T.P.P.). The authors thank Dr. Dai Fukumura, Dr. Jay Loeffler, Dr. Rakesh Jain, Dr. Vikash Chuahan, Julia Kahn, Anna Khachatryan, and members of the Edwin L. Steele Laboratories for support, technical assistance, and helpful discussions.
Glossary
- Ang
angiopoietin
- Akt
protein kinase B
- BEC
blood endothelial cell
- BSA
bovine serum albumin
- EC
endothelial cell
- HEK293T
human embryonic kidney 293 T
- HSD
honest significant difference
- IFP
interstitial fluid pressure
- LEC
lymphatic endothelial cell
- shRNA
short hairpin RNA
- shSCR
short hairpin scramble RNA
- TEK
gene encoding for TIE2
- TIE2
tyrosine kinase with Ig and endothelial growth factor homology domains-2
- VEGFR
VEGF receptor
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Kim K. T., Choi H. H., Steinmetz M. O., Maco B., Kammerer R. A., Ahn S. Y., Kim H. Z., Lee G. M., Koh G. Y. (2005) Oligomerization and multimerization are critical for angiopoietin-1 to bind and phosphorylate Tie2. J. Biol. Chem. 280, 20126–20131 [DOI] [PubMed] [Google Scholar]
- 2.Augustin H. G., Koh G. Y., Thurston G., Alitalo K. (2009) Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat. Rev. Mol. Cell Biol. 10, 165–177 [DOI] [PubMed] [Google Scholar]
- 3.Saharinen P., Eklund L., Miettinen J., Wirkkala R., Anisimov A., Winderlich M., Nottebaum A., Vestweber D., Deutsch U., Koh G. Y., Olsen B. R., Alitalo K. (2008) Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell-cell and cell-matrix contacts. Nat. Cell Biol. 10, 527–537 [DOI] [PubMed] [Google Scholar]
- 4.Fukuhara S., Sako K., Minami T., Noda K., Kim H. Z., Kodama T., Shibuya M., Takakura N., Koh G. Y., Mochizuki N. (2008) Differential function of Tie2 at cell-cell contacts and cell-substratum contacts regulated by angiopoietin-1. Nat. Cell Biol. 10, 513–526 [DOI] [PubMed] [Google Scholar]
- 5.Gavard J., Patel V., Gutkind J. S. (2008) Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev. Cell 14, 25–36 [DOI] [PubMed] [Google Scholar]
- 6.Maisonpierre P. C., Suri C., Jones P. F., Bartunkova S., Wiegand S. J., Radziejewski C., Compton D., McClain J., Aldrich T. H., Papadopoulos N., Daly T. J., Davis S., Sato T. N., Yancopoulos G. D., Yancopoulos G. D. (1997) Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277, 55–60 [DOI] [PubMed] [Google Scholar]
- 7.Bogdanovic E., Nguyen V. P., Dumont D. J. (2006) Activation of Tie2 by angiopoietin-1 and angiopoietin-2 results in their release and receptor internalization. J. Cell Sci. 119, 3551–3560 [DOI] [PubMed] [Google Scholar]
- 8.Lobov I. B., Brooks P. C., Lang R. A. (2002) Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc. Natl. Acad. Sci. USA 99, 11205–11210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gale N. W., Thurston G., Hackett S. F., Renard R., Wang Q., McClain J., Martin C., Witte C., Witte M. H., Jackson D., Suri C., Campochiaro P. A., Wiegand S. J., Yancopoulos G. D. (2002) Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev. Cell 3, 411–423 [DOI] [PubMed] [Google Scholar]
- 10.Valenzuela D. M., Griffiths J. A., Rojas J., Aldrich T. H., Jones P. F., Zhou H., McClain J., Copeland N. G., Gilbert D. J., Jenkins N. A., Huang T., Papadopoulos N., Maisonpierre P. C., Davis S., Yancopoulos G. D. (1999) Angiopoietins 3 and 4: diverging gene counterparts in mice and humans. Proc. Natl. Acad. Sci. USA 96, 1904–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee H. J., Cho C. H., Hwang S. J., Choi H. H., Kim K. T., Ahn S. Y., Kim J. H., Oh J. L., Lee G. M., Koh G. Y. (2004) Biological characterization of angiopoietin-3 and angiopoietin-4. FASEB J. 18, 1200–1208 [DOI] [PubMed] [Google Scholar]
- 12.Lund E. L., Høg A., Olsen M. W., Hansen L. T., Engelholm S. A., Kristjansen P. E. (2004) Differential regulation of VEGF, HIF1alpha and angiopoietin-1, -2 and -4 by hypoxia and ionizing radiation in human glioblastoma. Int. J. Cancer 108, 833–838 [DOI] [PubMed] [Google Scholar]
