

Abstract
Extracellular ATP binds to and signals through P2X7 receptors (P2X7Rs) to modulate immune function in both inflammasome-dependent and -independent manners. In this study, P2X7−/− mice, the pharmacological agonists ATP-magnesium salt (Mg-ATP; 100 mg/kg, EC50 ≈ 1.32 mM) and benzoylbenzoyl-ATP (Bz-ATP; 10 mg/kg, EC50 ≈ 285 μM), and antagonist oxidized ATP (oxi-ATP; 40 mg/kg, IC50 ≈ 100 μM) were used to show that P2X7R activation is crucial for the control of mortality, bacterial dissemination, and inflammation in cecal ligation and puncture–induced polymicrobial sepsis in mice. Our results with P2X7−/− bone marrow chimeric mice, adoptive transfer of peritoneal macrophages, and myeloid-specific P2X7−/− mice indicate that P2X7R signaling on macrophages is essential for the protective effect of P2X7Rs. P2X7R signaling protects through enhancing bacterial killing by macrophages, which is independent of the inflammasome. By using the connexin (Cx) channel inhibitor Gap27 (0.1 mg/kg, IC50 ≈ 0.25 μM) and pannexin channel inhibitor probenecid (10 mg/kg, IC50 ≈ 11.7 μM), we showed that ATP release through Cx is important for inhibiting inflammation and bacterial burden. In summary, targeting P2X7Rs provides a new opportunity for harnessing an endogenous protective immune mechanism in the treatment of sepsis.—Csóka, B., Németh, Z. H., Törő, G., Idzko, M., Zech, A., Koscsó, B., Spolarics, Z., Antonioli, L., Cseri, K., Erdélyi, K., Pacher, P., Haskó, G. Extracellular ATP protects against sepsis through macrophage P2X7 purinergic receptors by enhancing intracellular bacterial killing.
Keywords: connexin, inflammasome, inflammation, pannexin, phagocytosis
Sepsis is a serious medical condition caused by the microbial invasion of normally sterile body parts. It has been estimated that between 28 and 50% of the ∼700,000 people who develop sepsis annually die—far more than the number of U.S. deaths from prostate cancer, breast cancer, and AIDS combined (1–3). Patients with sepsis are generally hospitalized for extended periods, rarely leaving the intensive care unit for 2–3 wk (2). Accordingly, sepsis represents a major burden to the U.S. health care system, with costs estimated to be approximately $16.7 billion per year (4).
Current concepts suggest that organ failure and mortality in sepsis are caused by inappropriate regulation of the immune system (5, 6). This dysregulation manifests as an inability to control bacterial growth and dissemination and by excessive inflammation, processes that are interrelated and are caused, in large part, by macrophage dysfunction. There are 2 main signaling pathways that initiate macrophage activation in sepsis: one is triggered by pathogen-associated molecular patterns (PAMPs), the other one by host danger-associated molecular patterns (DAMPs) (7). Although PAMPs have been the usual focus of sepsis research, recent evidence points to a novel role for DAMPs (7). DAMPs comprise a diverse group of molecules that accumulate in the extracellular space in response to bacteria-mediated tissue destruction, trauma, and burns, all of which are associated with sepsis. Examples of DAMPs include heat shock proteins, high-mobility group box 1, hyaluronan fragments, uric acid, and ATP.
ATP is released from the intracellular into the extracellular space during inflammation, infection, and shock, all of which are associated with sepsis (8, 9). Multiple mechanisms have been proposed to mediate ATP release during immune activation, with connexin (Cx) hemichannels and pannexin (Panx) channels receiving major focus (10, 11). Detection of released ATP by P2 purinergic receptors on inflammatory cells alerts the immune system to danger and initiates and orchestrates host immunity and inflammation. P2 receptors fall into 2 classes: the ionotropic P2X receptors (P2X1–7R) and the G protein-coupled P2YRs (P2Y1, -2, -4, -6, -11, -12, -13, and -14) (9).
P2X7Rs are the principal immunoregulatory P2Rs and are expressed at the highest levels on cells of the immune system (12). Macrophages have 4 to 5 times greater expression of P2X7Rs than do B, T, and NK cells (12). The expression of P2X7Rs on neutrophils is controversial. Some studies have shown that it is present intracellularly but not on the cell surface (12, 13), whereas one study showed that it has antiapoptotic effects in neutrophils (14). Activation of macrophages is associated with increased expression of P2X7Rs (15).
One of the best characterized aspects of the P2X7R’s function is its ability to activate the nucleotide-binding domain leucine-rich repeat-containing receptor family member P3 (NLRP3) inflammasome assembly in macrophages, thereby initiating caspase-1-mediated IL-1β and -18 processing and release (16, 17). In addition, multiple lines of evidence document an emerging role for P2X7Rs in augmenting the recognition, phagocytosis, and killing of pathogens by macrophages (18–20). Recent studies have proposed that ATP controls pathogens through inflammasome-dependent pyroptosis (21). According to this concept, ATP causes the activation of caspase-1, which then leads to pyroptotic macrophage death, characterized by a rapid loss of cell membrane integrity and release of cytosolic contents, including bacteria. The released bacteria can then be ingested and killed by recruited neutrophils in a manner that is independent of both IL-1β and -18 (22).
Despite the mounting evidence supporting an important role of ATP and P2X7Rs in regulating inflammation and bacterial phagocytosis and elimination by macrophages in vitro, the role of ATP and P2X7Rs in regulating the host’s response to sepsis is unclear. In this study, we tested the hypothesis that endogenously released ATP controls the host’s inflammatory response to sepsis through P2X7 signaling on macrophages.
MATERIALS AND METHODS
Experimental animals, drugs, and cell culture
Breeders of P2X7-knockout (KO), caspase-1 KO, and LysM-Cre mice and C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and maintained at the animal facility at Rutgers New Jersey Medical School. The P2X7fl/fl mice were from the European Mouse Mutant Archive (EMMA ID 05116; Monterotondo, Italy) (23). P2X7fl/fl-LysM-Cre mice were generated by crossing the 2 strains. All mice were bred and all colonies were maintained in accordance with the recommendations of the U.S. National Institutes of Health’s Guide for the Care and Use of Laboratory Animals, and the experiments were approved by the New Jersey Medical School Animal Care Committee. Oxidized ATP (oxi-ATP), ATP-magnesium salt (Mg-ATP), 2′(3′)-O-(4-benzoylbenzoyl)adenosine 5′-triphosphate triethylammonium salt (Bz-ATP), YVAD-chloromethyl ketone (CMK), uricase, uric acid, and probenecid were from Sigma-Aldrich (St. Louis, MO, USA), and Gap27 was from Tocris Bioscience (Bristol, United Kingdom). Thioglycollate-elicited peritoneal macrophages from C57BL6/J mice were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 50 U/ml penicillin, 50 µg/ml streptomycin, and 1.5 mg/ml sodium bicarbonate in a humidified atmosphere of 95% air and 5% CO2.
Preparation of uric acid crystals
Uric acid (1.25 g; Sigma-Aldrich) was dissolved in 50 ml 1 M NaOH at 55°C and was continuously stirred with a magnetic stirrer. The hot, milky, white solution was filtered through a 0.2 µm filter, and the filtered solution was left at room temperature with slow stirring. After centrifugation at 500 g, the supernatant was discarded, and the crystal pellet was rinsed with ice-cold sterile water. The pellet was air-dried and dissolved in PBS at a 1 mg/ml concentration. After vigorous vortexing, the crystals were sonicated with an ultrasonic sonicator for 30 s (5 times). The uric acid crystals were examined under a phase/polarizing microscope to evaluate their uniformity.
Cecal ligation and puncture
Polymicrobial sepsis was induced by subjecting mice to cecal ligation and puncture (CLP) (24, 25). P2X7-KO, caspase-1-KO, wild-type (WT), P2X7fl/fl-LysM-Cre, and P2X7fl/fl male mice between the ages of 8 and 12 wk were anesthetized with pentobarbital (50 mg/kg i.p.). Under aseptic conditions, a 2-cm midline laparotomy was performed to allow exposure of the cecum with adjoining intestine. Approximately two-thirds of the cecum was tightly ligated with a 3-0 silk suture, and the ligated part of the cecum was perforated twice (through and through) with a 20 1/2-gauge needle (BD Biosciences, San Jose, CA, USA). The ligated cecum was gently squeezed to extrude a small amount of feces through the perforation site and was returned to the peritoneal cavity, and the laparotomy was closed in 2 layers with 4-0 silk sutures. Sham-operated animals underwent the same procedure without ligation or puncture of the cecum. After the operation, all mice were resuscitated with an injection of physiologic saline (1 ml s.c.) and returned to their cages, where they were provided free access to food and water. In experiments where biochemical, immunologic, and bacteriological analysis were performed, the mice were reanesthetized with pentobarbital (50 mg/kg i.p.) 6 or 16 h after the CLP procedure, and blood, peritoneal lavage fluid, and various organs were harvested. A separate set of WT and P2X7-KO mice were used in survival studies. The effect of oxi-ATP, Mg-ATP, Bz-ATP, uricase, uric acid, Gap27, and probenecid was evaluated in male C57BL/6J mice in a fashion similar to that described for the KO or WT mice. In these experiments, the mice were injected intraperitoneally with the various agents or their vehicle (physiologic saline for uricase and uric acid and DMSO for the other drugs) 30 min before the CLP operation (26).
ATP measurement
At 16 h after CLP or the sham operation, blood samples were collected into heparinized tubes. Serum was separated by centrifugation, and serum ATP was measured with the ATPlite Luminescence ATP Detection Assay System (PerkinElmer, Waltham, MA, USA).
Generation of P2X7-KO bone marrow chimeric mice
Bone marrow chimeras were generated as described elsewhere (27). In brief, male donor mice (8- to 10-wk-old WT or P2X7-KO) were euthanized, and bone marrow from the femur was harvested by flushing the marrow cavity with sterile isotonic NaCl solution. The bone marrow cells were centrifuged at 400 g for 5 min, resuspended, and counted. Recipient mice (8- to 10-wk-old WT mice) were irradiated with a total dose (in 2 doses) of 12 Gy delivered from a [137Cs] source. Bone marrow cells (107/recipient) were injected retro-orbitally in 0.2 ml physiologic saline. The resulting chimeric mice were housed for at least 8 wk before experimentation and were fed with water containing tetracycline (100 μg/ml) in the first 2 wk after bone marrow transplantation. The chimeric mice were subjected to CLP and euthanized 16 h later, as described above.
Adoptive transfer of peritoneal macrophages
Thioglycollate-elicited peritoneal cells (28) from donor WT and P2X7-KO mice were harvested in PBS. Purified CD11b+ cells were obtained by positive selection with magnetic beads coated with anti-CD11b Ab (Miltenyi Biotech, Auburn, CA, USA), according to the manufacturer’s protocol. Purified CD11b+ cells were resuspended in PBS, and 4.5 × 106 cells were injected intraperitoneally to separate groups of recipient WT mice 2 h before subjecting them to CLP.
Collection of blood, peritoneal lavage fluid, and organs
After opening the chest of each mouse, blood samples were obtained aseptically by cardiac puncture with heparinized syringes. The blood samples were placed into heparinized microcentrifuge tubes and kept on ice until further processing for bacteriological analysis. Serial dilutions for bacteriological analysis were made as described in several publications (24, 25, 27, 29). The blood samples were centrifuged at 2000 g for 10 min, and the recovered plasma was stored at –70°C until further use. For collection of peritoneal lavage fluid, the abdominal skin was cleansed with 70% ethanol, and the abdominal wall was exposed by opening the skin. Two milliliters of sterile physiologic saline was slowly injected into the peritoneal cavity via an 18 gauge needle. The abdomen was gently massaged for 1 min while the tip of the needle was kept in the peritoneum. In the next step, the peritoneal fluid was recovered through the needle and placed on ice until processing for bacteriological examination. After making serial dilutions of the lavage fluid and blood to determine the number of colony-forming units (CFUs), the fluids were centrifuged at 5000 g for 10 min, and the supernatant was stored at –70°C until further analysis. Heart, lung, kidney, and spleen samples were excised and snap frozen in liquid nitrogen.
Quantification of bacterial CFUs in blood and peritoneal lavage fluid
Blood and peritoneal lavage fluid were diluted serially in sterile physiologic saline. Fifty microliters of each dilution was aseptically plated and cultured on trypticase blood agar plates (BD Biosciences) at 37°C. After 12–16 h of incubation, the number of bacterial colonies was counted. The number of cultures is expressed as CFUs per milliliter of blood or peritoneal lavage fluid.
Ex vivo phagocytosis assay and flow cytometry of peritoneal cells
At 16 h after CLP, peritoneal exudates were harvested in 2 ml PBS and spun down at 150 g for 5 min, to remove most of the bacterial content. The peritoneal cells were resuspended in 1 ml PBS and passed through a 70 µm nylon mesh to remove tissue debris. After brief centrifugation at 250 g, the cells were resuspended in 1 ml PBS containing 107 FITC-labeled Escherichia coli (Life Technologies, Carlsbad, CA, USA). The cells were incubated with bacteria at 37°C for 30 min, after which phagocytosis was stopped by placing the samples on ice. Nonphagocytosed E. coli was removed by centrifugation at 150 g for 5 min, and the peritoneal cells were resuspended in 0.5 ml PBS containing 1% FBS for flow cytometry. After Fc blocking with an anti-CD16/CD32 antibody (eBioscience, San Diego, CA, USA), the peritoneal cells were labeled with phycoerythrin (PE)-conjugated anti-F4/80 and allophycocyanin-conjugated Ly6G antibody (eBioscience). The percentage of macrophages, neutrophils, and cell-specific phagocytosis in the lavage fluid was quantified by flow cytometry. At least 10,000 events were recorded for each analysis.
Assessment of MPO activity and histology of the lung
Lung samples obtained 6 h after CLP were homogenized in extraction buffer [20 mM acetate buffer (pH 4.7) containing 0.2 M NaCl, 0.5% cetyltrimethylammonium bromide, 10 μg/ml PMSF, and 1 mM EDTA] at a concentration of 100 mg/ml. A myeloperoxidase (MPO) activity assay kit (Northwest Life Science Specialties, Vancouver, WA, USA) was used for MPO determination, according to the instructions provided with the kit.