- 13.Yamakawa M., Liu L. X., Date T., Belanger A. J., Vincent K. A., Akita G. Y., Kuriyama T., Cheng S. H., Gregory R. J., Jiang C. (2003) Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ. Res. 93, 664–673 [DOI] [PubMed] [Google Scholar]
- 14.Olsen M. W. B., Ley C. D., Junker N., Hansen A. J., Lund E. L., Kristjansen P. E. (2006) Angiopoietin-4 inhibits angiogenesis and reduces interstitial fluid pressure. Neoplasia 8, 364–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim K. E., Cho C. H., Kim H. Z., Baluk P., McDonald D. M., Koh G. Y. (2007) In vivo actions of angiopoietins on quiescent and remodeling blood and lymphatic vessels in mouse airways and skin. Arterioscler. Thromb. Vasc. Biol. 27, 564–570 [DOI] [PubMed] [Google Scholar]
- 16.Leunig M., Goetz A. E., Dellian M., Zetterer G., Gamarra F., Jain R. K., Messmer K. (1992) Interstitial fluid pressure in solid tumors following hyperthermia: possible correlation with therapeutic response. Cancer Res. 52, 487–490 [PubMed] [Google Scholar]
- 17.Hoshida T., Isaka N., Hagendoorn J., di Tomaso E., Chen Y. L., Pytowski B., Fukumura D., Padera T. P., Jain R. K. (2006) Imaging steps of lymphatic metastasis reveals that vascular endothelial growth factor-C increases metastasis by increasing delivery of cancer cells to lymph nodes: therapeutic implications. Cancer Res. 66, 8065–8075 [DOI] [PubMed] [Google Scholar]
- 18.Goel S., Gupta N., Walcott B. P., Snuderl M., Kesler C. T., Kirkpatrick N. D., Heishi T., Huang Y., Martin J. D., Ager E., Samuel R., Wang S., Yazbek J., Vakoc B. J., Peterson R. T., Padera T. P., Duda D. G., Fukumura D., Jain R. K. (2013) Effects of vascular-endothelial protein tyrosine phosphatase inhibition on breast cancer vasculature and metastatic progression. J. Natl. Cancer Inst. 105, 1188–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chae S. S., Kamoun W. S., Farrar C. T., Kirkpatrick N. D., Niemeyer E., de Graaf A. M., Sorensen A. G., Munn L. L., Jain R. K., Fukumura D. (2010) Angiopoietin-2 interferes with anti-VEGFR2-induced vessel normalization and survival benefit in mice bearing gliomas. Clin. Cancer Res. 16, 3618–3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kadambi A., Mouta Carreira C., Yun C. O., Padera T. P., Dolmans D. E., Carmeliet P., Fukumura D., Jain R. K. (2001) Vascular endothelial growth factor (VEGF)-C differentially affects tumor vascular function and leukocyte recruitment: role of VEGF-receptor 2 and host VEGF-A. Cancer Res. 61, 2404–2408 [PubMed] [Google Scholar]
- 21.Skobe M., Hawighorst T., Jackson D. G., Prevo R., Janes L., Velasco P., Riccardi L., Alitalo K., Claffey K., Detmar M. (2001) Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat. Med. 7, 192–198 [DOI] [PubMed] [Google Scholar]
- 22.Padera T. P., Kadambi A., di Tomaso E., Carreira C. M., Brown E. B., Boucher Y., Choi N. C., Mathisen D., Wain J., Mark E. J., Munn L. L., Jain R. K. (2002) Lymphatic metastasis in the absence of functional intratumor lymphatics. Science 296, 1883–1886 [DOI] [PubMed] [Google Scholar]
- 23.Karnezis T., Shayan R., Caesar C., Roufail S., Harris N. C., Ardipradja K., Zhang Y. F., Williams S. P., Farnsworth R. H., Chai M. G., Rupasinghe T. W., Tull D. L., Baldwin M. E., Sloan E. K., Fox S. B., Achen M. G., Stacker S. A. (2012) VEGF-D promotes tumor metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium. Cancer Cell 21, 181–195 [DOI] [PubMed] [Google Scholar]
- 24.Mandriota S. J., Jussila L., Jeltsch M., Compagni A., Baetens D., Prevo R., Banerji S., Huarte J., Montesano R., Jackson D. G., Orci L., Alitalo K., Christofori G., Pepper M. S. (2001) Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. EMBO J. 20, 672–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karpanen T., Egeblad M., Karkkainen M. J., Kubo H., Ylä-Herttuala S., Jäättelä M., Alitalo K. (2001) Vascular endothelial growth factor C promotes tumor lymphangiogenesis and intralymphatic tumor growth. Cancer Res. 61, 1786–1790 [PubMed] [Google Scholar]