Determination of ALT and BUN
Alanine aminotransferase (ALT) and blood urea nitrogen (BUN) were analyzed with a clinical chemistry analyzer system (VetTest8008; IDEXX Laboratories, Totowa, NJ, USA) (25).
Protein extraction for cytokine ELISA and Western blot analysis
Organs were homogenized in a Dounce homogenizer (Kontes Glass Co., Vineland, NJ, USA) in modified radioimmunoprecipitation assay buffer [50 mM Tris HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 0.25% sodium deoxycholate, 1% Nonidet P-40, 1 μg/ml pepstatin, 1 μg/ml leupeptin, 1 mM PMSF, and 1 mM Na3VO4]. The lysates were centrifuged at 15,000 g for 15 min, and the supernatant was recovered. The Bio-Rad (Hercules, CA, USA) protein assay kit was used to determine the protein concentrations. Protein samples (20 μg) from spleen were separated on 8–12% Tris-glycine gels (Life Technologies) and transferred to nitrocellulose membranes. The membranes were probed with rabbit polyclonal anti-cleaved poly(ADP-ribose) polymerase (PARP) Ab from Cell Signaling Technology (Danvers, MA, USA). After several wash cycles, the nitrocellulose membranes were incubated with a secondary horseradish peroxidase (HRP)–conjugated anti-rabbit Ab (Santa Cruz Biotechnology, Santa Cruz, CA, USA). To assess equal protein loading, we used HRP-conjugated polyclonal goat anti-β-actin Ab from Santa Cruz Biotechnology. Bands were detected with ECL Western Blot Detection Reagent (GE Healthcare, Piscataway, NJ, USA).
In vivo and in vitro chemotaxis assays
In vitro migration of thioglycollate-elicited peritoneal macrophages obtained from WT and P2X7-KO mice toward 100 ng/ml monocyte chemotactic protein (MCP)-1 (R&D Systems, Minneapolis, MN, USA) was measured with a QCM Chemotaxis 96 well Cell Migration Assay (Millipore, Billerica, MA, USA), on migration filters with a pore size of 5 μm, according to the manufacturer’s protocol. In brief, 100 ng/ml MCP-1 or its vehicle was added in the feeder tray, and 105 peritoneal macrophages were seeded into the cell migration chamber. Thereafter, the migration chamber was placed into the feeding tray and the plates were incubated at 37°C for 4 h. After incubation, the transmigrated cells were lysed, and genomic DNA-specific fluorescent dye was added to the lysate. The fluorescence intensity of the cell extracts was measured at 480 nm/520 nm.
For the in vivo chemotaxis assay, WT and P2X7-KO mice were injected with 3% thioglycollate (1 ml i.p.), to elicit sterile peritonitis, with the macrophage count peaking on d 4 (30). After d 4, the mice were injected with 400 ng LPS per mouse as an inflammatory stimulus to induce efflux of leukocytes from the peritoneum to the draining lymph nodes. After 3 h, the peritoneal cells were collected by lavage and counted. The frequency of macrophages in the lavage fluid was quantified by flow cytometry with PE-conjugated antibody to F4/80 (eBioscience). At least 10,000 events were recorded for each analysis.
Ex vivo bacteria-killing assay
Peritoneal cells were recovered in 2 ml of saline after CLP. Extracellular bacteria were removed by centrifugation at 100 g for 10 min. Peritoneal cell counts were determined, and the density of the cells was adjusted to 106/ml with saline. The cells were incubated on ice for 2 h in the presence of 100 μg/ml gentamicin to kill the remaining extracellular and cell-surface–attached bacteria. Thereafter, gentamicin was removed by washing the cells 3 times with 1 ml saline, and the cells were lysed in 100 μl of 100 mM Tris-HCl/1% Triton-X100. The cell lysates were diluted serially in sterile physiologic saline, and 50 μl of each dilution was aseptically plated and cultured on trypticase blood agar plates (BD Biosciences) at 37°C. After 12–16 h of incubation, the number of bacterial colonies was counted. The number of cultures is expressed as CFUs per 106 peritoneal cells.
Determination of cytokine and chemokine levels
Concentrations of TNF-α; IL-1β, -10, -6, and -12p40; macrophage inflammatory proteins (MIP)-1α and -2; and MCP-1, in blood, peritoneal lavage fluid, organ extracts (heart, kidney, and lung), and supernatants from peritoneal macrophages were determined with commercially available ELISA kits (Tocris Bioscience) according to the manufacturer’s instructions.
Statistical Analysis
Survival statistics were determined by the Kaplan-Meier curve and log-rank test. To compare cytokine concentrations, the number of CFUs, and all other laboratory parameters, as well as densitometry read outs, we used Student’s t test or ANOVA followed by the Mann-Whitney test. Statistical significance was assigned to P < 0.05.
RESULTS
P2X7R signaling improves survival and decreases bacterial burden and the levels of inflammatory cytokines and chemokines in CLP-induced sepsis
To begin to examine the role of ATP in sepsis, we measured its extracellular accumulation after CLP, a clinically relevant model of polymicrobial sepsis (31). We found that plasma levels of ATP increased in response to CLP (Fig. 1A), which indicates that ATP accumulated in the extracellular space during sepsis. To study the role of this endogenously released ATP in sepsis, we subjected WT and P2rx7−/− (P2X7-KO) mice to CLP and monitored their survival. We observed that the survival rate of the WT mice was higher than that of the P2X7-KO animals, indicating that ATP interaction with P2X7Rs is protective (Fig. 1B). We then investigated the host’s immune response at 16 h after the CLP procedure, at which time bacterial dissemination and inflammation are at their maximum (29). We found that the increased survival of WT vs. P2X7-KO mice was associated with a decreased bacterial burden in the peritoneal cavity of WT vs. P2X7-KO animals (Fig. 1C) and a trend toward decreased bacterial load in the blood of WT vs. P2X7-KO mice, indicating that P2X7Rs control bacterial burden. We then assessed the inflammatory status of the mice by measuring levels of inflammatory mediators. We found that P2X7-KO mice had higher levels of inflammatory cytokines and chemokines in blood and peritoneum at 16 h (Fig. 1D–K). Similar results were found at 6 h (Supplemental Fig. S1B–D).
Figure 1.

P2X7Rs decrease mortality, bacterial load, and inflammatory cytokines in sepsis. A) ATP accumulates in the plasma following 16 h of polymicrobial sepsis induced by CLP. *P < 0.05 (WT and P2X7-KO; n = 5 and 8, respectively). B) P2X7-KO mice showed shorter survival time than WT mice. WT and P2X7-KO males were subjected to CLP, and survival was monitored for 7 d. *P < 0.05 (WT and P2X7-KO; n = 10 and 11, respectively). C) Bacterial burden was determined by counting the number of CFUs on blood agar plates after serial dilution of blood and peritoneal lavage samples. Blood and lavage fluid were collected at 16 h after CLP. *P < 0.05 vs. WT (WT and P2X7-KO; n = 7 and 6, respectively, for blood; n = 7 and 5, respectively, for lavage). WT and P2X7-KO animals were subjected to CLP and (D) TNF-α, (E) IL-1β, (F) IL-12p40, (G) IL-6, (H) IL-10, (I) MCP-1, (J) MIP-1α, and (K) MIP-2 levels were measured with ELISAs of blood and lavage fluid collected at 16 h after CLP. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. WT (WT and P2X7-KO; n = 12 and 8, respectively). Data are expressed as means ± sem.
P2X7Rs control sepsis-induced organ injury and inflammation
We next monitored organ inflammation and injury, which are direct causes of mortality in sepsis. Lung inflammatory cytokine levels were increased in P2X7-KO mice compared with those in WT mice (Fig. 2A–D). P2X7-KO mice also exhibited increased neutrophil influx, as detected by the MPO assay (32) (Fig. 2E). Liver (Fig. 2F) and kidney (Fig. 2G) injury increased, as indicated by the respective increases in plasma ALT and BUN (33). Apoptosis in the spleen was higher in the P2X7-KO mice than in their WT counterparts, as detected by measuring PARP cleavage (Fig. 2H). Flow cytometric analysis of splenocytes 16 h after CLP revealed a decrease in the number of CD4+, CD8+, and CD19+ cells in spleens of P2X7-KO mice when compared with those of WT mice (Fig. 2I–K) indicating that P2X7R signaling decreased CLP-induced lymphocyte apoptosis. Because there was no difference in inflammation of the heart between P2X7-KO and WT mice (data not shown), these data indicate that P2X7R controls organ injury and inflammation in an organ-specific manner.
Figure 2.

P2X7Rs protect against sepsis-induced organ injury and inflammation. IL-6 (A), MCP-1 (B), MIP-1α (C), and MIP-2 (D) levels in the lung of WT and P2X7-KO animals 16 h after CLP, as determined by ELISA (WT and P2X7-KO; n = 12 and 8, respectively). E) MPO levels in the lungs of WT and P2X7-KO animals 6 h after CLP. The level was determined in tissue lysates with a commercial kit (WT and P2X7-KO; n = 10 and 9, respectively). ALT (F) and BUN (G) levels in the plasma of P2X7-KO vs. WT mice 16 h after CLP (WT and P2X7-KO; n = 8 and 9, respectively, for ALT; n = 8 and 8, respectively, for BUN). H) The proteolytic cleavage of PARP in spleen was examined by Western blot analysis with a specific antibody. Percentage of CD4+ (I), CD8+ (J), and CD19+ (K) cells in the spleen of WT and P2X7-KO animals 16 h after CLP was determined by flow cytometry (WT and P2X7-KO; n = 8 and 9, respectively). *P < 0.05 and **P < 0.01 vs. WT. Data are expressed as means ± sem.
Pharmacological antagonism and agonism confirm the anti-inflammatory and antibacterial effects of P2X7Rs after CLP
To corroborate our findings with genetic inactivation (KO) of P2X7Rs, we inactivated the receptors by using the antagonist oxi-ATP (40 mg/kg i.p.). IL-1β levels were comparable in oxi-ATP- and vehicle-treated mice (Fig. 3A), but oxi-ATP-treated mice displayed increased inflammatory cytokine and chemokine levels and trended toward an increased bacterial burden when compared with that in vehicle-treated mice (Fig. 3B–F). We then hypothesized that activating P2X7Rs with exogenous agonists would have an effect opposite to that of inactivating these receptors with KO or an antagonist (i.e., decreased inflammation). To examine this hypothesis, we injected mice with the P2X7 agonists Mg-ATP (a nonspecific P2X7 agonist) or Bz-ATP (a specific P2X7 agonist) before performing CLP. Neither Mg-ATP nor Bz-ATP affected CLP-induced IL-1β production (Fig. 3G). Both Mg-ATP (100 mg/kg) and Bz-ATP (10 mg/kg) decreased inflammatory cytokine levels and CFUs (Fig. 3H–L). The data from the pharmacological antagonism and agonism experiments confirm the anti-inflammatory and antibacterial effects of P2X7Rs in sepsis.
Figure 3.
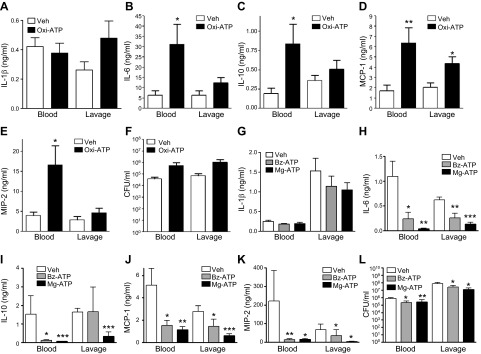
Pharmacological manipulation of P2X7Rs shows that they are anti-inflammatory in sepsis. The P2X7 antagonist oxi-ATP was administered 30 min before CLP, and IL-1β (A), IL-6 (B), IL-10 (C), MCP-1 (D), MIP-2 (E), and CFUs (F) were measured 16 h after CLP (vehicle and oxi-ATP treatment; n = 7 and 7, respectively). The P2X7 agonist Mg-ATP or Bz-ATP was injected 30 min before CLP, and IL-1β (G), IL-6 (H), IL-10 (I), MCP-1 (J), MIP-2 (K) (vehicle, Mg-ATP, and Bz-ATP; n = 8, 9, and 10, respectively), and CFUs (L) (vehicle, Mg-ATP, and Bz-ATP; n = 11, 12, and 11, respectively) were evaluated 16 h after CLP. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. vehicle. Data are expressed as means ± sem.
P2X7R signaling on macrophages suppresses sepsis-induced inflammation
P2X7Rs were initially thought to be restricted to hematopoietic cells; however, they have been found in a recent study on other cell types, including fibroblasts, endothelial cells, epithelial cells, glia, and neurons (15). Thus, to study whether P2X7Rs on hematopoietic cells or other cell types are necessary for controlling inflammation, we generated P2X7R bone marrow chimeric mice by transferring P2X7-KO or WT bone marrow into irradiated WT mice (27). P2X7-KO→P2X7 WT chimeras that have KO bone marrow and WT parenchyma exhibited higher cytokine and chemokine levels than did the P2X7 WT→P2X7 WT mice with WT parenchyma and WT bone marrow (Fig. 4A, C–E). The only exception was IL-1β, with comparable levels in the P2X7-KO→P2X7 WT and P2X7 WT→P2X7 WT mice (Fig. 4B). This observation demonstrates that P2X7R signaling on bone marrow-derived cells controls inflammation in sepsis.
Figure 4.

P2X7 receptor signaling on macrophages is anti-inflammatory. A–E) Blood and lavage fluid were obtained from WT→WT and P2X7-KO→WT bone marrow chimeric mice 16 h after CLP, and cytokines were measured by ELISA. *P < 0.05; **P < 0.01 vs. WT→WT bone-marrow chimeras (P2X7 WT→P2X7 WT and P2X7 KO→P2X7 WT; n = 8 and 7, respectively). F–J) CLP-induced sepsis increased cytokine levels in WT recipients of adoptively transferred P2X7-KO macrophages more than in WT recipients of WT macrophages (Mϕ). Cytokines were measured 16 h after CLP. *P < 0.05; **P < 0.01 vs. WT Mϕ→WT transfer (n = 10-10 recipient mice in each group). K–P) P2X7fl/−-LysM-Cre+/− and P2X7fl/−-LysM-Cre−/− mice were interbred to produce P2X7fl/fl-LysM-Cre+/− (P2X7fl/fl -LysM-Cre) mice and control P2X7fl/fl-LysM-Cre−/− (P2X7fl/fl) mice. Blood and peritoneal lavage fluid were collected at 16 h after CLP. *P < 0.05; **P < 0.01 vs. P2X7fl/fl (P2X7fl/fl and P2X7fl/fl-LysM-Cre mice; n = 8 and 10, respectively). Data are expressed as means ± sem.