- 26.Wong S. Y., Haack H., Crowley D., Barry M., Bronson R. T., Hynes R. O. (2005) Tumor-secreted vascular endothelial growth factor-C is necessary for prostate cancer lymphangiogenesis, but lymphangiogenesis is unnecessary for lymph node metastasis. Cancer Res. 65, 9789–9798 [DOI] [PubMed] [Google Scholar]
- 27.Achen M. G., Williams R. A., Minekus M. P., Thornton G. E., Stenvers K., Rogers P. A., Lederman F., Roufail S., Stacker S. A. (2001) Localization of vascular endothelial growth factor-D in malignant melanoma suggests a role in tumour angiogenesis. J. Pathol. 193, 147–154 [DOI] [PubMed] [Google Scholar]
- 28.Stacker S. A., Caesar C., Baldwin M. E., Thornton G. E., Williams R. A., Prevo R., Jackson D. G., Nishikawa S., Kubo H., Achen M. G. (2001) VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat. Med. 7, 186–191 [DOI] [PubMed] [Google Scholar]
- 29.Carlson T. R., Feng Y., Maisonpierre P. C., Mrksich M., Morla A. O. (2001) Direct cell adhesion to the angiopoietins mediated by integrins. J. Biol. Chem. 276, 26516–26525 [DOI] [PubMed] [Google Scholar]
- 30.Thomas M., Felcht M., Kruse K., Kretschmer S., Deppermann C., Biesdorf A., Rohr K., Benest A. V., Fiedler U., Augustin H. G. (2010) Angiopoietin-2 stimulation of endothelial cells induces alphavbeta3 integrin internalization and degradation. J. Biol. Chem. 285, 23842–23849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Padera T. P., Stoll B. R., So P. T., Jain R. K. (2002) Conventional and high-speed intravital multiphoton laser scanning microscopy of microvasculature, lymphatics, and leukocyte-endothelial interactions. Mol. Imaging 1, 9–15 [DOI] [PubMed] [Google Scholar]
- 32.Peters S., Cree I. A., Alexander R., Turowski P., Ockrim Z., Patel J., Boyd S. R., Joussen A. M., Ziemssen F., Hykin P. G., Moss S. E. (2007) Angiopoietin modulation of vascular endothelial growth factor: Effects on retinal endothelial cell permeability. Cytokine 40, 144–150 [DOI] [PubMed] [Google Scholar]
- 33.Yao L. C., Baluk P., Srinivasan R. S., Oliver G., McDonald D. M. (2012) Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am. J. Pathol. 180, 2561–2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gavrilovskaya I. N., Gorbunova E. E., Mackow E. R. (2013) Hypoxia induces permeability and giant cell responses of ANDV infected pulmonary endothelial cells by activating the mTOR-S6K signaling pathway. J. Virol. 87, 12999–13008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang H., Bhat A., Woodnutt G., Lappe R. (2010) Targeting the ANGPT-TIE2 pathway in malignancy. Nat. Rev. Cancer 10, 575–585 [DOI] [PubMed] [Google Scholar]
- 36.Andersen S., Donnem T., Al-Shibli K., Al-Saad S., Stenvold H., Busund L. T., Bremnes R. M. (2011) Prognostic impacts of angiopoietins in NSCLC tumor cells and stroma: VEGF-A impact is strongly associated with Ang-2. PLoS ONE 6, e19773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lyu T., Jia N., Wang J., Yan X., Yu Y., Lu Z., Bast R. C. Jr., Hua K., Feng W. (2013) Expression and epigenetic regulation of angiogenesis-related factors during dormancy and recurrent growth of ovarian carcinoma. Epigenetics 8, 1330–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Currie M. J., Gunningham S. P., Han C., Scott P. A., Robinson B. A., Harris A. L., Fox S. B. (2001) Angiopoietin-1 is inversely related to thymidine phosphorylase expression in human breast cancer, indicating a role in vascular remodeling. Clin. Cancer Res. 7, 918–927 [PubMed] [Google Scholar]
- 39.Brunckhorst M. K., Wang H., Lu R., Yu Q. (2010) Angiopoietin-4 promotes glioblastoma progression by enhancing tumor cell viability and angiogenesis. Cancer Res. 70, 7283–7293 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.