Macrophages are cells of hematopoietic origin and have been widely implicated in sepsis. In addition, macrophages are the primary P2X7R-expressing cell type (12). Thus, we hypothesized that P2X7R signaling on macrophages would be essential for controlling inflammation during sepsis. To address this hypothesis, we adoptively transferred peritoneal macrophages from donor WT and P2X7-KO mice to separate groups of recipient WT mice before subjecting the recipient mice to CLP. We observed that WT macrophage-recipient WT mice showed decreased levels of inflammatory cytokines when compared with WT mice receiving P2X7-KO macrophages (Fig. 4F–J). The exception was IL-1β, whose levels were comparable in WT and P2X7-KO macrophage-recipient mice (Fig. 4G). This finding corroborates the notion that the P2X7R on macrophages suppresses inflammation.
To further analyze the role of P2X7Rs within the hematopoietic cell compartment, we generated mice with the P2X7R gene deleted in myeloid cells (Supplemental Fig. S2) (P2X7fl/fl-LysM-Cre mice) by crossing mice expressing the Cre recombinase under the control of the myeloid-specific LysM promoter (LysM-Cre) and mice bearing “floxed” P2X7Rs (P2X7fl/fl). Our results demonstrated that, after CLP, the P2X7fl/fl-LysM-Cre mice had increased levels of cytokines and higher bacterial counts than those of the control P2X7fl/fl mice (Fig. 4K–P). Taken together, our results with bone marrow and macrophage transfer and cell-specific KO show that P2X7R signaling on macrophages suppresses inflammation.
The protective effect of P2X7Rs is independent of the NLRP3 inflammasome
Since P2X7Rs on macrophages are well-known activators of the NLRP3 inflammasome and are involved in caspase-1-mediated IL-1β release, we tested the role of the NLRP3 inflammasome. First, we compared IL-1β levels in WT and P2X7-KO mice after sepsis. Our results showed that IL-1β levels were increased in KO animals compared with WT animals in the peritoneum and that the IL-1β levels in blood were not significantly different (Supplemental Fig. S1A), indicating that P2X7Rs are not responsible for mediating IL-1β release in sepsis. To further study the role of the inflammasome, we subjected WT and caspase-1−/− (caspase-1-KO) mice to CLP. We found that, although IL-1β in the lavage was lower in caspase-1 KO mice, IL-6, IL-10, and MCP-1 levels were comparable between caspase-1-KO and WT mice (Supplemental Fig. S1B–H). Thus, although caspase-1 drives IL-1β in the peritoneum, it does not affect the production of other inflammatory cytokines. Using a caspase-1-specific inhibitor (YVAD-CMK; 10 mg/kg), we found that caspase-1 inhibition had no effect on the production of peritoneal inflammatory cytokines (Supplemental Fig. S1I–L). Moreover, inhibition of the endogenous inflammasome activator uric acid by uricase (100 U/mouse) or exogenous uric acid crystals (1 mg/mouse) failed to alter inflammation in sepsis (Supplemental Fig. S1M–P).
P2X7Rs on myeloid cells augments killing of intracellular bacteria in sepsis
We then studied the mechanisms by which P2X7Rs decrease inflammation in sepsis. First we tested the possibility that P2X7R activation decreases macrophage activation directly. We pretreated LPS-stimulated peritoneal macrophages with Bz-ATP and measured cytokine production and found that it failed to affect their expression (Fig. 5A). In addition, cytokine production by naïve or E. coli-stimulated peritoneal cells obtained from septic P2X7-KO and WT mice was comparable (Supplemental Fig. S3A–D). We next hypothesized that P2X7Rs decrease inflammation by reducing inflammatory cell recruitment to the inflammatory focus, in this case, the peritoneum. We found that P2X7-KO mice had increased macrophage but not neutrophil counts in the peritoneal cavity (Fig. 5B, C), whereas no differences were detected in the number of cells in blood (data not shown). This result indicates that P2X7Rs decrease macrophage infiltration into the peritoneum. We next investigated whether P2X7Rs cause decreased macrophage recruitment to the peritoneum by decreasing chemotaxis. Using both an in vivo and in vitro chemotaxis assay, we found that P2X7Rs failed to affect macrophage chemotaxis (Fig. 5D, E). Because we found that P2X7Rs controlled bacteria in the peritoneum, we posited that P2X7Rs decrease macrophage recruitment by decreasing bacterial growth. One possibility is that this decrease occurs through increased phagocytosis by macrophages or neutrophils, the 2 major phagocytic cell types in sepsis. We found that whereas macrophages from septic WT and P2X7-KO mice phagocytosed bacteria equally efficiently, P2X7-KO neutrophils were actually more phagocytic than WT neutrophils (Fig. 5F, G). Thus, it is unlikely that P2X7Rs control bacterial counts by interfering with phagocytosis, which led us to hypothesize that P2X7Rs decrease bacteria by increasing their killing by macrophages. To test this hypothesis, we obtained peritoneal cells from WT and P2X7-KO animals after sepsis and enumerated live bacteria in the recovered cells. We found increased counts of live bacteria in P2X7-KO vs. WT cells (Fig. 5H). Moreover, our data demonstrate that P2X7fl/fl-LysM-Cre mice had higher intracellular bacterial counts than did control P2X7fl/fl mice after CLP (Fig. 5I). Altogether, our results indicate that P2X7R signaling on myeloid cells augments intracellular killing of bacteria in sepsis.
Figure 5.

Role of P2X7Rs in regulating macrophage cytokine production, chemotaxis, and phagocytosis. A) Peritoneal macrophages were stimulated with LPS (100 ng/ml) in the presence or absence of Bz-ATP (100 μM) for 6 h (n = 4 wells/ group). IL-6 from the supernatants was measured by ELISA. Macrophage (F4/80+) (B) and neutrophil (C) (Ly-6G+) counts in the peritoneum after 16 h of CLP were determined by flow cytometry. *P < 0.05 vs. WT (WT and P2X7-KO; n = 8). D) WT and P2X7-KO mice had comparable emigration of macrophages from the peritoneal cavity in vivo. Macrophage emigration was induced by intraperitoneal LPS injection. ***P < 0.001 vs. vehicle (vehicle- and LPS-treated macrophages; n = 4 and 6, respectively). E) In vitro chemotaxis of macrophages toward MCP-1. Chemotaxis of peritoneal macrophages from WT or P2X7-KO mice was assessed with a commercial chemotaxis assay kit. *P < 0.05 vs. vehicle (n = 6). F, G) Ex vivo phagocytosis of labeled E. coli by macrophages and neutrophils. At 16 h after CLP, peritoneal cells were obtained and incubated with labeled E. coli. Phagocytosed E. coli in macrophages (F) (F4/80+) or neutrophils (G) (Ly-6G+) was detected by flow cytometry. *P < 0.05 vs. WT (WT and P2X7-KO; n = 8 and 8, respectively). H, I) Ex vivo determination of bacterial killing ability of peritoneal cells. At 16 h after CLP, peritoneal cells were obtained from P2X7-KO and WT mice (H) and P2X7fl/fl -LysM-Cre and P2X7fl/fl (I), and mice and intracellular CFUs were determined. *P < 0.05 vs. WT; **P < 0.01 vs. P2X7fl/fl (P2X7fl/fl and P2X7fl/fl -LysM-Cre or WT and P2X7-KO; n = 9 and 11, respectively). Data are expressed as means ± sem.
Cx channel inhibition increases bacterial burden and the levels of inflammatory cytokines and chemokines in CLP-induced sepsis
Because Cx and Panx channels have been shown to be the main conduits for extracellular release of ATP during inflammation, we began to assess the role of these ATP-release channels in regulating sepsis. We found that blocking Cx channels with the peptide mimetic blocker Gap27 reproduced the effect of P2X7 blockade, as it increased CFUs and IL-6 and -10 concentrations in sepsis (Fig. 6). However, Panx blockade with probenecid (1 and 10 mg/kg) was without effect (data not shown).
Figure 6.

Blockade of Cx channels increases cytokines and bacterial counts in sepsis. At 30 min before CLP, Gap27 was injected (0.1 mg/kg i.p.), and cytokines (A–D) and CFUs (E) were determined in the peritoneal lavage fluid 16 h after CLP. *P < 0.05; **P < 0.01 vs. vehicle (veh) (vehicle- and Gap27-treated; n = 8 and 10, respectively). Data are expressed as means ± sem.
DISCUSSION
Inefficient elimination of bacteria and dysregulation of the immune system in sepsis lead to a primarily macrophage-mediated systemic inflammatory response syndrome, which describes the underlying massive inflammatory reaction of the body that contributes to the development of organ failure and shock (34, 35). A dysregulated immune response coexisting with increased inflammation also occurs in trauma or burn patients in whom symptoms of sepsis develop, but no infectious agent is detected (36). In these patients, leukocytes display a prolonged decrease in the expression of genes involved in antigen presentation, antibacterial defense mechanisms, and a prolonged increase in the expression of inflammatory genes (37). Knowing that P2X7Rs regulate the macrophage inflammatory response and control bacteria, we tested the hypothesis that ATP release and subsequent activation of P2X7Rs are important in curbing excessive inflammation and the growth of bacteria in sepsis. Data yielded by both loss-of-function and gain-of-function approaches showed that P2X7R activation is crucial for the control of mortality, systemic inflammation, and bacterial growth in sepsis. Moreover, our results with P2X7-KO bone marrow chimeric animals, adoptive transfer of macrophages, and myeloid-specific P2X7-KO mice indicate that P2X7R signaling on macrophages is necessary for the protective effect of P2X7Rs.
ATP is degraded to AMP by ectonucleoside triphosphate diphosphohydrolase 1 (CD39) (38). We recently demonstrated that CD39 decreases inflammation in sepsis (25, 39). This is counterintuitive to our current results, which suggest that ATP itself and not its degradation is anti-inflammatory. However, it should be noted that, in the current study, we looked only at the role of one ATP receptor, the P2X7R. It is possible that other P2 receptors affect sepsis differently. Therefore, more studies are needed to delineate the exact role of ATP and P2 receptors in controlling inflammation in sepsis.
We found increased ATP levels in the plasma of septic mice, indicating increased extracellular ATP release. ATP release in response to immune activation occurs mostly through Cx and Panx channels, and immune cells are a rich source of extracellular ATP. Macrophages release ATP in response to Bacillus anthracis infection, which is blocked by both pharmacological Cx inhibition and siRNA-mediated Cx43 depletion (40). Eltzschig et al. (41) showed that neutrophils liberate ATP in response to N-formyl-Met-Leu-Phe (fMLP) or leukotriene B4 and that this ATP release is blocked by a peptide inhibitor of Cx43 or in Cx43-KO neutrophils. In another study, fMLP-induced ATP release by neutrophils was Panx1 dependent (42). T cells release ATP during their activation via Panx1 channels (43). Our data showing that Cx inhibition had an effect similar to that of P2X7R blockade indicate that Cx channels may be the primary conduits of ATP release. One caveat is that the inhibitors we used may not be absolutely specific for Cx or Panx channels. Further experiments are necessary to uncover the precise mechanisms, cell, and tissue origins of ATP release in sepsis.
P2X7Rs have been shown to mediate effective defense against several pathogens, both in vitro and in vivo, through different mechanisms (18–20). P2X7Rs have been documented to be important for macrophage phagocytosis of pathogens (44). However, it is unlikely that such a mechanism accounts for protection in sepsis, as our ex vivo results failed to demonstrate a stimulatory effect of P2X7Rs on macrophage or neutrophil phagocytosis. It has been shown that P2X7Rs regulate bacterial dissemination and inflammation by stimulating the emigration of macrophages and neutrophils to the site of inflammation (20). In our study, this emigration effect was not the case, as we did not find differences in the migration of WT and P2X7R-deficient macrophages or neutrophil infiltration of organs.
It has been shown in in vitro studies that P2X7Rs can augment the killing of obligate intracellular pathogens, such as Mycobacterium tuberculosis (19), Toxoplasma gondii (45), and Leishmania amazonensis (46) by macrophages (18). However, the role of P2X7Rs in the killing of bacteria that cause polymicrobial sepsis has been unknown. Our results now show that P2X7R can also augment the killing of bacteria that cause polymicrobial gut-origin sepsis and that are nonobligate intracellular pathogens. P2X7Rs can augment killing of bacteria through caspase-1-mediated macrophage inflammasome activation, pyroptosis, and subsequent neutrophil-mediated toxicity (21, 22). However, caspase-1 KO does not phenocopy P2X7-KO, and other inflammasome pathway modulators fail to alter septic inflammation, all of which suggests that P2X7 control of bacteria is independent of caspase-1, the inflammasome, or pyroptosis.
The suppressive effect of P2X7Rs on the systemic inflammatory response may also be through P2X7R signaling on macrophages, but independent of bacteria. However, our in vitro data argue against a direct macrophage-mediated effect, as P2X7R ligation failed to suppress macrophage cytokine production. Again, more studies are necessary to delineate the mechanisms by which P2X7Rs control bacteria and inflammation in the complex setting of sepsis.
While this paper was in preparation, another research group reported that P2X7Rs contribute to the development of the exacerbated inflammatory response in polymicrobial sepsis (47). The opposing results of our study and theirs may be caused by differences in experimental conditions.
In summary, a critical barrier to progress in the field of sepsis research is our incomplete understanding of endogenous immune regulatory mechanisms that have evolved to protect the host from attacks by pathogens. Our data indicate that ATP release and P2X7R activation are critical in limiting the dissemination of bacteria by enhanced killing of these pathogens and in preventing deadly inflammation during abdominal sepsis. Our results thus indicate that targeting P2X7Rs provides a new opportunity for harnessing an endogenous protective immune mechanism in the treatment of sepsis.
Supplementary Material
Acknowledgments
This work was supported by U.S. National Institutes of Health (NIH) Grant R01GM66189 from the National Institute of General Medical Sciences, U.S. Army Medical Research and Materiel Command Grant 09065004/W81XWH-10-1-1015, and Hungarian Scientific Research Fund Grants (OTKA) CK 78275 and K109178 (all to G.H.) and by funding from the Intramural Research Program of the NIH, National Institute on Alcohol Abuse and Alcoholism (to P.P.). The authors declare no conflicts of interests.
Glossary
- ALT
alanine aminotransferase
- BUN
blood urea nitrogen
- Bz-ATP
2′(3′)-O-(4-benzoylbenzoyl)adenosine 5′-triphosphate triethylammonium salt
- CFU
colony-forming unit
- CLP
cecal ligation and puncture
- CMK
chloromethyl ketone
- Cx
connexin hemichannel
- DAMPs
danger-associated molecular patterns
- FBS
fetal bovine serum
- KO
knockout
- fMLP
N-formyl-Met-Leu-Phe
- HRP
horseradish peroxidase
- MCP
monocyte chemotactic protein
- Mg-ATP
ATP-magnesium salt
- MIP
macrophage inflammatory protein
- MPO
myeloperoxidase
- NLRP3
nucleotide-binding domain leucine-rich repeat-containing receptor family member P3
- oxi-ATP
oxidized ATP
- P2X7R
P2X7 receptor
- PAMPs
pathogen-associated molecular patterns
- Panx
pannexin channel
- PARP
poly(ADP-ribose) polymerase
- PE
phycoerythrin
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Stearns-Kurosawa D. J., Osuchowski M. F., Valentine C., Kurosawa S., Remick D. G. (2011) The pathogenesis of sepsis. Annu. Rev. Pathol. 6, 19–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riedemann N. C., Guo R. F., Ward P. A. (2003) The enigma of sepsis. J. Clin. Invest. 112, 460–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van der Poll T., Opal S. M. (2008) Host-pathogen interactions in sepsis. Lancet Infect. Dis. 8, 32–43 [DOI] [PubMed] [Google Scholar]
- 4.Angus D. C., Linde-Zwirble W. T., Lidicker J., Clermont G., Carcillo J., Pinsky M. R. (2001) Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 29, 1303–1310 [DOI] [PubMed] [Google Scholar]
- 5.Benjamim C. F., Hogaboam C. M., Kunkel S. L. (2004) The chronic consequences of severe sepsis. J. Leukoc. Biol. 75, 408–412 [DOI] [PubMed] [Google Scholar]
- 6.Oberholzer A., Oberholzer C., Moldawer L. L. (2001) Sepsis syndromes: understanding the role of innate and acquired immunity. Shock 16, 83–96 [DOI] [PubMed] [Google Scholar]
- 7.King E. G., Bauzá G. J., Mella J. R., Remick D. G. (2014) Pathophysiologic mechanisms in septic shock. Lab. Invest. 94, 4–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.La Sala A., Ferrari D., Di Virgilio F., Idzko M., Norgauer J., Girolomoni G. (2003) Alerting and tuning the immune response by extracellular nucleotides. J. Leukoc. Biol. 73, 339–343 [DOI] [PubMed] [Google Scholar]
- 9.Idzko M., Ferrari D., Eltzschig H. K. (2014) Nucleotide signalling during inflammation. Nature 509, 310–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lohman A. W., Isakson B. E. (2014) Differentiating connexin hemichannels and pannexin channels in cellular ATP release. FEBS Lett. 588, 1379–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eugenin E. A. (2014) Role of connexin/pannexin containing channels in infectious diseases. FEBS Lett. 588, 1389–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gu B. J., Zhang W. Y., Bendall L. J., Chessell I. P., Buell G. N., Wiley J. S. (2000) Expression of P2X(7) purinoceptors on human lymphocytes and monocytes: evidence for nonfunctional P2X(7) receptors. Am. J. Physiol. Cell Physiol. 279, C1189–C1197 [DOI] [PubMed] [Google Scholar]
- 13.Martel-Gallegos G., Rosales-Saavedra M. T., Reyes J. P., Casas-Pruneda G., Toro-Castillo C., Pérez-Cornejo P., Arreola J. (2010) Human neutrophils do not express purinergic P2X7 receptors. Purinergic Signal. 6, 297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christenson K., Björkman L., Tängemo C., Bylund J. (2008) Serum amyloid A inhibits apoptosis of human neutrophils via a P2X7-sensitive pathway independent of formyl peptide receptor-like 1. J. Leukoc. Biol. 83, 139–148 [DOI] [PubMed] [Google Scholar]
- 15.Bartlett R., Stokes L., Sluyter R. (2014) The P2X7 receptor channel: recent developments and the use of P2X7 antagonists in models of disease. Pharmacol. Rev. 66, 638–675 [DOI] [PubMed] [Google Scholar]
- 16.Dubyak G. R. (2012) P2X7 receptor regulation of non-classical secretion from immune effector cells. Cell. Microbiol. 14, 1697–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bours M. J., Swennen E. L., Di Virgilio F., Cronstein B. N., Dagnelie P. C. (2006) Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 112, 358–404 [DOI] [PubMed] [Google Scholar]
- 18.Wiley J. S., Sluyter R., Gu B. J., Stokes L., Fuller S. J. (2011) The human P2X7 receptor and its role in innate immunity. Tissue Antigens 78, 321–332 [DOI] [PubMed] [Google Scholar]
- 19.Lammas D. A., Stober C., Harvey C. J., Kendrick N., Panchalingam S., Kumararatne D. S. (1997) ATP-induced killing of mycobacteria by human macrophages is mediated by purinergic P2Z(P2X7) receptors. Immunity 7, 433–444 [DOI] [PubMed] [Google Scholar]
- 20.Morandini A. C., Savio L. E., Coutinho-Silva R. (2014) The role of P2X7 receptor in infectious inflammatory diseases and the influence of ectonucleotidases. Biomed. J. 37, 169–177 [DOI] [PubMed] [Google Scholar]
- 21.Chen Q., Jin Y., Zhang K., Li H., Chen W., Meng G., Fang X. (2014) Alarmin HNP-1 promotes pyroptosis and IL-1β release through different roles of NLRP3 inflammasome via P2X7 in LPS-primed macrophages. Innate Immun. 20, 290–300 [DOI] [PubMed] [Google Scholar]
- 22.Miao E. A., Leaf I. A., Treuting P. M., Mao D. P., Dors M., Sarkar A., Warren S. E., Wewers M. D., Aderem A. (2010) Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skarnes W. C., Rosen B., West A. P., Koutsourakis M., Bushell W., Iyer V., Mujica A. O., Thomas M., Harrow J., Cox T., Jackson D., Severin J., Biggs P., Fu J., Nefedov M., de Jong P. J., Stewart A. F., Bradley A. (2011) A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Csóka B., Németh Z. H., Mukhopadhyay P., Spolarics Z., Rajesh M., Federici S., Deitch E. A., Bátkai S., Pacher P., Haskó G. (2009) CB2 cannabinoid receptors contribute to bacterial invasion and mortality in polymicrobial sepsis. PLoS ONE 4, e6409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haskó G., Csóka B., Koscsó B., Chandra R., Pacher P., Thompson L. F., Deitch E. A., Spolarics Z., Virág L., Gergely P., Rolandelli R. H., Németh Z. H. (2011) Ecto-5′-nucleotidase (CD73) decreases mortality and organ injury in sepsis. J. Immunol. 187, 4256–4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grenz A., Zhang H., Hermes M., Eckle T., Klingel K., Huang D. Y., Müller C. E., Robson S. C., Osswald H., Eltzschig H. K. (2007) Contribution of E-NTPDase1 (CD39) to renal protection from ischemia-reperfusion injury. FASEB J. 21, 2863–2873 [DOI] [PubMed] [Google Scholar]
- 27.Csóka B., Németh Z. H., Rosenberger P., Eltzschig H. K., Spolarics Z., Pacher P., Selmeczy Z., Koscsó B., Himer L., Vizi E. S., Blackburn M. R., Deitch E. A., Haskó G. (2010) A2B adenosine receptors protect against sepsis-induced mortality by dampening excessive inflammation. J. Immunol. 185, 542–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Csóka B., Németh Z. H., Virág L., Gergely P., Leibovich S. J., Pacher P., Sun C. X., Blackburn M. R., Vizi E. S., Deitch E. A., Haskó G. (2007) A2A adenosine receptors and C/EBPbeta are crucially required for IL-10 production by macrophages exposed to Escherichia coli. Blood 110, 2685–2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Németh Z. H., Csóka B., Wilmanski J., Xu D., Lu Q., Ledent C., Deitch E. A., Pacher P., Spolarics Z., Haskó G. (2006) Adenosine A2A receptor inactivation increases survival in polymicrobial sepsis. J. Immunol. 176, 5616–5626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Gils J. M., Derby M. C., Fernandes L. R., Ramkhelawon B., Ray T. D., Rayner K. J., Parathath S., Distel E., Feig J. L., Alvarez-Leite J. I., Rayner A. J., McDonald T. O., O’Brien K. D., Stuart L. M., Fisher E. A., Lacy-Hulbert A., Moore K. J. (2012) The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat. Immunol. 13, 136–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Remick D. G., Newcomb D. E., Bolgos G. L., Call D. R. (2000) Comparison of the mortality and inflammatory response of two models of sepsis: lipopolysaccharide vs. cecal ligation and puncture. Shock 13, 110–116 [DOI] [PubMed] [Google Scholar]
- 32.Gallardo-Soler A., Gómez-Nieto C., Campo M. L., Marathe C., Tontonoz P., Castrillo A., Corraliza I. (2008) Arginase I induction by modified lipoproteins in macrophages: a peroxisome proliferator-activated receptor-gamma/delta-mediated effect that links lipid metabolism and immunity. Mol. Endocrinol. 22, 1394–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Senthil M., Watkins A., Barlos D., Xu D. Z., Lu Q., Abungu B., Caputo F., Feinman R., Deitch E. A. (2007) Intravenous injection of trauma-hemorrhagic shock mesenteric lymph causes lung injury that is dependent upon activation of the inducible nitric oxide synthase pathway. Ann. Surg. 246, 822–830 [DOI] [PubMed] [Google Scholar]
- 34.Bone R. C., Balk R. A., Cerra F. B., Dellinger R. P., Fein A. M., Knaus W. A., Schein R. M., Sibbald W. J.; ACCP/SCCM Consensus Conference Committee (2009) Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. 1992. Chest 136(5, Suppl)e28. [DOI] [PubMed] [Google Scholar]
- 35.Ulloa L., Tracey K. J. (2005) The “cytokine profile”: a code for sepsis. Trends Mol. Med. 11, 56–63 [DOI] [PubMed] [Google Scholar]
- 36.Ulloa L., Brunner M., Ramos L., Deitch E. A. (2009) Scientific and clinical challenges in sepsis. Curr. Pharm. Des. 15, 1918–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiao W., Mindrinos M. N., Seok J., Cuschieri J., Cuenca A. G., Gao H., Hayden D. L., Hennessy L., Moore E. E., Minei J. P., Bankey P. E., Johnson J. L., Sperry J., Nathens A. B., Billiar T. R., West M. A., Brownstein B. H., Mason P. H., Baker H. V., Finnerty C. C., Jeschke M. G., López M. C., Klein M. B., Gamelli R. L., Gibran N. S., Arnoldo B., Xu W., Zhang Y., Calvano S. E., McDonald-Smith G. P., Schoenfeld D. A., Storey J. D., Cobb J. P., Warren H. S., Moldawer L. L., Herndon D. N., Lowry S. F., Maier R. V., Davis R. W., Tompkins R. G.; Inflammation and Host Response to Injury Large-Scale Collaborative Research Program (2011) A genomic storm in critically injured humans. J. Exp. Med. 208, 2581–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Antonioli L., Pacher P., Vizi E. S., Haskó G. (2013) CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 19, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Csóka B., Németh Z. H., Törő G., Koscsó B., Kókai E., Robson S. C., Enjyoji K., Rolandelli R. H., Erdélyi K., Pacher P., Haskó G. (2015) CD39 improves survival in microbial sepsis by attenuating systemic inflammation. FASEB J. 29, 25–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ali S. R., Timmer A. M., Bilgrami S., Park E. J., Eckmann L., Nizet V., Karin M. (2011) Anthrax toxin induces macrophage death by p38 MAPK inhibition but leads to inflammasome activation via ATP leakage. Immunity 35, 34–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eltzschig H. K., Eckle T., Mager A., Küper N., Karcher C., Weissmüller T., Boengler K., Schulz R., Robson S. C., Colgan S. P. (2006) ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ. Res. 99, 1100–1108 [DOI] [PubMed] [Google Scholar]
- 42.Bao Y., Chen Y., Ledderose C., Li L., Junger W. G. (2013) Pannexin 1 channels link chemoattractant receptor signaling to local excitation and global inhibition responses at the front and back of polarized neutrophils. J. Biol. Chem. 288, 22650–22657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schenk U., Westendorf A. M., Radaelli E., Casati A., Ferro M., Fumagalli M., Verderio C., Buer J., Scanziani E., Grassi F. (2008) Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci. Signal. 1, ra6 [DOI] [PubMed] [Google Scholar]
- 44.Gu B. J., Baird P. N., Vessey K. A., Skarratt K. K., Fletcher E. L., Fuller S. J., Richardson A. J., Guymer R. H., Wiley J. S. (2013) A rare functional haplotype of the P2RX4 and P2RX7 genes leads to loss of innate phagocytosis and confers increased risk of age-related macular degeneration. FASEB J. 27, 1479–1487 [DOI] [PubMed] [Google Scholar]
- 45.Lees M. P., Fuller S. J., McLeod R., Boulter N. R., Miller C. M., Zakrzewski A. M., Mui E. J., Witola W. H., Coyne J. J., Hargrave A. C., Jamieson S. E., Blackwell J. M., Wiley J. S., Smith N. C. (2010) P2X7 receptor-mediated killing of an intracellular parasite, Toxoplasma gondii, by human and murine macrophages. J. Immunol. 184, 7040–7046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chaves M. M., Marques-da-Silva C., Monteiro A. P., Canetti C., Coutinho-Silva R. (2014) Leukotriene B4 modulates P2X7 receptor-mediated Leishmania amazonensis elimination in murine macrophages. J. Immunol. 192, 4765–4773 [DOI] [PubMed] [Google Scholar]
- 47.Santana P. T., Benjamim C. F., Martinez C. G., Kurtenbach E., Takiya C. M., Coutinho-Silva R. (2015) The P2X7 Receptor contributes to the development of the exacerbated inflammatory response associated with sepsis [E-pub ahead of print]. J. Innate Immun. DOI:10.1159/000371388. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.